Effect of Polymers on the Physicochemical Properties and Biological Performance of Fenoprofen Calcium Dihydrate-Triacetyl-β-Cyclodextrin Complex

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Polymer-Drug Complex Blend
2.2.2. In Vitro Dissolution Studies
2.2.3. Characterization of Drug Complex and Polymer-Drug Complex Blends
Differential Scanning Calorimetry (DSC)
Fourier-Transform Infrared Spectroscopy (FT-IR)
X-ray Diffractometry (XRD)
Scanning Electron Microscopy (SEM)
2.3. In Vivo Studies
2.3.1. Assessment of Anti-Inflamamtory Activity (Carrageenan-Induced Edema)
2.3.2. Assessment of the Analgesic Activity (Writhing Test)
3. Results and Discussion
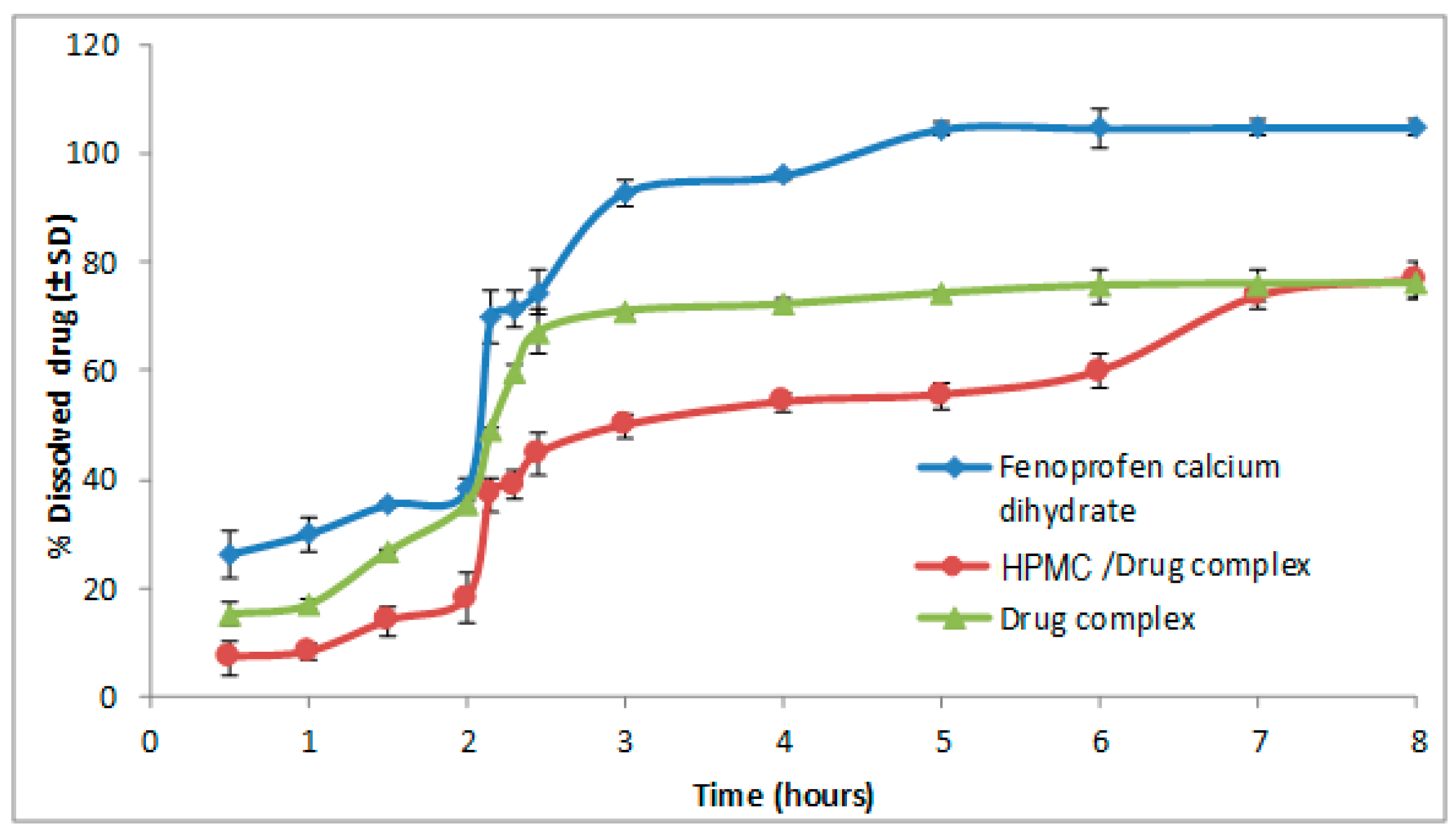
3.1. Effect of Polymers on the Dissolution of Fenoprofen Calcium Dihydrate
3.2. Characterization of Drug Complex and Polymers/Drug Complex
3.2.1. Differential Scanning Calorimetry (DSC)
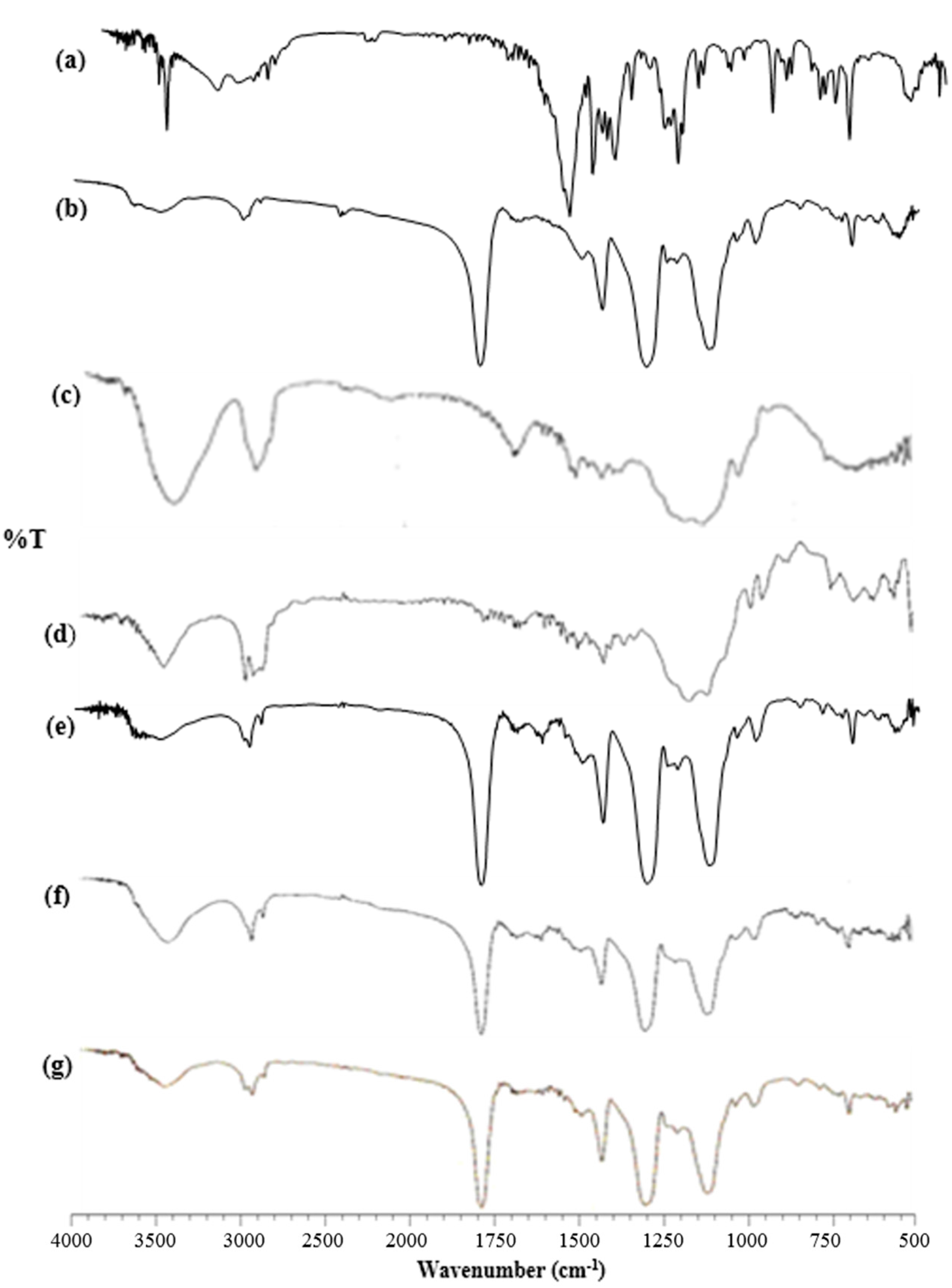
3.2.2. Fourier Transform-Infrared Spectroscopy (FT-IR)
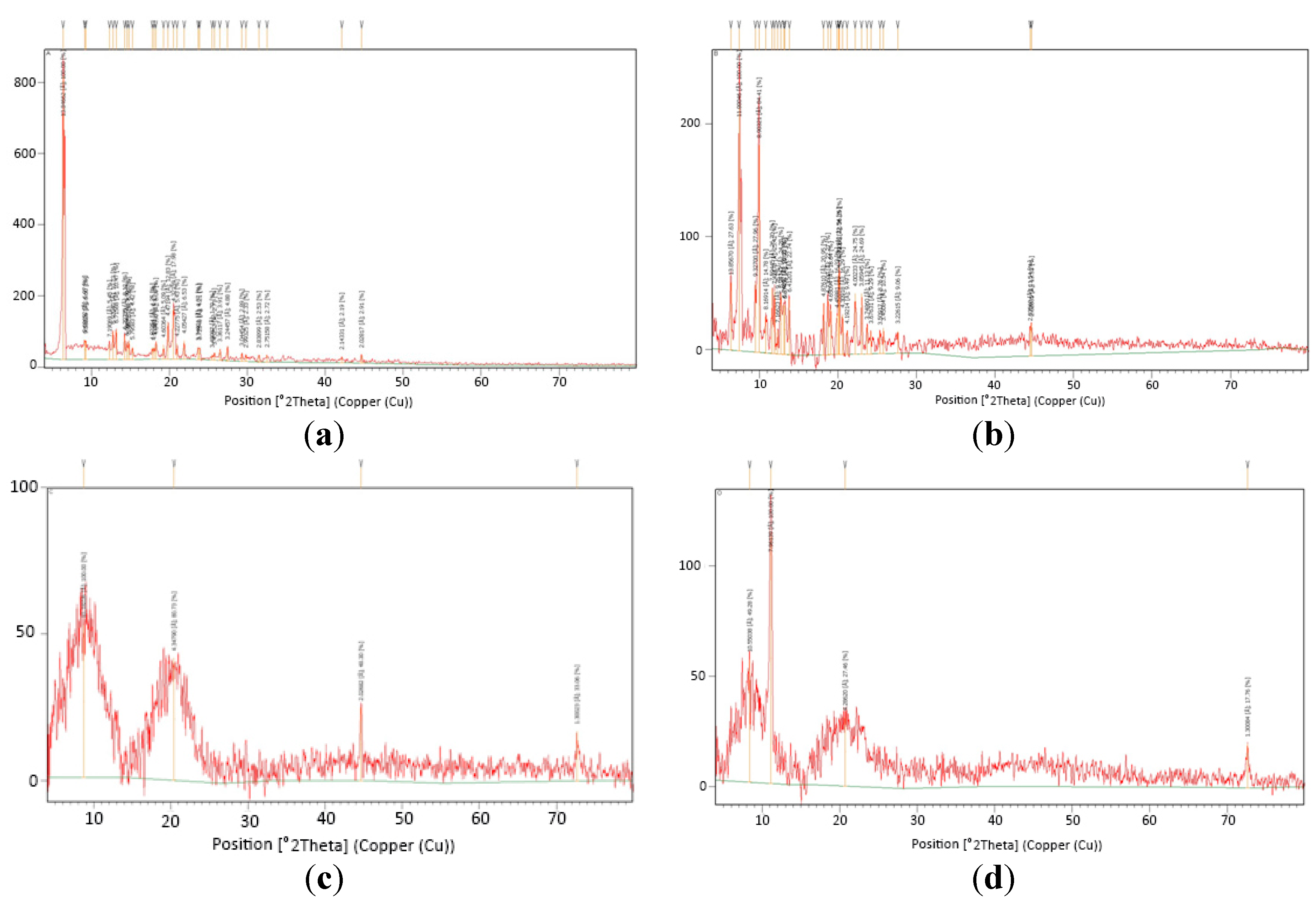
3.2.3. X-ray Diffractometry (XRD)
3.2.4. Scanning Electron Microscopy (SEM)
3.3. In Vivo Studies
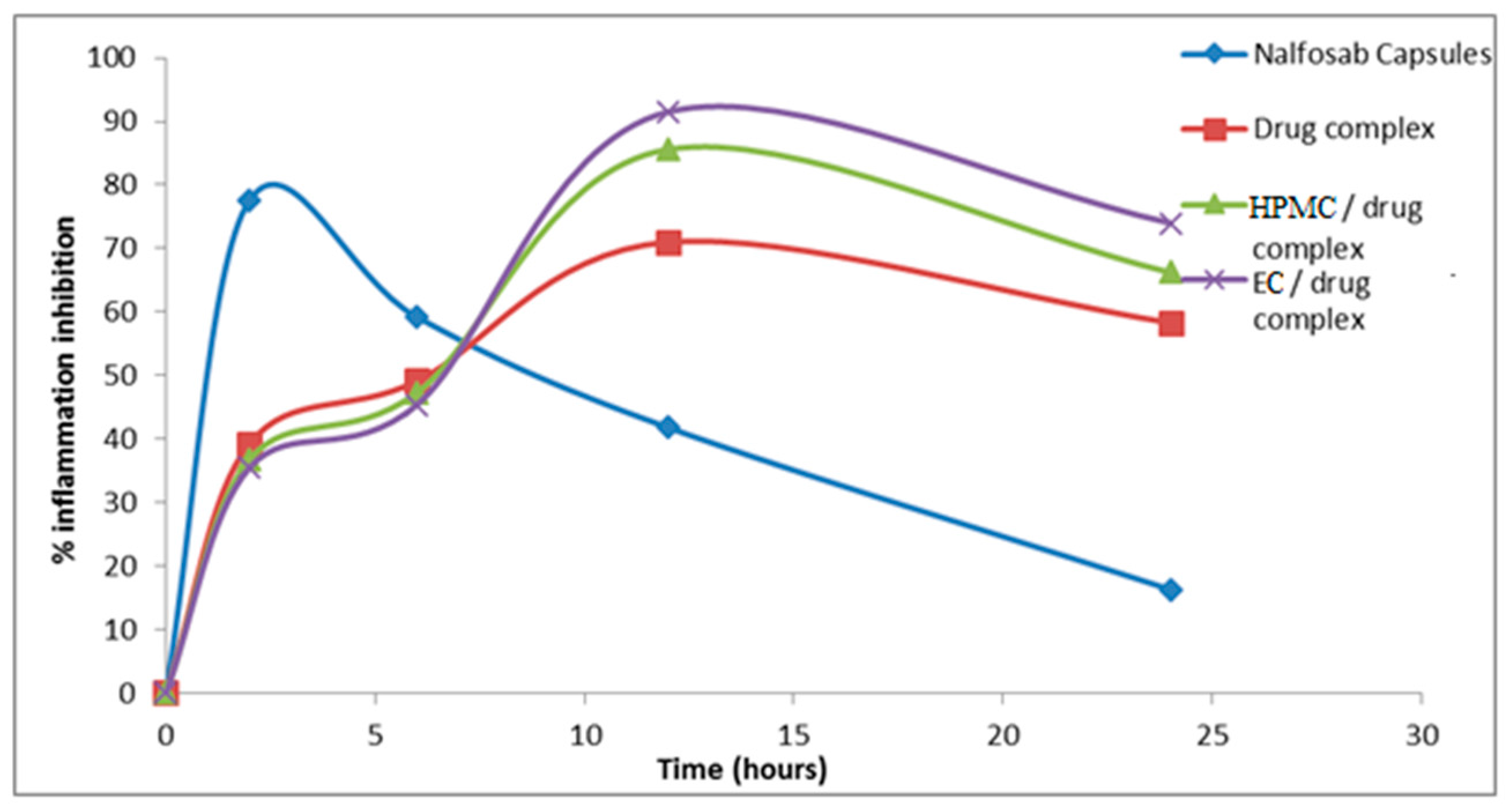
3.3.1 Assessment of the Anti-inflammatory Activity
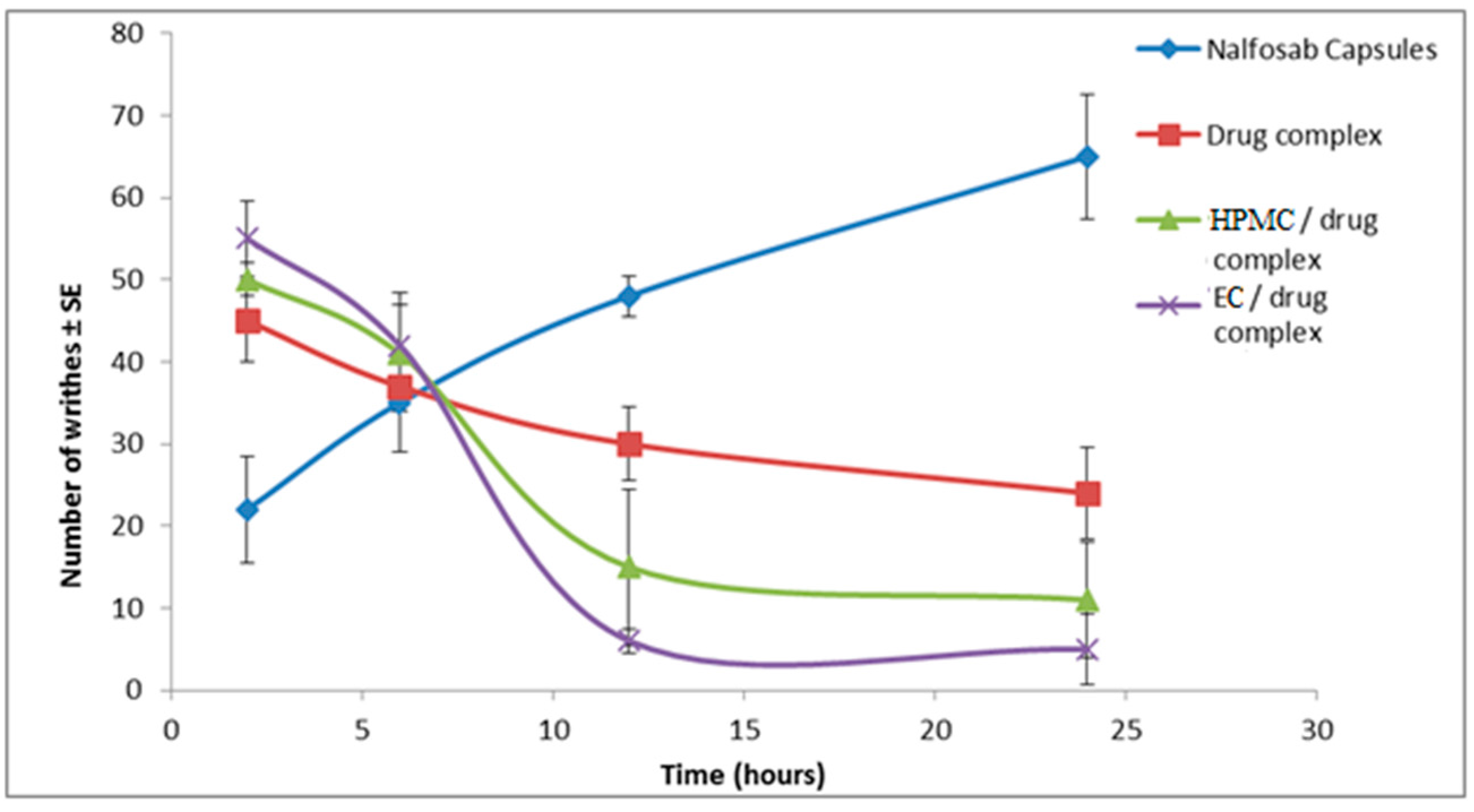
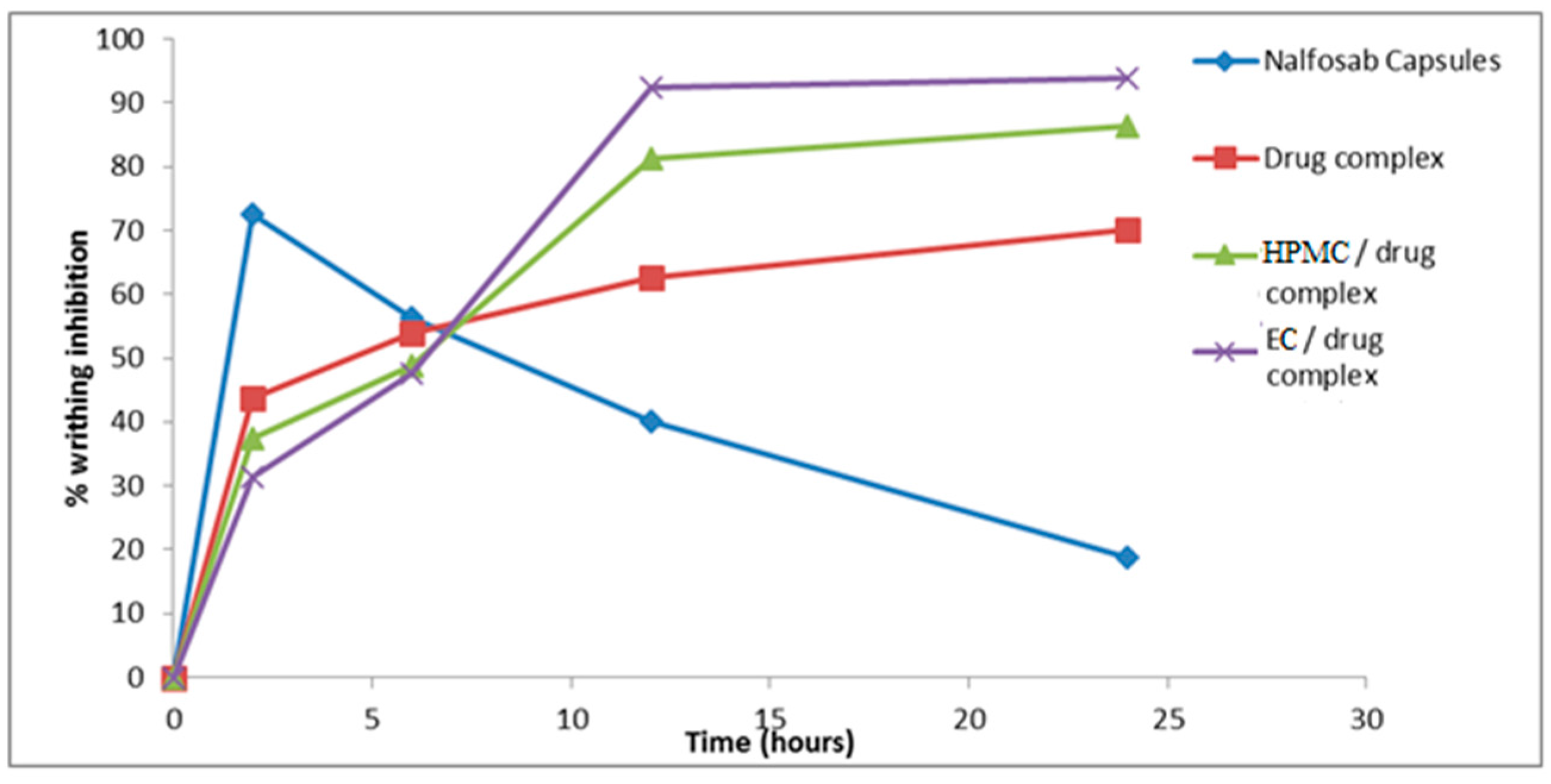
