
Current Trends in Development of Liposomes for Targeting Bacterial Biofilms

Abstract
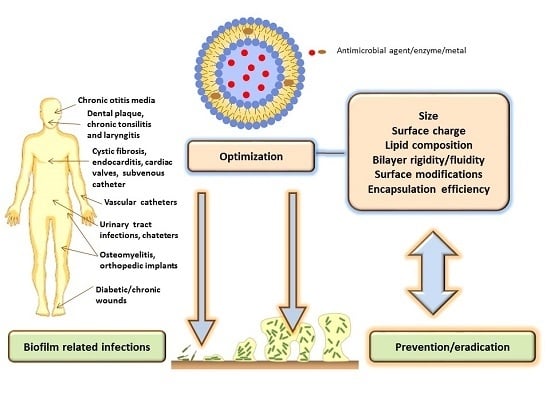
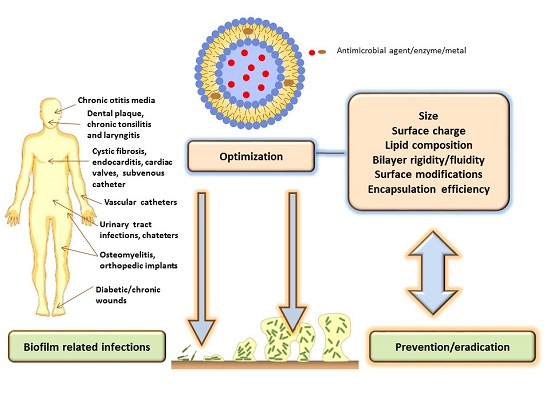
:
1. Introduction
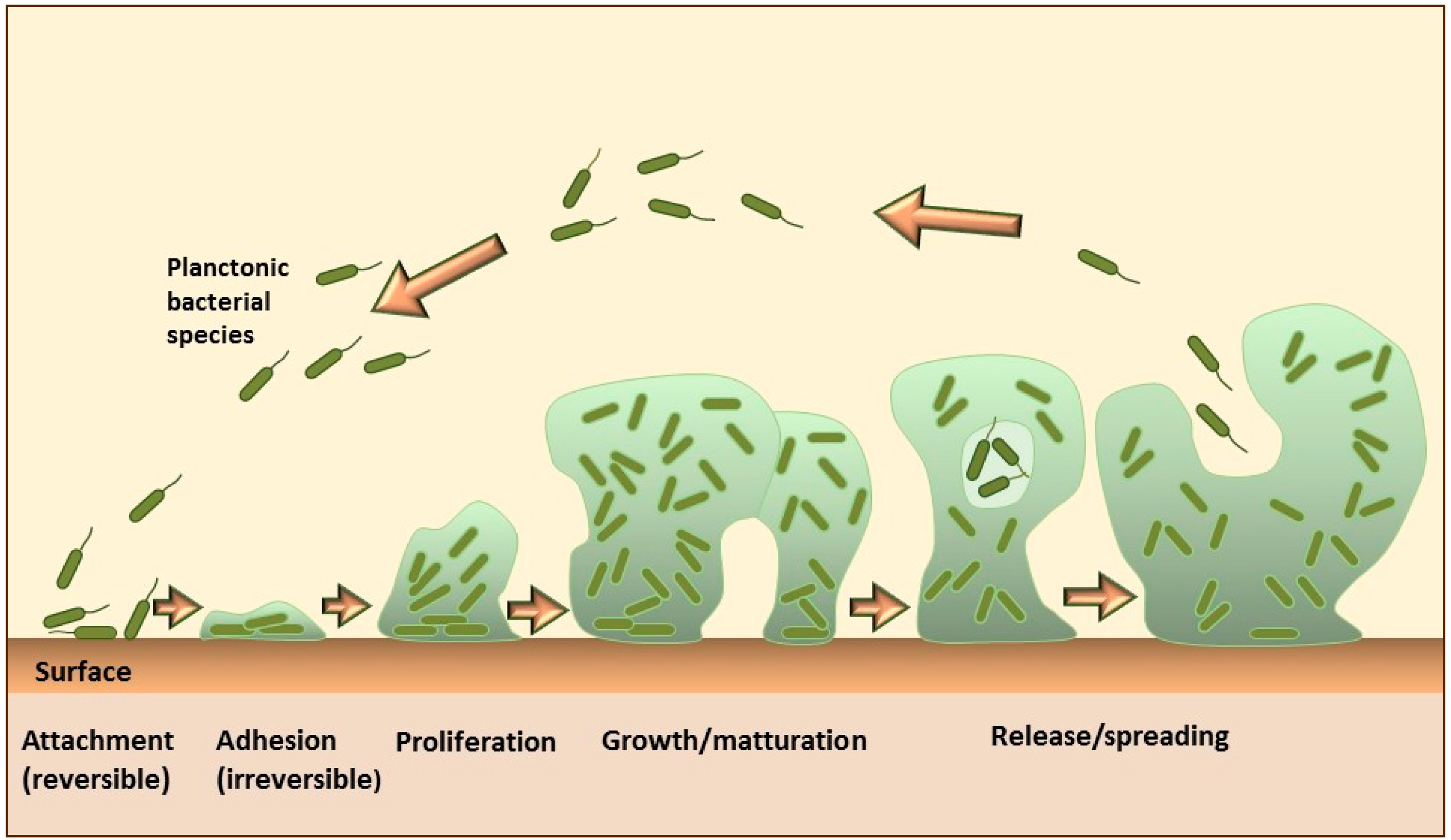
2. Liposomes for Targeting Bacterial Biofilms
2.1. General Considerations
2.2. The Role of Liposomal Physicochemical Properties
2.3. Conventional Liposomes
2.4. Fusogenic Liposomes
2.5. Surface-Modified Liposomes
2.6. Reactive Liposomes Encapsulating Enzyme(s)
2.7. Antibiotic-Metal Co-Encapsulating Liposomes
2.8. Liposomes-in-Hydrogel
2.9. Solid Supported Liposomes (SSLs) and Liposome Loaded Scaffolds (LLSs)
2.10. Miscellaneous
3. Toxicity Aspects
4. Limitations of Liposomal Formulations
5. Perspectives
6. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| ALN-TEG-Chol | alendronate-tri(ethyleneglycol)-cholesterol conjugate |
| BBLs | biomineral-binding liposomes |
| βTCP | β-tricalcium phosphate |
| BiEDT | bismuth-ethanedithiol |
| CF | cystic fibrosis |
| CHM | cholesteryl mannan |
| Chol | cholesterol |
| CHEMS | cholesteryl hemisuccinate |
| ConA | concanavalin A |
| CPO | chloroperoxidase |
| CS | calcium sulfate |
| CS/KGM | chitosan/konjac glucomannan |
| DC-Chol | dimethylammonium ethane carbamoyl cholesterol |
| DCP | dicetylphosphate |
| DDAB | dimethyldioctadecylammonium bromide |
| DMPC | dimyristoylphosphatidylcholine |
| DMPG | dimyristoylphosphatidylglycerol |
| DOPC | dioleoylphophatidylcholine |
| DOPE | dioleoylphosphatidylethanolamine |
| DOTAP | 1,2-oleoyloxy-3-trimethylammonium-propan |
| DPPC | dipalmitoylphosphatidylcholine |
| DPPE | diplalmitoylphosphatidylethanolamine |
| DPPEMBS | maleimidobenzoyl-N-hydroxysuccinimide (MBS) derivative of DPPE |
| DPPG | dipalmitoylphosphatidylglycerol |
| DSPC | distearoylphosphatidylcholine |
| GO | glucose oxidase |
| GO-HRP | glucose oxidase-horse radish peroxidase |
| HSPC | hydrogenated soy phosphatidylcholine |
| LipoBiEDT-TOB | liposomal BiEDT-loaded tobramycin |
| Lipo-Ga-GEN | liposomal formulation with gentamicin and gallium |
| LLS | liposome loaded scaffold |
| LPO | lactoperoxidase |
| MBC | minimal bactericidal concentration |
| MBEC | minimal biofilm eradication concentration |
| MIC | minimal inhibitory concentration |
| n-HA/CS/KGM | nano-hydroxyapatite/chitosan/konjac glucomannan |
| PC | phosphatidylcholine |
| PEG | poly(ethylene) glycol |
| PI | phosphatidylinositol |
| QS | quorum sensing |
| SA | stearylamine |
| SCh | sodium cholate |
| SM | sialo-mannan |
| SPC | soy phosphatidylcholine (lecithin) |
| SSLs | solid supported liposomes |
References
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, E.P. Bacterial communication and group behavior. J. Clin. Investig. 2003, 112, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Givskov, M. Pharmacological inhibition of quorum sensing for the treatment of chronic bacterial infections. J. Clin. Investig. 2003, 112, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Hoiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, P.; Sauer, K.; Davies, D.G.; Costerton, J.W. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef] [PubMed]
- Forier, K.; Raemdonck, K.; De Smedt, S.C.; Demeester, J.; Coenye, T.; Braeckmans, K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J. Control. Release 2014, 190, 607–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nafee, N. Nanocarriers against bacterial biofilms: Current status and future perspectives. In Nanotechnology in Diagnosis, Treatment and Prophylaxis of Infectious Diseases; Rai, M., Kon, K., Eds.; Academic Press, Elsevier: London, UK, 2015; pp. 167–189. [Google Scholar]
- Nickel, J.C.; McLean, R.J.C. Bacterial biofilms in urology. Infect. Urol. 1998, 11, 169–175. [Google Scholar]
- Percival, S.L.; Bowler, P.G. Biofilms and. their potential role in wound healing. Wounds 2004, 16, 234–240. [Google Scholar]
- Scheie, A.A.; Petersen, F.C. The biofilm concept: Consequences for future prophylaxis of oral diseases? Crit. Rev. Oral Biol. Med. 2004, 15, 4–12. [Google Scholar] [CrossRef]
- Singh, P.K.; Schaefer, A.L.; Parsek, M.R.; Moninger, T.O.; Welsh, M.J.; Greenberg, E.P. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 2000, 407, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Post, J.C. Direct evidence of bacterial biofilms in otitis media. Laryngoscope 2001, 111, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, A.R.; Leid, J.G.; Hunsaker, D. Bacterial Biofilms on the sinus mucosa of human subjects with chronic rhinosinusitis. Laryngoscope 2006, 116, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Boumis, E.; Gesu, G.; Menichetti, F.; Ranieri, M.; Rinaldi, M.; Suter, F.; Nicastri, E.; Lauria, F.N.; Carosi, G.; Moroni, M.; et al. Consensus document on controversial issues in the diagnosis and treatment of bloodstream infections and endocarditis. Int. J. Infect. Dis. 2010, 14, S23–S38. [Google Scholar] [CrossRef] [PubMed]
