
Sarcomeric versus Non-Sarcomeric HCM
 , ,
, ,
Abstract
:1. Introduction
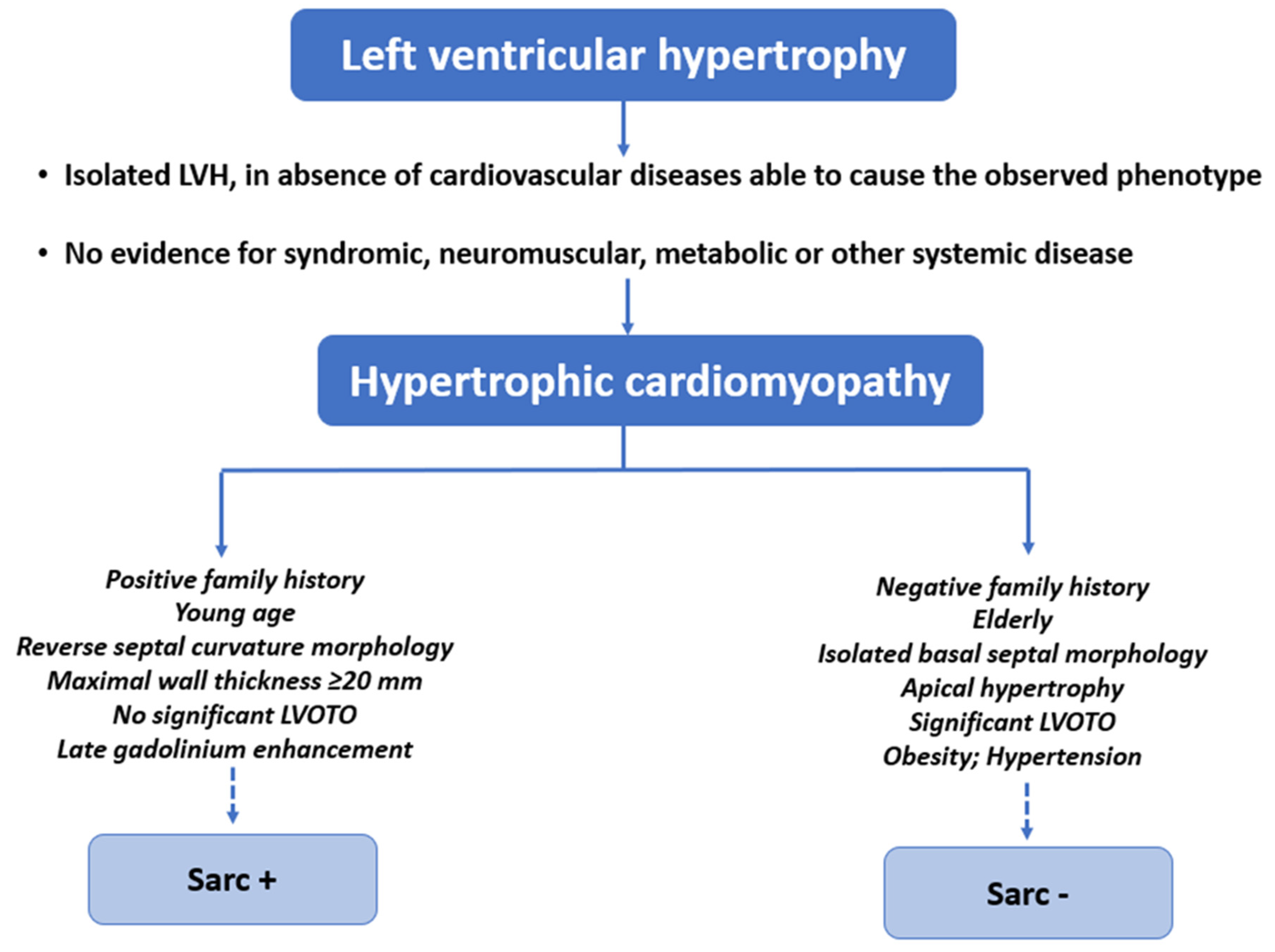
2. Definitions of Sarcomeric and Non-Sarcomeric HCM
3. Differential Diagnosis with Phenocopies
4. Genetics of Sarc+ and Sarc− HCM
- the genetic variant co-segregates with the HCM phenotype in the family and is absent in the phenotype-negative individuals;
- the genetic variant has prior evidence of pathogenicity, which means it has been documented as a disease-causing mutation in ≥1 patient in the published literature;
- the genetic variant is absent in the healthy population;
- the genetic variant is predicted (in silico or by functional studies) to cause major disruptions of the structure and function of the encoded protein.