3.3.2 Assessment of the Analgesic Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations:
| Abbreviation | Meaning |
| % | Percentage |
| C | Degree Centigrade |
| µg | Microgram |
| µm | Micrometer |
| AUC | Area Under the Concentration Curve |
| CD | Cyclodextrin |
| C–H | Carbon Hydrogen bond |
| DE | Dissolution Efficiency |
| DSC | Differential Scanning Calorimetry |
| EC | Ethyl cellulose |
| FCD | Fenoprofen Calcium Dehydrate |
| FT-IR | Fourier Transform Infrared Spectroscopy |
| HPMC | Hydroxypropylmethyl Cellulose |
| Ip | Intraperitoneal |
| KBr | Potassium Bromide |
| KV | Kill Voltage |
| MC | Methylcellulose |
| M | Mole |
| MCE | Methocel Cellulose Ester |
| MW | Molecular Weight |
| Ma | Milliamper |
| Mwt/Mwt | Molecular Weight/Molecular Weight |
| N | Normal |
| Nm | Nanometer |
| OH | Hydroxyl |
| Ph | Hydrogen Ion Concentration |
| r.p.m | Rotation Per Minute |
| Tmax | Time Of Maximum Plasma Concentration. |
| TA-β-CD | Triacetyl-β-Cyclodextrin |
| USP | United States Pharmacopeia |
| V | Vibration |
| V–Me | Vibrational–Methyl |
| V–OH | Vibrational–OH |
| W | Watt |
| W/V | Wight/Volume |
| W/V | Wight/Volume |
| W/W | Weight/Weight |
| λmax | Maximum Wavelength |
References
- Poggi, J.C.; Barissa, G.R.; Donadi, E.A.; Foss, M.C.; de Queiroz Cuha, F.; Lanchote, V.L.; Reis, M.L.D. Pharmacodynamics, chiral pharmacokinetics and pharmacokinetic-pharmacodynamic modeling of Fenoprofen in patients with diabetes mellitus. J. Clin. Pharmacol. 2006, 46, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, K. Martindale the Complete Drug Reference: Thirty-Second ed.; Pharmaceutical Press: St. Louis, MO, USA, 1999; p. 2315. [Google Scholar]
- MethocelTM Cellulose Ethers Technical Handbook: The Dow Chemical Company. Available online: http://msdssearch.dow.com/PublishedLiteratureDOWCOM/dh_096d/0901b8038096db95.pdf?filepath=productsafety/pdfs/noreg/233-00327.pdf&fromPage=GetDoc (accessed on 19 September 2013).