- Costerton, W.; Veeh, R.; Shirtliff, M.; Pasmore, M.; Post, C.; Ehrlich, G. The application of biofilm science to the study and control of chronic bacterial infections. J. Clin. Investig. 2003, 112, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Mah, T.F.C.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Smith, A.W. Biofilms and antibiotic therapy: Is there a role for combating bacterial resistance by the use of novel drug delivery systems? Adv. Drug Deliver. Rev. 2005, 57, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Sihorkar, V.; Vyas, S.P. Biofilm consortia on biomedical and biological surfaces: Delivery and targeting strategies. Pharm. Res. 2001, 18, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Bothello, C.; Oliveria, R. Nanotechnology applied to medical biofilms control. In Science against Microbial Pathogens: Communicating Current Research and Technological Advances; Medez-Vilas, A., Ed.; Formatex: Badajoz, Spain, 2011; Volume 2, pp. 878–888. [Google Scholar]
- Sun, F.J.; Qu, F.; Ling, Y.; Mao, P.Y.; Xia, P.Y.; Chen, H.P.; Zhou, D.S. Biofilm-associated infections: Antibiotic resistance and novel therapeutic strategies. Future Microbiol. 2013, 8, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2001, 15, 167–193. [Google Scholar] [CrossRef]
- Donelli, G.; Francolini, I.; Piozzi, A.; Di Rosa, R.; Marconi, W. New polymer-antibiotic systems to inhibit bacterial biofilm formation: A suitable approach to prevent central venous catheter-associated infections. J. Chemother. 2002, 14, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Piozzi, A.; Francolini, I.; Occhiaperti, L.; Venditti, M.; Marconi, W. Antimicrobial activity of polyurethanes coated with antibiotics: A new approach to the realization of medical devices exempt from microbial colonization. Int. J. Pharm. 2004, 280, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Breuing, K.H.; Bayer, L.; Neuwalder, J.; Orgill, D.P. Early experience using low-frequency ultrasound in chronic wounds. Ann. Plast. Surg. 2005, 55, 183–187. [Google Scholar] [CrossRef]
- Stanisic, M.M.; Provo, B.J.; Larson, D.L.; Kloth, L.C. Wound debridement with 25 kHz ultrasound. Adv. Skin Wound Care 2005, 18, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, R.D.; Rumbaugh, K.P.; James, G.; Schultz, G.; Phillips, P.; Yang, Q.; Watters, C.; Stewart, P.S.; Dowd, S.E. Biofilm maturity studies indicate sharp debridement opens a time-dependent therapeutic window. J. Wound Care 2010, 19, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.B.; Givskov, M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int. J. Med. Microbiol. 2006, 296, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Matl, F.D.; Obermeier, A.; Zlotnyk, J.; Friess, W.; Stemberger, A.; Burgkart, R. Augmentation of antibiotic activity by low-frequency electric and electromagnetic fields examining Staphylococcus aureus in broth media. Bioelectromagnetics 2011, 32, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Ensing, G.T.; Roeder, B.L.; Nelson, J.L.; van Horn, J.R.; van der Mei, H.C.; Busscher, H.J.; Pitt, W.G. Effect of pulsed ultrasound in combination with gentamicin on bacterial viability in biofilms on bone cements in vivo. J. Appl. Microbiol. 2005, 99, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Soukos, N.S.; Socransky, S.S.; Mulholland, S.E.; Lee, S.; Doukas, A.G. Photomechanical drug delivery into bacterial biofilms. Pharm. Res. 2000, 17, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Kasimanickam, R.K.; Ranjan, A.; Asokan, G.V.; Kasimanickam, V.R.; Kastelic, J.P. Prevention and treatment of biofilms by hybrid- and nanotechnologies. Int. J. Nanomed. 2013, 8, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Tamilvanan, S.; Venkateshan, N.; Ludwig, A. The potential of lipid- and polymer-based drug delivery carriers for eradicating biofilm consortia on device-related nosocomial infections. J. Control. Release 2008, 128, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A. Liposomes as delivery systems for antibiotics. Int. J. Pharm. 2010, 387, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Alhariri, M.; Azghani, A.; Omri, A. Liposomal antibiotics for the treatment of infectious diseases. Expert Opin. Drug Deliv. 2013, 10, 1515–1532. [Google Scholar] [CrossRef] [PubMed]
- Basnet, P.; Škalko-Basnet, N. Nanodelivery systems for improved topical antimicrobial therapy. Curr. Pharm. Des. 2013, 19, 7237–7243. [Google Scholar] [CrossRef] [PubMed]