5. HCM Caused by Mutations in Non-Sarcomeric Genes
6. Patients’ Demographic and Clinical Characteristics
7. Echocardiography and Cardiac Magnetic Resonance Findings
8. Clinical Presentation and Prognosis
- significant LGE at CMR (usually ≥15% of LV mass);
- LVEF < 50%;
- abnormal blood pressure response during exercise test;
- LV apical aneurysm;
- presence of a sarcomeric pathogenic variant [76].
9. Phenotypic Variability and Personalized Clinical Approach
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy. Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisterfer-Lowrance, A.A.T.; Kass, S.; Tanigawa, G.; Vosberg, H.-P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A β Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Seidman, C.E.; Seidman, J.G. Identifying Sarcomere Gene Mutations in Hypertrophic Cardiomyopathy. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, W.E.; Elliott, P.M. Changing Concepts in Heart Muscle Disease: The Evolving Understanding of Hypertrophic Car-diomyopathy. Heart 2022, 108, 768–773. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Zenovich, A.G.; Link, M.S.; Pandian, N.G.; Kuvin, J.T.; Nistri, S.; Cecchi, F.; Udelson, J.E.; Maron, B.J. Hypertrophic Cardiomyopathy Is Predominantly a Disease of Left Ventricular Outflow Tract Obstruction. Circulation 2006, 114, 2232–2239. [Google Scholar] [CrossRef] [Green Version]
- Daw, E.W.; Chen, S.N.; Czernuszewicz, G.; Lombardi, R.; Lu, Y.; Ma, J.; Roberts, R.; Shete, S.; Marian, A.J. Genome-Wide Mapping of Modifier Chromosomal Loci for Human Hypertrophic Cardiomyopathy. Hum. Mol. Genet. 2007, 16, 2463–2471. [Google Scholar] [CrossRef]
- Kumar, A.; Rani, B.; Sharma, R.; Kaur, G.; Prasad, R.; Bahl, A.; Khullar, M. ACE2, CALM3 and TNNI3K Polymorphisms as Potential Disease Modifiers in Hypertrophic and Dilated Cardiomyopathies. Mol. Cell. Biochem. 2018, 438, 167–174. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Ex-pressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Tadros, R.; Francis, C.; Xu, X.; Vermeer, A.M.C.; Harper, A.R.; Huurman, R.; Kelu Bisabu, K.; Walsh, R.; Hoorntje, E.T.; te Rijdt, W.P.; et al. Shared Genetic Pathways Contribute to Risk of Hypertrophic and Dilated Cardiomyopathies with Opposite Di-rections of Effect. Nat. Genet. 2021, 53, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Udelson, J.E.; Maron, M.S. Clinical Spectrum and Management of Heart Failure in Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018, 6, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 76, e159–e240. [Google Scholar] [CrossRef]
- Neubauer, S.; Kolm, P.; Ho, C.Y.; Kwong, R.Y.; Desai, M.Y.; Dolman, S.F.; Appelbaum, E.; Desvigne-Nickens, P.; DiMarco, J.P.; Friedrich, M.G.; et al. Distinct Subgroups in Hypertrophic Cardiomyopathy in the NHLBI HCM Registry. J. Am. Coll. Cardiol. 2019, 74, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of Clinical Genetic Testing of 2,912 Probands with Hypertrophic Cardiomyopathy: Expanded Panels Offer Limited Additional Sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [Green Version]