- Reza, M.S.; Quadir, M.A.; Haid, S.S. Comparative evaluation of plastic, hydrophobic and hydrophilic polymers as matrices for controlled-release drug delivery. J. Pharm. Pharm. Sci. 2003, 6, 274–291. [Google Scholar]
- Study of effect of polymer viscosity and polymer: Excipient ratio on drug-release patterns from swellable matrices. Drug Deliv. Technol. 2005, 5, 1–7.
- Mallipeddi, R.; Saripella, K.K.; Neau, S.H. Use of coarse ethylcellulose and PEO in beads produced by extrusion–spheronization. Int. J. Pharm. 2010, 385, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, R.; Saripella, K.K.; Neau, S.H. Use of fine particle ethylcellulose as the diluent in the production of pellets by extrusion-spheronization. Saudi Pharm. J. 2014, 22, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Hamedelniel, I.E.; Bajdik, J.; Sovány, T.; Pintye-Hódi, K. Delayed release matrix pellet 1006–1010 preparation containing an alkalizing pore-former agent. Chem. Eng. Res. Des. 2010, 89, 1006–1010. [Google Scholar] [CrossRef]
- Pearnchob, N.; Bodmeier, R. Coating of pellets with micronized ethylcellulose particles by a dry powder coating technique. Int. J. Pharm. 2003, 268, 1–11. [Google Scholar]
- Sadeghi, F.; Ford, J.L.; Rajabi-Siahboomi, A. The influence of drug type on the release profiles from Surelease-coated pellets. Int. J. Pharm. 2003, 254, 123–135. [Google Scholar] [CrossRef]
- McConnell, E.L.; Short, M.D.; Basit, A.W. An in vivo comparison of intestinal pH and bacteria as physiological trigger mechanisms for colonic targeting in man. J. Control. Release 2008, 130, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.O.; Nahdy, M.A.; Makram, T.S.; Mosallam, S. Effect of the hydrophobic nature of triacetyl-β-cyclodextrin on the dissolution properties of Fenoprofen Calcium Dihydrate prepared by kneading and co-evaporating methods. Eur. J. Biomed. Pharm. Sci. 2016, 3, 127–130. [Google Scholar]
- Wang, Z.; Horikawa, T.; Hirayama, F.; Uekama, K. Design and in vitro evaluation of a modified-release oral dosage form of nifedipine by hybridization of hydroxypropyl-beta-cyclodextrin and hydroxypropyl-cellulose. J. Pharm. Pharmacol. 1993, 45, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.M.; Teresa Viera, M.; Veiga, F.J. Physicochemical characterization and in vitro dissolution behavior of nicardipine-cyclodextrins inclusion compounds. Eur. J. Pharm. Sci. 2002, 15, 79–88. [Google Scholar] [CrossRef]
- Chowdary, K.P.R.; Reddy, G.K. Complexes of nifedipine with β-and hydroypropyl-β-cyclodextrin in the design of nifedipine SR tablets. Ind. J. Pharm. Sci. 2002, 64, 142–146. [Google Scholar]
- Burgos, A.E.; Belchior, J.C.; Sinisterra, R.D. Controlled release of rhodium (II) carboxylates and their association complexes with cyclodextrins from hydroxyapatite matrix. Biomaterials 2002, 23, 2519–2526. [Google Scholar] [CrossRef]
- Corti, G.; Cirri, M.; Maestrelli, F.; Mennini, N.; Mura, P. Sustained-release matrix tablets of metformin hydrochloride in combination with tiracetyl β-cyclodextrin. Eur. J. Pharm. Biopharm. 2008, 68, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.M.; Baptista, F.J. Effect of the hydrophobic nature of triacetyl-β-cyclodextrin on the complexation with nicardipine hydrochloride: Physicochemical and dissolution properties of the kneaded and spray-dried complexes. Chem. Pharm. Bull. 2002, 50, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Nitalikar, M.M.; Sakarkar, D.M.; Jain, P.V. The cyclodextrins. J. Curr. Pharm. Res. 2012, 10, 1–6. [Google Scholar]
- The United States Pharmacopeia 30: NF 25; US pharmacopeial Convention: Twinbrook Parkway, Rockville, MD, USA, 2007.