- Fenske, D.B.; Cullis, P.R. Liposomal nanomedicines. Expert Opin. Drug Deliv. 2008, 5, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Vanić, Ž.; Holaeter, A.M.; Škalko-Basnet, N. (Phospho)lipid-based nanosystems for skin administration. Curr. Pharm. Des. 2015, 21, 4174–4192. [Google Scholar] [CrossRef] [PubMed]
- Abed, N.; Couvreur, P. Nanocarriers for antibiotics: A promising solution to treat intracellular bacterial infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Low, W.L.; Gupta, A.; Amin, M.C.; Radecka, I.; Britland, S.T.; Raj, P.; Kenward, K.M. Strategies for antimicrobial drug delivery to biofilm. Curr. Pharm. Des. 2015, 21, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pornpattananangku, D.; Hu, C.M.; Huang, C.M. Development of nanoparticles for antimicrobial drug delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Aljuffali, I.A.; Huang, C.H.; Fang, J.Y. Nanomedical strategies for targeting skin microbiomes. Curr. Drug Metab. 2015, 16, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Vanić, Ž.; Hurler, J.; Ferderber, K.; Golja Gašparović, P.; Škalko-Basnet, N.; Filipović-Grčić, J. Novel vaginal drug delivery system: Deformable propylene glycol liposomes-in-hydrogel. J. Liposome Res. 2014, 24, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Vanić, Ž.; Škalko-Basnet, N. Nanopharmaceuticals for improved topical vaginal therapy: Can they deliver? Eur. J. Pharm. Sci. 2013, 50, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, D.; Shekunov, B.; Blanchard, J.; Hickey, A. Lipid-based carriers for pulmonary products: Preclinical development and case studies in humans. Adv. Drug Deliv. Rev. 2014, 75, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, K.; Cheow, W.S. Nano-antibiotics in chronic lung infection therapy against Pseudomonas aeruginosa. Colloids Surf. B 2014, 116, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.P.; Bagui, M.; Tamboli, V.; Mitra, A.K. Recent applications of liposomes in ophthalmic drug delivery. J. Drug Deliv. 2011, 2011, 863734. [Google Scholar] [CrossRef] [PubMed]
- Sachetelli, S.; Khalil, H.; Chen, T.; Beaulac, C.; Senechal, S.; Lagace, J. Demonstration of a fusion mechanism between a fluid bactericidal liposomal formulation and bacterial cells. BBA-Biomembranes 2000, 1463, 254–266. [Google Scholar] [CrossRef]
- Robinson, A.M.; Bannister, M.; Creeth, J.E.; Jones, M.N. The interaction of phospholipid liposomes with mixed bacterial biofilms and their use in the delivery of bactericide. Colloid. Surface A 2001, 186, 43–53. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A.; Gubernator, J.; Gula, G.; Bocer, T.; Doroszkiewicz, W. The interaction between Pseudomonas aeruginosa cells and cationic PC:Chol:DOTAP liposomal vesicles versus outer-membrane structure and envelope properties of bacterial cell. Int. J. Pharm. 2009, 367, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Thomas, N.; Thierry, B.; Vreugde, S.; Clive, A.; Prestidge, C.A.; Wormald, P.-J. Distribution and inhibition of liposomes on Staphylococcus aureus and Pseudomonas aeruginosa biofilm. PLoS ONE 2015, 10, e0131806. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, A.S.; Forier, K.; Nelis, H.; Braeckmans, K.; Coenye, T. Transport of nanoparticles and tobramycin-loaded liposomes in Burkholderia cepacia complex biofilms. PLoS ONE 2013, 8, e79220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.M.; Creeth, J.E.; Jones, M.N. The use of immunoliposomes for specific delivery of antimicrobial agents to oral bacteria immobilized on polystyrene. J. Biomat. Sci. Polym. Ed. 2000, 11, 1381–1393. [Google Scholar] [CrossRef]
- Vyas, S.P.; Sihorkar, V.; Dubey, P.K. Preparation, characterization and in vitro antimicrobial activity of metronidazole bearing lectinized liposomes for intra-periodontal pocket delivery. Pharmazie 2001, 56, 554–560. [Google Scholar] [PubMed]
- Strathmann, M.; Wingender, J.; Flemming, H.C. Application of fluorescently labelled lectins for the visualization and biochemical characterization of polysaccharides in biofilms of Pseudomonas aeruginosa. J. Microbiol. Methods 2002, 50, 237–248. [Google Scholar] [CrossRef]
- Gubernator, J.; Drulis-Kawa, Z.; Dorotkiewicz-Jach, A.; Doroszkiewicz, W.; Kozubek, A. In vitro antimicrobial activity of liposomes containing ciprofloxacin, meropenem and gentamicin against gram-negative clinical bacterial strains. Lett. Drug Des. Discov. 2007, 4, 297–304. [Google Scholar] [CrossRef]
- Sanderson, N.M.; Jones, M.N. Targeting of cationic liposomes to skin-associated bacteria. Pestic. Sci. 1996, 46, 255–261. [Google Scholar] [CrossRef]
- Sanderson, N.M.; Guo, B.Q.; Jacob, A.E.; Handley, P.S.; Cunniffe, J.G.; Jones, M.N. The interaction of cationic liposomes with the skin-associated bacterium Staphylococcus epidermidis: Effects of ionic strength and temperature. BBA-Biomembranes 1996, 1283, 207–214. [Google Scholar] [CrossRef]