- Pieroni, M.; Ciabatti, M.; Saletti, E.; Tavanti, V.; Santangeli, P.; Martinese, L.; Liistro, F.; Olivotto, I.; Bolognese, L. Beyond Sarcomeric Hypertrophic Cardiomyopathy: How to Diagnose and Manage Phenocopies. Curr. Cardiol. Rep. 2022, 24, 1567–1585. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, e212–e260. [Google Scholar] [CrossRef] [Green Version]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Gimeno-Blanes, J.; Helio, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic Work-up in Cardiomyopathies: Bridging the Gap between Clinical Phenotypes and Final Diagnosis. A Position Statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- Losi, M.A.; Nistri, S.; Galderisi, M.; Betocchi, S.; Cecchi, F.; Olivotto, I.; Agricola, E.; Ballo, P.; Buralli, S.; D’Andrea, A.; et al. Echo-cardiography in patients with hypertrophic cardiomyopathy: Usefulness of old and new techniques in the diagnosis and pathophysiological assessment. Cardiovasc Ultrasound. 2010, 17, 7. [Google Scholar] [CrossRef]
- Hoffmann, B.; Beck, M.; Sunder-Plassmann, G.; Borsini, W.; Ricci, R.; Mehta, A. Nature and Prevalence of Pain in Fabry Disease and Its Response to Enzyme Replacement Therapy—A Retrospective Analysis From the Fabry Outcome Survey. Clin. J. Pain 2007, 23, 535–542. [Google Scholar] [CrossRef]
- Karen, J.K.; Hale, E.K.; Ma, L. Angiokeratoma Corporis Diffusum (Fabry Disease). Dermatol. Online J. 2005, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Review: Danon Disease: Review of Natural History and Recent Advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef]
- Cipriani, A.; de Michieli, L.; Porcari, A.; Licchelli, L.; Sinigiani, G.; Tini, G.; Zampieri, M.; Sessarego, E.; Argirò, A.; Fumagalli, C.; et al. Low QRS Voltages in Cardiac Amyloidosis. JACC CardioOncol. 2022, 4, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Melas, M.; Beltsios, E.T.; Adamou, A.; Koumarelas, K.; McBride, K.L. Molecular Diagnosis of Hypertrophic Cardiomyopathy (HCM): In the Heart of Cardiac Disease. J. Clin. Med. 2022, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef] [PubMed]
- Osio, A.; Tan, L.; Chen, S.N.; Lombardi, R.; Nagueh, S.F.; Shete, S.; Roberts, R.; Willerson, J.T.; Marian, A.J. Myozenin 2 Is a Novel Gene for Human Hypertrophic Cardiomyopathy. Circ. Res. 2007, 100, 766–768. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Lombardi, R.; Mirra, B.; Barretta, F.; Esposito, M.V.; Uomo, F.; Caiazza, M.; Monda, E.; Losi, M.A.; Limongelli, G.; et al. Next-Generation Sequencing Gene Panels in Inheritable Cardiomyopathies and Channelopathies: Prevalence of Pathogenic Variants and Variants of Unknown Significance in Uncommon Genes. Biomolecules 2022, 12, 1417. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.v.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the Genetic Architecture of Hypertrophic Cardiomyopathy: Re-Evaluating the Role of Non-Sarcomeric Genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Lipari, M.; Wypasek, E.; Karpinski, M.; Tomkiewicz-Pajak, L.; Laino, L.; Binni, F.; Giannarelli, D.; Rubis, P.; Petkow-Dimitrow, P.; Undas, A.; et al. Identification of a Variant Hotspot in MYBPC3 and of a Novel CSRP3 Autosomal Recessive Alteration in a Cohort of Polish Cases Affected by Hypertrophic Cardiomyopathy. Pol. Arch. Intern. Med. 2020, 130, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janin, A.; Bessière, F.; Chauveau, S.; Chevalier, P.; Millat, G. First Identification of Homozygous Truncating CSRP3 Variants in Two Unrelated Cases with Hypertrophic Cardiomyopathy. Gene 2018, 676, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Delgado, M.; Gonzalez-Lopez, E.; Garcia-Guereta, L.; Ortega-Molina, M.; Gonzalez-Vioque, E.; Cobo-Marcos, M.; Alonso-Pulpon, L.; Garcia-Pavia, P. Adverse Clinical Course and Poor Prognosis of Hypertrophic Cardiomyopathy Due to Mutations in FHL1. Int. J. Cardiol. 2015, 191, 194–197. [Google Scholar] [CrossRef]