- Kshirsagar, S.J.; Bhalekar, M.R.; Sable, P.N.; Wadekar, S.; Madgulkar, A.R. Dissolution improvement of poorly water soluble drug using dry emulsion and solid dispersion. J. Pharm. Res. 2009, 2, 1780–1785. [Google Scholar]
- Mahapatra, A.K.; Murthy, P.N.; Swadeep, B.; Prasad, R. Self-emulsifying drug delivery systems (SEDDS): An update from formulation development to therapeutic strategies. Int. J. Pharm. Tech. Res. 2014, 6, 546–568. [Google Scholar]
- Beg, S.; Swain, S.; Singh, H.P.; Patra, C.N.; Rao, M.E.B. Development, optimization and characterization of solid self-nano emulsifying drug delivery systems of valsartan using porous carriers. AAPS PharmSciTech 2012, 13, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Naveen, C.; Shastri, N.; Tadikonda, R.R. Use of liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm. Sin. B 2012, 2, 502–508. [Google Scholar]
- Barakat, N.S. Etodolac-liquid-filled dispersion into hard gelatin capsules: An approach to improve dissolution and stability of etodolac formulation: Letters in drug design and discovery. J. Inf. Healthc. 2009, 32, 865–876. [Google Scholar]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.W.; Flanner, H. Mathematical comparison of dissolution profiles. Pharm. Tech. 1996, 20, 64–74. [Google Scholar]
- Santos, J.V.; Batista de Carvalho, I.A.E.; Pina, M.E.T. The influence of the compression force on zidovudine release from matrix tablets. AAPS Pharm. Sci. Tech. 2010, 2, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, N.M.; Khan, H.; Naseem, S. Encapsulation and characterization of controlled release flurbiprofen-loaded microspheres using bees wax as an encapsulating agent. J. Mater. Sci. 2010, 21, 1621–1630. [Google Scholar]
- Wang, Y.; Jiang, Z.T.; Li, R. Complexation and molecular microcapsules of Litsea cubeba essential oil with β-cyclodextrin and its derivatives. Eur. Food Res. Technol. 2009, 228, 865–873. [Google Scholar] [CrossRef]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenan-induced edema in hind Paw of the rat as an essay for anti-inflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Anti-inflammatory and antipyretic activities of indomethacin, 1-(p-chlorobenzoyl)-5-methoxy-2-methyl-indol-3-acetic acid. J.Exp. Biol. Ther. 1963, 141, 369–376. [Google Scholar]
- Devi, B.P.; Boominathan, R.; Mandal, S.C. Anti-inflammatory, analgesic and antipyretic properties of Clitoria ternatea root. Fitoterapia 2003, 74, 345–349. [Google Scholar] [CrossRef]
- Panthogen, A.; Kanjanapothi, D.; Taesotikul, T.; Phankummoon, A.; Panthong, K.; Reutrakul, V. Anti-inflammatory activity of methanolic extracts from Ventilago harmandiana Pierre. J. Ethnopharmacol. 2004, 91, 237–242. [Google Scholar]
- Thambi, P.T.; Kuzhivelil, B.; Sabu, M.C.; Jolly, C.I. Antioxidant and anti-inflammatory activities of flowers of tabernaemontana coronaria (L.) R Br. Indian J. Pharm. Sci. 2006, 68, 352–355. [Google Scholar]
- Vasudevan, M.; Gunnam, K.K.; Parle, M. Antinociceptive and anti-inflammatory effects of Thespesia populnea bark extract. J. Ethnopharmacol. 2007, 109, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.; Manthery, B.; Vernon-Roberts, B.; Brooks, P.M. Assessment of non-steroidal anti-inflammatory drug combinations by the polyurethane sponge implantation model in the rat. Ann. Rheum. Dis. 1983, 42, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Lanhers, M.C.; Fleurentin, J.; Mortier, F.; Vinche, A.; Younosm, C. Anti-inflammatory and analgesic effects of an aqueous extract of Harpagophytum procumbens. Planta Med. 1992, 58, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Trivedi, P.; Chaturvedi, S.C. Dextran-etodolac conjugates: Synthesis in vitro and in vivo evaluation. J. Acta Poloniae Pharm. 2009, 66, 201–206. [Google Scholar]
- Baboota, S.; Shakeel, F.; Ahuja, A.; Ali, J.; Shafiq, S. Design, development and evaluation of novel nano-emulsion formulations for transdermal potential of celecoxib. Acta. Pharm. 2007, 57, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Amita, M.; Goel, H. In vivo bioavailability and therapeutic assessment of host-guest inclusion phenomena for the hydrophobic molecule etodolac: Pharmacodynamic and pharmacokinetic evaluation. J. Scientia Pharm. 2010, 78, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Kaster, R.; Anderson, M.; De-Beer, E.J. Acetic acid analgesic screen. Fed. Proc. 1959, 18, 418–420. [Google Scholar]
- Adzu, B.; Amos, S.; Wambebe, C.; Gamaniel, K. Anti-nociceptive activity of aqueous extract of zixyphus spinachristi rootbark. Fitoterapia 2001, 72, 344–350. [Google Scholar] [CrossRef]
- Taber, R.I.; Greenhuouse, D.D.; Rendell, J.K.; Irwin, S. Agonist and antagonist interactions of opioids and acetic acid-induced abdominal stretching of mice. J. Pharmacol. Exp. Ther. 1969, 69, 29–37. [Google Scholar]