- Sanderson, N.M.; Jones, M.N. Encapsulation of vancomycin and gentamicin within cationic liposomes for inhibition of growth of Staphylococcus epidermidis. J. Drug Target. 1996, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Gias, E.L.M.; Jones, M.N. The adsorption of cationic liposomes to Staphylococcus aureus biofilms. Colloid. Surface A 1999, 149, 561–570. [Google Scholar] [CrossRef]
- Jones, M.N.; Song, Y.H.; Kaszuba, M.; Reboiras, M.D. The interaction of phospholipid liposomes with bacteria and their use in the delivery of bactericides. J. Drug Target. 1997, 5, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jones, M.N. The delivery of benzyl penicillin to Staphylococcus aureus biofilms by use of liposomes. J. Liposome Res. 2004, 14, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Meers, P.; Neville, M.; Malinin, V.; Scotto, A.W.; Sardaryan, G.; Kurumunda, R.; Mackinson, C.; James, G.; Fisher, S.; Perkins, W.R. Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections. J. Antimicrob. Chemother. 2008, 61, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Dupont, L.; Konstan, M.W.; Billings, J.; Fustik, S.; Goss, C.H.; Lymp, J.; Minic, P.; Quittner, A.L.; Rubenstein, R.C.; et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013, 68, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Bilton, D.; Pressler, T.; Fajac, I.; Clancy, J.P.; Sands, D.; Minic, P.; Cipolli, M.; LaRosa, M.; Galeva, I.; Amparo Sole, A.; et al. Phase 3 efficacy and safety data from randomized, multicenter study of liposomal amikacin for inhalation (Arikace®) compared with TOBI® in cystic fibrosis patients with chronic infection due to Pseudomonas aeruginosa. In Proceedings of the North American Cystic Fibrosis Conference (NACFC), Salt Lake City, UT, USA, 17–19 October 2013.
- Waters, V.; Ratjen, F. Inhaled liposomal amikacin. Expert Rev. Respir. Med. 2014, 8, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Bruinenberg, P.; Blanchard, J.D.; Cipolla, D.; Dayton, F.; Mudumga, S.; Gonda, I. Inhaled liposomal ciprofloxacin: Once a day management of respiratory infections. In Respiratory Drug Delivery; Byron, P.R., Dalby, R.N., Peart, J., Suman, J.D., Farr, S.J., Young, P.M., Eds.; Davis Haelthcare Int’l Publishing IL: Orlando, FL, USA, 2010; Volume 1, pp. 73–82. [Google Scholar]
- Cipolla, D.; Blanchard, J.; Gonda, I. Development of liposomal ciprofloxacin to treat lung infections. Pharmaceutics 2016, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Alhajlan, M.; Alhariri, M.; Omri, A. Efficacy and safety of liposomal clarithromycin and its effect on Pseudomonas aeruginosa virulence factors. Antimicrob. Agents Chemother. 2013, 57, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Solleti, V.S.; Alhariri, M.; Halwani, M.; Omri, A. Antimicrobial properties of liposomal azithromycin for Pseudomonas infections in cystic fibrosis patients. J. Antimicrob. Chemoth. 2015, 70, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Cevc, G. How membrane chain-melting phase-transition temperature is affected by the lipid chain asymmetry and degree of unsaturation: An effective chain-length model. Biochemistry 1991, 30, 7186–7193. [Google Scholar] [CrossRef] [PubMed]
- Beaulac, C.; Clement-Major, S.; Hawari, J.; Lagace, J. Eradication of mucoid Pseudomonas aeruginosa with fluid liposome-encapsulated tobramycin in an animal model of chronic pulmonary infection. Antimicrob. Agents Chemother. 1996, 40, 665–669. [Google Scholar] [PubMed]
- Sachetelli, S.; Beaulac, C.; Riffon, R.; Lagace, J. Evaluation of the pulmonary and systemic immunogenicity of Fluidosomes, a fluid liposomal-tobramycin formulation for the treatment of chronic infections in lungs. BBA-Gen. Subjects 1999, 1428, 334–340. [Google Scholar] [CrossRef]
- Beaulac, C.; Sachetelli, S.; Lagace, J. In-vitro bactericidal efficacy of sub-MIC concentrations of liposome-encapsulated antibiotic against gram-negative and gram-positive bacteria. J. Antimicrob. Chemother. 1998, 41, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Beaulac, C.; Sachetelli, S.; Lagace, J. Aerosolization of low phase transition temperature liposomal tobramycin as a dry powder in an animal model of chronic pulmonary infection caused by Pseudomonas aeruginosa. J. Drug Target. 1993, 7, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Marier, J.F.; Lavigne, J.; Ducharme, M.P. Pharmacokinetics and efficacies of liposomal and conventional formulations of tobramycin after intratracheal administration in rats with pulmonary Burkholderia cepacia infection. Antimicrob. Agents Chemother. 2002, 46, 3776–3781. [Google Scholar] [CrossRef] [PubMed]
- Inhaled Liposomal Tobramycin—Axentis Pharma AG. Available online: http://adisinsight.springer.com/drugs/800025294 (accessed on 28 January 2016).