- Friedrich, F.W.; Wilding, B.R.; Reischmann, S.; Crocini, C.; Lang, P.; Charron, P.; Müller, O.J.; McGrath, M.J.; Vollert, I.; Hansen, A.; et al. Evidence for FHL1 as a Novel Disease Gene for Isolated Hypertrophic Cardiomyopathy. Hum. Mol. Genet. 2012, 21, 3237–3254. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Wang, J.; Zhang, C.; Wu, G.; Zhu, C.; Tang, B.; Zou, Y.; Huang, X.; Hui, R.; Song, L.; et al. Mutation Profile of FLNC Gene and Its Prognostic Relevance in Patients with Hypertrophic Cardiomyopathy. Mol. Genet. Genomic. Med. 2018, 6, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Gómez, J.; Lorca, R.; Reguero, J.R.; Morís, C.; Martín, M.; Tranche, S.; Alonso, B.; Iglesias, S.; Alvarez, V.; Díaz-Molina, B.; et al. Screening of the Filamin C Gene in a Large Cohort of Hypertrophic Cardiomyopathy Patients. Circ. Cardiovasc. Genet. 2017, 10, e001584. [Google Scholar] [CrossRef] [Green Version]
- Verdonschot, J.A.J.; Vanhoutte, E.K.; Claes, G.R.F.; Helderman-van den Enden, A.T.J.M.; Hoeijmakers, J.G.J.; Hellebrekers, D.M.E.I.; Haan, A.; Christiaans, I.; Lekanne Deprez, R.H.; Boen, H.M.; et al. A Mutation Update for the FLNC Gene in Myo-pathies and Cardiomyopathies. Hum. Mutat. 2020, 41, 1091–1111. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, J.P.; Lopes, L.R.; Perez-Barbeito, M.; Cazón-Varela, L.; Torre-Carpente, M.M.; Sonicheva-Paterson, N.; de Uña-Iglesias, D.; Quinn, E.; Kuzmina-Krutetskaya, S.; Garrote, J.A.; et al. Deletions of Specific Exons of FHOD3 Detected by Next-generation Sequencing Are Associated with Hypertrophic Cardiomyopathy. Clin. Genet. 2020, 98, 86–90. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef]
- Vanninen, S.U.M.; Leivo, K.; Seppälä, E.H.; Aalto-Setälä, K.; Pitkänen, O.; Suursalmi, P.; Annala, A.-P.; Anttila, I.; Alastalo, T.-P.; Myllykangas, S.; et al. Heterozygous Junctophilin-2 (JPH2) p.(Thr161Lys) Is a Monogenic Cause for HCM with Heart Failure. PLoS ONE 2018, 13, e0203422. [Google Scholar] [CrossRef] [PubMed]
- Parisi, V.; Chiti, C.; Graziosi, M.; Pasquale, F.; Ditaranto, R.; Minnucci, M.; Biffi, M.; Potena, L.; Girolami, F.; Baldovini, C.; et al. Phospholamban Cardiomyopathy: Unveiling a Distinct Phenotype Through Heart Failure Stages Progression. Circ. Cardiovasc. Imaging 2022, 15, e014232. [Google Scholar] [CrossRef] [PubMed]
- Medin, M.; Hermida-Prieto, M.; Monserrat, L.; Laredo, R.; Rodriguez-Rey, J.C.; Fernandez, X.; Castro-Beiras, A. Mutational Screening of Phospholamban Gene in Hypertrophic and Idiopathic Dilated Cardiomyopathy and Functional Study of the PLN -42 C>G Mutation. Eur. J. Heart Fail. 2007, 9, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Czernuszewicz, G.; Tan, Y.; Lombardi, R.; Jin, J.; Willerson, J.T.; Marian, A.J. Human Molecular Genetic and Functional Studies Identify TRIM63, Encoding Muscle RING Finger Protein 1, as a Novel Gene for Human Hypertrophic Cardiomyopathy. Circ. Res. 2012, 111, 907–919. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Mendiguchía, J.; Ochoa, J.P.; Palomino-Doza, J.; Domínguez, F.; Díez-López, C.; Akhtar, M.; Ramiro-León, S.; Cle-mente, M.M.; Pérez-Cejas, A.; Robledo, M.; et al. Mutations in TRIM63 Cause an Autosomal-Recessive Form of Hypertrophic Cardiomyopathy. Heart 2020, 106, 1342–1348. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Abramsson, A.; Osborn, D.P.S.; Danielsson, O.; Fazlinezhad, A.; Nilipour, Y.; Hübbert, L.; Nennesmo, I.; Visuttijai, K.; Bharj, J.; et al. Cardiomyopathy with Lethal Arrhythmias Associated with Inactivation of KLHL24. Hum. Mol. Genet. 2019, 28, 1919–1929. [Google Scholar] [CrossRef]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Non-familial Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [Green Version]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.; Arscott, P.; Concannon, M.; Saberi, S.; Day, S.M.; Yashar, B.M.; Helms, A.S. Genetic Testing Impacts the Utility of Prospective Familial Screening in Hypertrophic Cardiomyopathy through Identification of a Nonfamilial Subgroup. Genet. Med. 2018, 20, 69–75. [Google Scholar] [CrossRef] [Green Version]
- de Feria, A.E.; Kott, A.E.; Becker, J.R. Sarcomere Mutation Negative Hypertrophic Cardiomyopathy Is Associated with Ageing and Obesity. Open Heart 2021, 8, e001560. [Google Scholar] [CrossRef]
- Wong, C.Y.; O’Moore-Sullivan, T.; Leano, R.; Byrne, N.; Beller, E.; Marwick, T.H. Alterations of Left Ventricular Myocardial Characteristics Associated With Obesity. Circulation 2004, 110, 3081–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodiwiss, A.J.; Libhaber, C.D.; Majane, O.H.I.; Libhaber, E.; Maseko, M.; Norton, G.R. Obesity Promotes Left Ventricular Concentric Rather Than Eccentric Geometric Remodeling and Hypertrophy Independent of Blood Pressure. Am. J. Hypertens 2008, 21, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Aurigemma, G.P.; de Simone, G.; Fitzgibbons, T.P. Cardiac Remodeling in Obesity. Circ. Cardiovasc. Imaging 2013, 6, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, C.; Maurizi, N.; Day, S.M.; Ashley, E.A.; Michels, M.; Colan, S.D.; Jacoby, D.; Marchionni, N.; Vincent-Tompkins, J.; Ho, C.Y.; et al. Association of Obesity With Adverse Long-Term Outcomes in Hypertrophic Cardiomyopathy. JAMA Cardiol. 2020, 5, 65. [Google Scholar] [CrossRef]
- Ippisch, H.M.; Inge, T.H.; Daniels, S.R.; Wang, B.; Khoury, P.R.; Witt, S.A.; Glascock, B.J.; Garcia, V.F.; Kimball, T.R. Reversibility of Cardiac Abnormalities in Morbidly Obese Adolescents. J. Am. Coll. Cardiol. 2008, 51, 1342–1348. [Google Scholar] [CrossRef] [Green Version]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.M.; Will, M.L.; Gersh, B.J.; Kruisselbrink, T.M.; Ommen, S.R.; Ackerman, M.J. Characterization of a Phenotype-Based Genetic Test Prediction Score for Unrelated Patients With Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2014, 89, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Binder, J.; Ommen, S.R.; Gersh, B.J.; van Driest, S.L.; Tajik, A.J.; Nishimura, R.A.; Ackerman, M.J. Echocardiography-Guided Genetic Testing in Hypertrophic Cardiomyopathy: Septal Morphological Features Predict the Presence of Myofilament Mutations. Mayo Clin. Proc. 2006, 81, 459–467. [Google Scholar] [CrossRef]
- Pelliccia, F.; Alfieri, O.; Calabrò, P.; Cecchi, F.; Ferrazzi, P.; Gragnano, F.; Kaski, J.P.; Limongelli, G.; Maron, M.; Rapezzi, C.; et al. Multidisciplinary Evaluation and Management of Obstructive Hypertrophic Cardiomyopathy in 2020: Towards the HCM Heart Team. Int. J. Cardiol. 2020, 304, 86–92. [Google Scholar] [CrossRef]