- Young, J.M.; Luo, Y.; Cheng, H.; Hsieh, W.C.; Liao, J.C.; Peng, W.H. Analgesic and anti-inflammatory activities of 6-gingerol. J. Ethnopharmacol. 2005, 96, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Bettinetti, G.; Sorrenti, M.; Catenacci, L.; Setti, M.; Ferrari, F.; Rossi, S.; Carraro, P. Solid-state properties of triacetyl α-, β- and γ-cyclodextrins and potential use for prolonged release of vancomycin. In Proceedings of the 12th International Cyclodextrin Symposium, Montpellier, France, 16–19 May 2004. [Google Scholar]
- Williams, R.O., III; Mahaguna, V.; Sriwongjanya, M. Characterization of an inclusion complex of cholesterol and hydroxypropyl-β-cyclodextrin. Eur. J. Pharm. Biopharm. 1998, 46, 355–360. [Google Scholar] [CrossRef]
- Ozdemir, N.; Ordu, S. Improvement of dissolution properties of furosemide by complexation with β-cyclodextrin. Drug Dev. Ind. Pharm. 1998, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Florey, K.; Christine, K.W.; Roger, E.S. Fenoprofen calcium. In Analytical Profiles Drug Substances; Elsevier: Burlington, VT, USA, 1977; pp. 161–182. [Google Scholar]
- Bratu, I.; Veiga, F.; Fernandes, C.; Hernanz, A.; Gavira, M. Infrared spectroscopic study of triacetyl-β-cyclodextrin and its inclusion complex with nicardipine. Spectroscopy 2004, 18, 459–467. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X. Spectroscopic Identification of Organic Compounds, 6th ed.; John Wiley and Sons: New York, NY, USA, 2002; pp. 72–77. [Google Scholar]
- Sappidi, S.; Thadkala, K.; Kota, S.; Aukunuru, J. Preparation and characterization of ethyl cellulose microspheres encapsulating metformin hydrochloride and glipizide. Der. Pharm. Lett. 2014, 6, 213–226. [Google Scholar]
- Rajamanickam, D.; Rajappan, M.; Varadharajan, M.; Srinivasan, B. Development and evaluation of aceclofenac-loaded ethyl cellulose microshperes. Asian J. Pham. Sci. 2012, 7, 50–57. [Google Scholar]
- Ammar, H.O.; Ghorab, M.; El-Nahhas, S.A.; Emara, L.H.; Makram, T.S. Effect of cyclodextrins on the physicochemical characteristics of furosemide. Pharmazie 1999, 54, 142–144. [Google Scholar]
- Zhu, H.; Zu, J.; Varlashkin, P.; Long, S.; Kidd, C. Dehydration, hydration behavior, and structural analysis of Fenoprofen Calcium. J. Pharm. Sci. 2001, 90, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Spulber, M.; Pinteala, M.; Fifere, A.; Moldoveanu, C.; Mangolagiu, I.; Harabagiu, V.; Simionescu, B.C. Water soluble complexes of methyl-β-cyclodextrin and sulconazole nitrate. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 135–142. [Google Scholar] [CrossRef]
- Sinha, V.R.; Anitha, R.; Ghosh, S.; Nanda, A.; Kumria, R. Complexation of celecoxib with β-cyclodextrin: Characterization of the interaction in solution and in solid state. J. Pharm. Sci. 2005, 94, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Caccia, F.; Dispenza, R.; Fronja, G.; Fuganti, C.; Malpezzi, L.; Mele, A. Structure of neohesperidin dihydrochalconc/β-cyclodextrin inclusion complex: NMR, MS and X-ray spectroscopic investigation. J. Agric. Food Chem. 1998, 46, 1500–1505. [Google Scholar] [CrossRef]
- Moyano, J.R.; Arias, M.J.; Gines, J.M.; Pérez, J.I.; Rabasco, A.M. Dissolution behavior of oxazepam in presence of cyclodextrins: Evaluation of oxazepam-dimeb binary systems. Drug Dev. Ind. Pharm. 1997, 23, 379–385. [Google Scholar] [CrossRef]
- Kedzierewicz, F.; Hoffman, M.; Maincent, P. Comparison of tolbutamide β-cyclodextrin inclusion compounds and solid dispersions. Int. J. Pharm. 1990, 58, 221–227. [Google Scholar] [CrossRef]
- Hassan, M.A.; Suleiman, M.S.; Najib, N.M. Improvement of the in vitro dissolution characteristics of famotidine by inclusion in β-cyclodextrin. Int. J. Pharm. 1990, 58, 19–24. [Google Scholar] [CrossRef]
- Ammar, H.O.; Salama, H.A.; Ghorab, M.; Mahmoud, A.A. Formulation and biological evaluation of glimepride-cyclodextrin-polymer systems. Int. J. Pharm. 2006, 309, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Hoag, S.W.; Hussain, A.S. The impact of formulation on bioavailability: Summary of workshop discussion. J. Nutr. 2001, 131, 1389S–1391S. [Google Scholar] [PubMed]
- Agrawal, V.; Gupta, V.; Ramteke, S.; Trivedi, P. Preparation and evaluation of tubular micelles of pleuronic lecithin organogel for transdermal deliver of sumatriptan. AAPS PharmSciTech 2010, 11, 1718–1725. [Google Scholar] [CrossRef] [PubMed]












© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ammar, H.O.; Makram, T.S.; Mosallam, S. Effect of Polymers on the Physicochemical Properties and Biological Performance of Fenoprofen Calcium Dihydrate-Triacetyl-β-Cyclodextrin Complex. Pharmaceutics 2017, 9, 23. https://doi.org/10.3390/pharmaceutics9030023
Ammar HO, Makram TS, Mosallam S. Effect of Polymers on the Physicochemical Properties and Biological Performance of Fenoprofen Calcium Dihydrate-Triacetyl-β-Cyclodextrin Complex. Pharmaceutics. 2017; 9(3):23. https://doi.org/10.3390/pharmaceutics9030023
Chicago/Turabian StyleAmmar, Hussein O., Tarek S. Makram, and Shaimaa Mosallam. 2017. "Effect of Polymers on the Physicochemical Properties and Biological Performance of Fenoprofen Calcium Dihydrate-Triacetyl-β-Cyclodextrin Complex" Pharmaceutics 9, no. 3: 23. https://doi.org/10.3390/pharmaceutics9030023