- Nicolosi, D.; Scalia, M.; Nicolosi, V.M.; Pignatello, R. Encapsulation in fusogenic liposomes broadens the spectrum of action of vancomycin against Gram-negative bacteria. Int. J. Antimicrob. Agents 2010, 35, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Gubernator, J.; Dorotkiewicz-Jach, A.; Doroszkiewicz, W.; Kozubek, A. In vitro antimicrobial activity of liposomal meropenem against Pseudomonas aeruginosa strains. Int. J. Pharm. 2006, 315, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, X.; Huang, X.; Wang, X.; Liao, G.; Chen, Z. Preparation and characterization of flexible nanoliposomes loaded with daptomycin, a novel antibiotic, for topical skin therapy. Int. J. Nanomed. 2013, 8, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Suntres, Z.E.; Omri, A. Importance of DNase and alginate lyase for enhancing free and liposome encapsulated aminoglycoside activity against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2009, 64, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Muiruri, P.W.; Jones, G.H.; Scott, M.J.; Jones, M.N. The effect of grafted poly(ethylene glycol) on the electrophoretic properties of phospholipid liposomes and their adsorption to bacterial biofilms. Colloids Surface A 2001, 194, 287–296. [Google Scholar] [CrossRef]
- Moghadas-Sharif, N.; Fazly Bazzaz, B.S.; Khameneh, B.; Malaekeh-Nikouei, B. The effect of nanoliposomal formulations on Staphylococcus epidermidis biofilm. Drug Dev. Ind. Pharm. 2015, 41, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Creeth, J.E.; Jones, M.N. The specificity and affinity of immunoliposome targeting to oral bacteria. BBA-Biomembranes 1998, 1369, 278–286. [Google Scholar] [CrossRef]
- Kaszuba, M.; Robinson, A.M.; Song, Y.H.; Creeth, J.E.; Jones, M.N. The visualisation of the targeting of phospholipid liposomes to bacteria. Colloids Surface B 1997, 8, 321–332. [Google Scholar] [CrossRef]
- Vyas, S.P.; Sihorkar, V.; Jain, S. Mannosylated liposomes for bio-film targeting. Int. J. Pharm. 2007, 330, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.J.; Kaszuba, M.; Creeth, J.E.; Jones, M.N. Reactive liposomes encapsulating a glucose oxidase-peroxidase system with antibacterial activity. BBA-Biomembranes 1997, 1326, 37–46. [Google Scholar] [CrossRef]
- Jones, M.N.; Hill, K.J.; Kaszuba, M.; Creeth, J.E. Antibacterial reactive liposomes encapsulating coupled enzyme systems. Int. J. Pharm. 1998, 162, 107–117. [Google Scholar] [CrossRef]
- Halwani, M.; Yebio, B.; Suntres, Z.E.; Alipour, M.; Azghani, A.O.; Omri, A. Co-encapsulation of gallium with gentamicin in liposomes enhances antimicrobial activity of gentamicin against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2008, 62, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Halwani, M.; Blomme, S.; Suntres, Z.E.; Alipour, M.; Azghani, A.O.; Kumar, A.; Omri, A. Liposomal bismuth-ethanedithiol formulation enhances antimicrobial activity of tobramycin. Int. J. Pharm. 2008, 358, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Halwani, M.; Hebert, S.; Suntres, Z.E.; Lafrenie, R.M.; Azghani, A.O.; Omri, A. Bismuth-thiol incorporation enhances biological activities of liposomal tobramycin against bacterial biofilm and quorum sensing molecules production by Pseudomonas aeruginosa. Int. J. Pharm. 2009, 373, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Dorval, C.; Suntres, Z.E.; Omri, A. Bismuth-ethanedithiol incorporated in a liposome-loaded tobramycin formulation modulates the alginate levels in mucoid Pseudomonas aeruginosa. J. Pharm. Pharmacol. 2011, 63, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Suntres, Z.E.; Lafrenie, R.M.; Omri, A. Attenuation of Pseudomonas aeruginosa virulence factors and biofilms by co-encapsulation of bismuth-ethanedithiol with tobramycin in liposomes. J. Antimicrob. Chemother. 2010, 65, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Alhariri, M.; Omri, A. Efficacy of liposomal bismuth-ethanedithiol-loaded tobramycin after intratracheal administration in rats with pulmonary Pseudomonas aeruginosa infection. Antimicrob. Agents Chemother. 2013, 57, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Catuogno, C.; Jones, M.N. The antibacterial properties of solid supported liposomes on Streptococcus oralis biofilms. Int. J. Pharm. 2003, 257, 125–140. [Google Scholar] [CrossRef]
- Tang, H.; Xu, Y.Q.; Zheng, T.; Li, G.; You, Y.G.; Jiang, M.Y.; Li, J.; Ding, J. Treatment of osteomyelitis by liposomal gentamicin-impregnated calcium sulfate. Arch. Orthop. Traum. Surg. 2009, 129, 1301–1308. [Google Scholar]
- Zhu, C.T.; Xu, Y.Q.; Shi, J.A.; Li, J.; Ding, J. Liposome combined porous beta-TCP scaffold: Preparation, characterization, and anti-biofilm activity. Drug Deliv. 2010, 17, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Shang, B.C.; Tang, H.; Zhou, T.H.; Xu, G.L.; Li, H.L.; Chen, Q.H.; Xu, Y.Q. Nano-hydroxyapatite/chitosan/konjac glucomannan scaffolds loaded with cationic liposomal vancomycin: Preparation, in vitro release and activity against Staphylococcus aureus biofilms. J. Biomat. Sci. Polym. Ed. 2011, 22, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.H.; Su, M.; Shang, B.C.; Ma, T.; Xu, G.L.; Li, H.L.; Chen, Q.H.; Sun, W.; Xu, Y.Q. Nano-hydroxyapatite/beta-tricalcium phosphate ceramics scaffolds loaded with cationic liposomal ceftazidime: preparation, release characteristics in vitro and inhibition to Staphylococcus aureus biofilms. Drug Dev. Ind. Pharm. 2012, 38, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Zhang, Y.J.; Chen, F.; Khutsishvili, I.; Fehringer, E.V.; Marky, L.A.; Bayles, K.W.; Wang, D. Prevention of orthopedic device-associated osteomyelitis using oxacillin-containing biomineral-binding liposomes. Pharm. Res. 2012, 29, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Hurler, J.; Sorensen, K.K.; Fallarero, A.; Vuorela, P.; Škalko-Basnet, N. Liposomes-in-hydrogel delivery system with mupirocin: In vitro antibiofilm studies and in vivo evaluation in mice burn model. Biomed. Res. Int. 2013, 2013, 498485. [Google Scholar] [CrossRef] [PubMed]