- Sakamoto, T.; Tei, C.; Murayama, M.; Ichiyasu, H.; Hada, Y.; Hayashi, T.; Amano, K. Giant T Wave Inversion as a Manifestation of Asymmetrical Apical Hypertrophy (AAH) of the Left Ventricle. Jpn. Heart J. 1976, 17, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Kitaoka, H.; Okawa, M.; Hirota, T.; Hoshikawa, E.; Hayato, K.; Yamasaki, N.; Matsumura, Y.; Yabe, T.; Nishinaga, M.; et al. Clinical Profiles of Hypertrophic Cardiomyopathy With Apical Phenotype Comparison of Pure-Apical Form and Distal-Dominant Form. Circ. J. 2009, 73, 2330–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarich, K.W.; Attenhofer Jost, C.H.; Binder, J.; Connolly, H.M.; Scott, C.G.; Freeman, W.K.; Ackerman, M.J.; Nishimura, R.A.; Tajik, A.J.; Ommen, S.R. Risk of Death in Long-Term Follow-Up of Patients With Apical Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2013, 111, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- Basso, C. Hypertrophic Cardiomyopathy and Sudden Death in the Young: Pathologic Evidence of Myocardial Ischemia. Hum. Pathol. 2000, 31, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Shirani, J.; Pick, R.; Roberts, W.C.; Maron, B.J. Morphology and Significance of the Left Ventricular Collagen Network in Young Patients with Hypertrophic Cardiomyopathy and Sudden Cardiac Death. J. Am. Coll. Cardiol. 2000, 35, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, L.; Mahrholdt, H.; Wagner, A.; Choi, K.M.; Elliott, M.D.; Klocke, F.J.; Bonow, R.O.; Judd, R.M.; Kim, R.J. Myocardial Scarring in Asymptomatic or Mildly Symptomatic Patients with Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Varnava, A.M.; Elliott, P.M.; Mahon, N.; Davies, M.J.; McKenna, W.J. Relation between Myocyte Disarray and Outcome in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2001, 88, 275–279. [Google Scholar] [CrossRef]
- Varnava, A.M. Hypertrophic Cardiomyopathy: The Interrelation of Disarray, Fibrosis, and Small Vessel Disease. Heart 2000, 84, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Abbasi, S.A.; Neilan, T.G.; Shah, R.v.; Chen, Y.; Heydari, B.; Cirino, A.L.; Lakdawala, N.K.; Orav, E.J.; González, A.; et al. T1 Measurements Identify Extracellular Volume Expansion in Hypertrophic Cardiomyopathy Sarcomere Mutation Car-riers With and Without Left Ventricular Hypertrophy. Circ. Cardiovasc. Imaging 2013, 6, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, M.S.; Chan, R.H.; Hausvater, A.; Wang, W.; Rastegar, H.; Maron, B.J. Interaction of Adverse Disease Related Pathways in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2017, 120, 2256–2264. [Google Scholar] [CrossRef]
- Hodges, K.; Rivas, C.G.; Aguilera, J.; Borden, R.; Alashi, A.; Blackstone, E.H.; Desai, M.Y.; Smedira, N.G. Surgical Management of Left Ventricular Outflow Tract Obstruction in a Specialized Hypertrophic Obstructive Cardiomyopathy Center. J. Thorac. Cardiovasc. Surg. 2019, 157, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Evolution of Risk Stratification and Sudden Death Prevention in Hypertrophic Cardi-omyopathy: Twenty Years with the Implantable Cardioverter-Defibrillator. Heart Rhythm. 2021, 18, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Rowin, E.J.; Hausvater, A.; Link, M.S.; Abt, P.; Gionfriddo, W.; Wang, W.; Rastegar, H.; Estes, N.A.M.; Maron, M.S.; Maron, B.J. Clinical Profile and Consequences of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Circulation 2017, 136, 2420–2436. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, B.J.; Carrick, R.T.; Patel, P.P.; Koethe, B.; Wells, S.; Maron, M.S. Outcomes in Patients With Hypertrophic Cardiomyopathy and Left Ventricular Systolic Dysfunction. J. Am. Coll. Cardiol. 2020, 75, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Watkins, H. Time to Think Differently About Sarcomere-Negative Hypertrophic Cardiomyopathy. Circulation 2021, 143, 2415–2417. [Google Scholar] [CrossRef]