- DiTizio, V.; Ferguson, G.W.; Mittelman, M.W.; Khoury, A.E.; Bruce, A.W.; DiCosmo, F. A liposomal hydrogel for the prevention of bacterial adhesion to catheters. Biomaterials 1998, 19, 1877–1884. [Google Scholar] [CrossRef]
- Pugach, J.L.; DiTizio, V.; Mittelman, M.W.; Bruce, A.W.; DiCosmo, F.; Khoury, A.E. Antibiotic hydrogel coated Foley catheters for prevention of urinary tract infection in a rabbit model. J. Urol. 1999, 162, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Kaszuba, M.; Jones, M.N. Hydrogen peroxide production from reactive liposomes encapsulating enzymes. BBA-Biomembranes 1999, 1419, 221–228. [Google Scholar] [CrossRef]
- Olakanmi, O.; Britigan, B.E.; Schlesinger, L.S. Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect. Immun. 2000, 68, 5619–5627. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.R.; Martens, R.J.; Cohen, N.D.; Bernstein, L.R. Antimicrobial activity of gallium against virulent Rhodococcus equiin vitro and in vivo. J. Vet. Pharmacol. Ther. 2006, 29, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B.E.; Singh, P.K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Investig. 2007, 117, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Antunes, L.C.S.; Imperi, F.; Minandri, F.; Visca, P. In Vitro and in vivo antimicrobial activities of gallium nitrate against multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 5961–5970. [Google Scholar] [CrossRef] [PubMed]
- Domenico, P.; Baldassarri, L.; Schoch, P.E.; Kaehler, K.; Sasatsu, M.; Cunha, B.A. Activities of bismuth thiols against staphylococci and staphylococcal biofilms. Antimicrob. Agents Chemother. 2001, 45, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Domenico, P.; Reich, J.; Madonia, W.; Cunha, B.A. Resistance to bismuth among gram-negative bacteria is dependent upon iron and its uptake. J. Antimicrob. Chemother. 1996, 38, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Veloira, W.G.; Domenico, P.; LiPuma, J.J.; Davis, J.M.; Gurzenda, E.; Kazzaz, J.A. In vitro activity and synergy of bismuth thiols and tobramycin against Burkholderia cepacia complex. J. Antimicrob. Chemother. 2003, 52, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Stewart, P.S. Reduction of polysaccharide production in Pseudomonas aeruginosa biofilms by bismuth dimercaprol (BisBAL) treatment. J. Antimicrob. Chemother. 1999, 44, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Folsom, J.P.; Baker, B.; Stewart, P.S. In vitro efficacy of bismuth thiols against biofilms formed by bacteria isolated from human chronic wounds. J. Appl. Microbiol. 2011, 111, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Varposhti, M.; Abdi Ali, A.; Mohammadi, P. Synergistic effects of bismuth thiols and barious antibiotics against Pseudomonas aeruginosa biofilm. Jundishapur J. Microbiol. 2014, 7, e9142. [Google Scholar] [CrossRef] [PubMed]
- Hurler, J.; Berg, O.A.; Skar, M.; Conradi, A.H.; Johnsen, P.J.; Škalko-Basnet, N. Improved burns therapy: Liposomes-in-hydrogel delivery system for mupirocin. J. Pharm. Sci. 2012, 101, 3906–3915. [Google Scholar] [CrossRef] [PubMed]
- Palac, Z.; Hurler, J.; Škalko-Basnet, N.; Filipović-Grčić, J.; Vanić, Ž. Elastic liposomes-in-vehicle formulations destined for skin therapy: The synergy between type of liposomes and vehicle. Drug Dev. Ind. Pharm. 2015, 41, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavelić, Ž.; Škalko-Basnet, N.; Filipović-Grčić, J.; Martinac, A.; Jalšenjak, I. Development and in vitro evaluation of a liposomal vaginal delivery system for acyclovir. J. Control. Release 2005, 106, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, K.K.; Borden, E.; Omtri, R.S.; Boyapati, S.P.; Smith, M.; Lebby, K.; Mulpuru, M.; Gadde, M. Ability of chitosan gels to disrupt bacterial biofilms and their applications in the treatment of bacterial vaginosis. J. Pharm. Sci. 2013, 102, 2096–2101. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.P.; Espiga, A.; Silva, D.; Baptista, P.; Henriques, J.; Ferreira, C.; Silva, J.C.; Borges, J.P.; Pires, E.; Chaves, P.; et al. Development of a new chitosan hydrogel for wound dressing. Wound Repair Regen. 2009, 17, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, D.J.; Marsh, P.D.; Watson, G.K.; Cummins, D. The effects of triclosan and zinc citrate, alone and in combination, on a community of oral bacteria grown-in vitro. J. Dent. Res. 1993, 72, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, Y.; Zhang, L.; Zuo, Y.; Jansen, J.A. Preparation and characterization of nano-hydroxyapatite/chitosan/konjac glucomannan composite. J. Biomed. Mater. Res. A 2007, 83, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Zhu, J.A.F. Study on antimicrobial activity of chitosan with different molecular weights. Carbohyd. Polym. 2003, 54, 527–530. [Google Scholar] [CrossRef]
- No, H.K.; Park, N.Y.; Lee, S.H.; Meyers, S.P. Antibacterial activity of chitosans and chitosan oligomers with different molecular weights. Int. J. Food Microbiol. 2002, 74, 65–72. [Google Scholar] [CrossRef]
- Yamakami, K.; Tsumori, H.; Sakurai, Y.; Shimizu, Y.; Nagatoshi, K.; Sonomoto, K. Sustainable inhibition efficacy of liposome-encapsulated nisin on insoluble glucan-biofilm synthesis by Streptococcus mutans. Pharm. Biol. 2013, 51, 267–270. [Google Scholar] [CrossRef] [PubMed]
- New, R.R.C. Liposomes: A Practical Approach; IRL Press: Oxford, UK, 1990; pp. 1–31. [Google Scholar]
- Škalko-Basnet, N.; Vanić, Ž. Lipid-based nanopharmaceuticals in antimicrobial therapy. In Functionalized Nanomaterials for the Management of Microbial Infection; Boukherroub, R., Szunerits, S., Drider, D., Eds.; Elsevier: London, UK, in press.