- Tadros, R.; Zheng, S.L.; Grace, C.; Jordà, P.; Francis, C.; Jurgens, S.J.; Thomson, K.L.; Harper, A.R.; Ormondroyd, E.; West, D.M.; et al. Large Scale Genome-Wide Association Analyses Identify Novel Genetic Loci and Mechanisms in Hypertrophic Car-diomyopathy. medRxiv 2023. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Function of the Encoded Protein | Level of Evidence for HCM Association | Phenotypes and Mode of Inheritance | References |
|---|---|---|---|---|
| CSRP3 | regulation of myogenesis; maintenance of myocyte cytoskeleton; mechano-signaling and transduction | Moderate | HCM and rarely DCM (AD and AR) | [32,33] |
| FHL1 | Biomechanical sensing; regulation of sarcomere stiffness, hypertrophy, ion channels | Moderate | HCM and FHL1-related myopathies (X-linked) | [34,35] |
| FLNC | Crosslinking of actin filaments and interaction with Z-disc and sarcolemma | Limited | HCM, DCM and ACM (AD) | [36,37,38] |
| FHOD3 | Promoting polymerization of actin thin filaments | Limited | HCM and DCM (AD) | [39,40] |
| JPH2 | Coupling of transverse tubule associated L-type Ca2+ channels with RYR2 | Moderate | HCM (AD) and DCM (AR) | [41] |
| PLN | Regulation of sarco/endoplasmic reticulum Ca2+ ATPase activity | Limited | HCM, DCM and ACM (AD and AR) | [42,43] |
| TRIM63 | Regulation of sarcomeric protein degradation | Limited | HCM (AD and AR) | [44,45] |
| KLHL24 | Regulation of the balance between intermediate filament stability and degradation | Limited | Epidermolysis bullosa simplex with DCM (AD) HCM (AR) | [46] |
| Patient Characteristics | Sarc+ | Sarc− |
|---|---|---|
| Male sex | +− | ++ |
| Age at diagnosis | 46 ± 12 | 51 ± 10 |
| Hypertension | +− | ++ |
| Obesity | +− | ++ |
| Comorbidities | + | ++ |
| Susteined VT | ++ | +− |
| Sudden death event | ++ | − |
| High HCM−risk score | ++ | − |
| ICD | ++ | − |
| Atrial fibrillation | +− | +− |
| Echocardiography | ||
| Maximal wall thickness | ++ | +− |
| Isolated basal septal hypertrophy | +− | ++ |
| Apical hypertrophy | +− | ++ |
| Reverse septal morphology | ++ | − |
| LVOTO | +− | ++ |
| Dilated LA | +− | +− |
| Cardiac MRI | ||
| Presence of LGE | ++ | −+ |
| ECV expansion | ++ | −+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrelli, F.; Losi, M.A.; Canciello, G.; Todde, G.; Perillo, E.F.; Ordine, L.; Frisso, G.; Esposito, G.; Lombardi, R. Sarcomeric versus Non-Sarcomeric HCM. Cardiogenetics 2023, 13, 92-105. https://doi.org/10.3390/cardiogenetics13020009
Borrelli F, Losi MA, Canciello G, Todde G, Perillo EF, Ordine L, Frisso G, Esposito G, Lombardi R. Sarcomeric versus Non-Sarcomeric HCM. Cardiogenetics. 2023; 13(2):92-105. https://doi.org/10.3390/cardiogenetics13020009
Chicago/Turabian StyleBorrelli, Felice, Maria Angela Losi, Grazia Canciello, Gaetano Todde, Errico Federico Perillo, Leopoldo Ordine, Giulia Frisso, Giovanni Esposito, and Raffaella Lombardi. 2023. "Sarcomeric versus Non-Sarcomeric HCM" Cardiogenetics 13, no. 2: 92-105. https://doi.org/10.3390/cardiogenetics13020009