- Kirby, C.; Gregoriadis, G. Liposomes. In Encyclopedia of Controlled Drug Delivery; Mathiowith, E., Ed.; Wiley: New York, NY, USA, 1999; pp. 461–492. [Google Scholar]
- Ulrich, A.S. Biophysical aspects of using liposomes as delivery vehicles. Biosci. Rep. 2002, 22, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Briones, E.; Colino, C.I.; Lanao, J.M. Delivery systems to increase the selectivity of antibiotics in phagocytic cells. J. Control. Release 2008, 125, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Vorauer-Uhl, K. Liposome technology for industrial purposes. J. Drug Deliv. 2011, 2011, 591325. [Google Scholar] [CrossRef] [PubMed]
- Storm, G.; Crommelin, D.J.A. Liposomes: Quo vadis? Pharm. Sci. Technol. Today 1998, 1, 19–31. [Google Scholar] [CrossRef]
- Silva-Dias, A.; Palmeira-De-Oliveira, A.; Miranda, I.M.; Branco, J.; Cobrado, L.; Monteiro-Soares, M.; Queiroz, J.A.; Pina-Vaz, C.; Rodrigues, A.G. Anti-biofilm activity of low-molecular weight chitosan hydrogel against Candida species. Med. Microbiol. Immun. 2014, 203, 25–33. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Liposome Type | Lipid Composition (Molar Ratio) | Drug | Average Size (nm) | Biofilm | Findings | Reference |
|---|---|---|---|---|---|---|
| Conventional liposomes | DPPC/Chol/SA (1:0.49:0.4) | - | 126 | S. epidermidis | Strong affinity for biofilm; lipid concentration and ionic strength of medium influenced the liposome adsorption to the biofilm | [59,60] |
| DPPC/Chol (1:0.21) DPPC/Chol/DDAB (1:0.28:0.22) | Vancomycin, gentamicin | ~120 | Pronounced biofilm-inhibition effect with cationic liposomes | [61] | ||
| DPPC/Chol/SA (variable mol% SA) DPPC/DPPG (variable mol% DPPG) DPPC/PI (variable mol% PI) DPPC/Chol/DDAB (11 mol% DDAB) DPPC/DC-Chol (32.3 mol% DC-Chol) | Triclosan, chlorhexidine | ~100 | S. epidermidis P. vulgaris S. mutans S. sanguis | Anionic and cationic liposomes superior in bactericide delivery as compared with the free bactericides; diffusion mechanism responsible for the bactericide delivery | [63] | |
| DPPC/Chol/SA (1:0.49:0.20–1.01) DPPC/DC-Chol (1:0.49:0.11–0.65) DPPC/Chol/DDAB (1:0.49:0.17–0.52) | Vancomycin | ~120 | S. aureus | Strong adsorption to the biofilm related to concentration of cationic lipid; DDAB-based liposomes inhibited bacterial growth more effectively than the free drug | [62] | |
| DMPC/Chol/DDAB (58.5:23:18.5) DMPC/PI (82.5:17.5) DPPC/Chol/SA (52.8:26:21.2) | Triclosan | 108–129 | S. salivarius S. sanguis | Linear relationship between liposome adsorption and mixed biofilm composition; anionic liposomes more effective than cationic | [51] | |
| DPPC/Chol/DC-Chol (1:0.49:0.43) | Benzyl penicillin G | ~140 | S. aureus | Effective delivery into biofilm at low drug concentrations and short exposure time | [64] | |
| DPPC/Chol (2:1) | Amikacin | ~300 | P. aeruginosa | Penetration into the biofilm and infected sputum; superior in vivo efficacy of inhaled liposomal amikacin in comparison to the free drug | [65] | |
| DMPC/Chol (2:1) | Tobramycin, gentamicin, amikacin | 345–512 | 8–32-fold increased MBECs (non-mucoid strains) and 64–256-fold increased MBECs (mucoid strains), as compared to the corresponding MICs | [83] | ||
| DPPC/Chol (6:1) DPPC/Chol/DCP (2:1:4) DPPC/Chol/DDAB (2:1:4) | Clarithro-mycin | 170–220 | Complete biofilm eradication with cationic and anionic liposomes; anionic liposomes superior (encapsulation efficiency, safety profile) | [71] | ||
| DPPC/Chol (2:1) DOPC/DPPG (8:1) | Tobramycin | 230–430 | B. cepacia | No increased anti-biofilm activity relative to the free drug | [54] | |
| DPPC/Chol (6:1) | Azithromycin | 406 | P. aeruginosa | Significant reduction of bacterial growth in the biofilm and attenuation of virulence factors production | [72] | |
| Fusogenic liposomes | DPPC/DMPG (15:1; 10:1) | Tobramycin | ~400 | P. aeruginosa | High bactericidal activity in vivo (rats) | [74] |
| DPPC/DMPG (15:1; 18:1) | ~400 | P. aeruginosa S. maltophilia B. cepacia S. aureus E. coli | Strong activity at sub-MICs doses; increased penetration attained with dry powder preparations | [76,77] | ||
| DPPC/DMPG (18:1) | ~400 | P. aeruginosa | Confirmed liposome fusion with bacterial cells | [50] | ||
| DPPC/DMPG (10:1) | 230–400 | B. cepacia | Superior activity in vivo (rats); sustained drug release | [78] | ||
| DOPE/DPPC/CHEMS (4:2:4) | Vancomycin | ~100 | E. coli P. aeruginosa A. baumannii | Enhanced penetration of the drug (adhesion and fusion mechanism) | [80] | |
| DPPC/DMPG liposomes (18:1) PC/DOPE/SA (1:1:0.7) PC/DOTAP/Chol (2.2:1:2.6) | Meropenem | 107–142 | P. aeruginosa | Fluidosomes showed higher MICs than the free drug did; increased activity of cationic liposomes (2–4-fold lower MICs than for the free drug) | [81] | |
| SPC/SCh (9.3:1) | Daptomycin | 55 | S. aureus | Powerful antimicrobial action in vivo; permeation into the skin; effective therapeutic concentrations maintained for several hours | [82] | |
| Surface-modified liposomes | DMPC/DDAB/DPPE-PEG-2000 (80:20-11:0-9) DPPC/DDAB/DPPE-PEG-2000 (80:20-11:0-9) DSPC/DDAB/DPPE-PEG-2000 (80:20-11:0-9) DSPC/PI/DPPE-PEG-2000 (80:20-11:0-9) | - | ~120 | S. aureus | PEGylation reduced the liposomal anti-biofilm activity | [84] |
| DPPC/SA/Chol/PEG (1:0.5:0.5:0.02) DPPC/DCP/Chol/PEG (1:0.5:0.5:0.02) | Vancomycin, rifampin | 120–140 | S. epidermidis | PEGylation did not reduce liposome anti-biofilm activity; increasing of incubation time enhanced the biofilm eradication effect | [85] | |
| DPPC/PI/DPPEMBS- anti- oralis antibody (82.8:2.6:14.6; 92.6:3.6:4.8) DPPC/Chol/SA/DPPEMBS- anti-oralis antibody (59:24.9:3.6:12.5) | - | ~100 | S. oralis S. epidermidis | Immunoliposomes showed greater antimicrobial affinity than the antibody-free liposomes | [86] | |
| DPPC/PI/DPPEMBS- anti- oralis antibody (1:0.03:0.14) | - | ~80–120 | Immunoliposomes adsorbed to the surface of S. oralis; cationic liposomes had greater affinity for S. epidermidis biofilm than anionic liposomes | [87] | ||
| DPPC/PI/DPPEMBS-anti- oralis antibody (82.8:2.6:14.6; 92.6:3.6:4.8) | Triclosan chlorhexidine | ~100 | Greater antibacterial activity than with the free drug; the extent of growth inhibition linearly related to the number of liposomes targeted to biofilm surface | [55] | ||
| PC/Chol/SA (2:1:0.1) (PC/Chol/SA)/ConA (0.1):1 | Metronidazole | ~3000 | S. mutans | Targeting the surface “glyco-calyx” of biofilm; inhibition of bacterial growth | [56] | |
| PC/Chol/SA (7:2:1) (PC/Chol/SA)/CHM (5):1 (PC/Chol/SA)/SM (7):1 | 400–450 | S. aureus | Mannosylated liposomes showed increased ability to target biofilms; superior targeting ability with SM-based liposomes | [88] | ||
| Reactive liposomes | DPPC/PI (4.85:1) | GO GO+HRP CPO+GO LPO+GO | 97–224 | S. gordonii | Inhibition increases with liposome-biofilm and substrate-biofilm incubation time and extent of enzyme encapsulation | [89,90] |
| Metal co-encapsulating liposomes | DPPC/DMPG (1:1) | Gentamicin-gallium | ~300 | P. aeruginosa | Eradication of the biofilm and interruption of QS signaling | [91] |
| DSPC/Chol (2:1) | Tobramycin- BiEDT | ~900 | P. aeruginosa | Eradication of the biofilm at significantly lower concentrations than with the free BiEDT; less toxicity of the liposomal formulation; QS suppressing properties; deeper penetration into the biofilm; attenuation of the alginate production; reduction of bacterial counts in the lungs of infected rats in vivo but without complete eradication | [92,93,94,95,96] | |
| SSLs | DPPC/PI (PI-14 mol%), DPPC/DDAB/Chol (DDAB-14 mol%) adsorbed on zinc citrate particles | Triclosan, penicillin G | ~100 | S. oralis | Activity affected by the amount of the lipid adsorbed onto zinc citrate particles; no particular advancement in comparison to individual constituents | [97] |
| DPPC/Chol/SA (1:0.43:0.49), impregated in CS | Gentamicin | n.d. | S. aureus | Significantly increased efficacy as compared to liposomal gentamicin; complete sterilization of bone tissues; prolonged drug release | [98] | |
| LLSs | DPPC/Chol (3:1), β-TCP | Gentamicin | ~110–5200 | S. aureus | Superior antibiofilm activity achieved with 800 nm-sized liposomes; controlled drug delivery | [99] |
| SPC/SA/Chol (7:3:1), n-HA/CS/KGM | Vancomycin | ~200 | Successful inhibition of the biofilm formation; sustained release from LLS | [100] | ||
| SPC/SA/Chol (7:1:1), β-TCP | Ceftazimide | ~160 | Significant in vitro anti-biofilm activity during the longer incubation time; sustained drug release | [101] | ||
| BBLs | DSPC/Chol/ALN-TEG-Chol | Oxacillin | ~100 | S. aureus | Significantly higher antibacterial effects than with DSPC/Chol liposomes; fast and strong binding to hydroxyapatite | [102] |
| Liposomes-in-hydrogel | SPC, chitosan hydrogel | Mupirocin | 920 | S. aureus | Prevention of biofilm formation; reduced cytotoxicity; in vivo efficacy comparable to the marketed product | [103] |
| DPPC/Chol/PEG-DSPE/rhodamine-DPPE (1:1:0.05:0.001), PEG-gelatin hydrogel | Ciprofloxacin | ~100 | P. aeruginosa | Prevention of bacterial adhesion and biofilm formation on urinary catheters; prolonged drug release; improved biocompatibility of coated catheters; effective delay of the bateriuria in vivo | [104,105] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rukavina, Z.; Vanić, Ž. Current Trends in Development of Liposomes for Targeting Bacterial Biofilms. Pharmaceutics 2016, 8, 18. https://doi.org/10.3390/pharmaceutics8020018
Rukavina Z, Vanić Ž. Current Trends in Development of Liposomes for Targeting Bacterial Biofilms. Pharmaceutics. 2016; 8(2):18. https://doi.org/10.3390/pharmaceutics8020018
Chicago/Turabian StyleRukavina, Zora, and Željka Vanić. 2016. "Current Trends in Development of Liposomes for Targeting Bacterial Biofilms" Pharmaceutics 8, no. 2: 18. https://doi.org/10.3390/pharmaceutics8020018