
Challenges, Solutions, and Quality Metrics of Personal Genome Assembly in Advancing Precision Medicine


Abstract
:1. Introduction
2. History of Human Genome Sequencing
3. Evolution of Sequencing Platforms
3.1. Illumina Platforms
3.2. Roche 454
3.3. Life Technology Ion Torrent
3.4. Qiagen Intelligent Biosystems
3.5. Pacific Biosciences
3.6. Oxford Nanopore
4. Current Solutions for De Novo Assembly and Post-Assembly
4.1. De Novo Assembly Approaches
4.2. Post-Assembly Approach
- (1)
- (2)
- Extending contigs and filling gaps. Tools such as GAA program and Reconciliator, integrate several different generated assemblies to extend or merge contigs [80].
- (3)
- Reads error correction. Tools such as ICORN, Artemis and AutoEditor are used to improve base calling accuracy, correct indels and deal with repeated regions [81].
- (4)
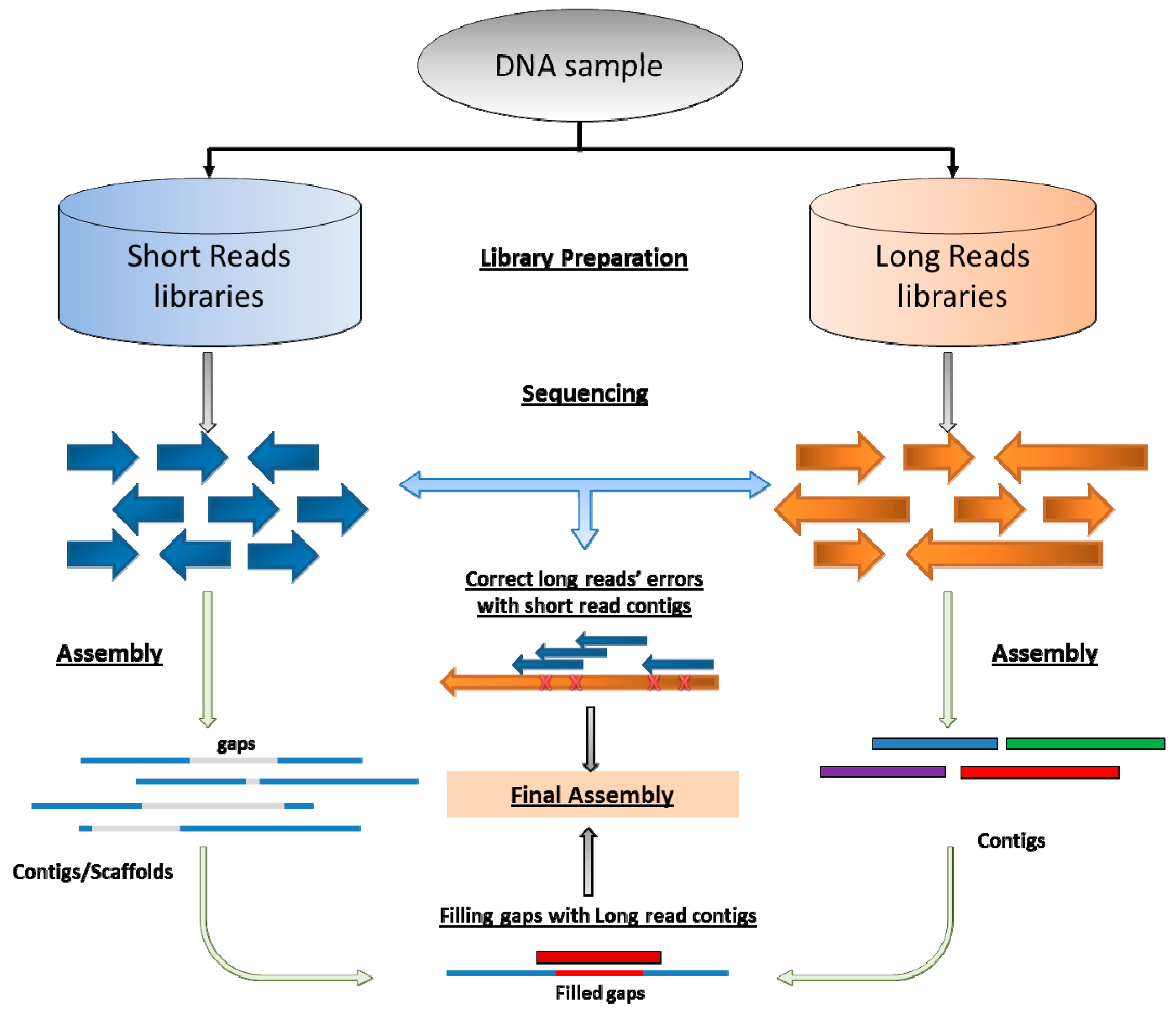
5. Coming Era of Long-Reads Sequencing and Hybrid Assembly
6. Computer Infrastructure Needs for Genome Assembly
6.1. Data Storage
- (1)
- The HPC clusters require a dedicated facility along with computer hardware and IT personnel, which is expensive. Additionally, ever growing number of associated data types require databases to store and manipulate metadata efficiently. This creates a need for database administration, which is an added cost.
- (2)
- In the near future, the scope of sequencing projects is expected to grow due to the reduced sequencing costs; hence, more samples will be sequenced within each project. In that case, the NGS data will become overwhelming and it will not be possible to efficiently store the data in a limited number of disks. Moreover, the need to keep backup copies of the data sets, which should be maintained to prevent accidental data loss, imposes extra data management and storage costs. Given the fact that enterprise level NGS platforms currently hold petabytes of data, the cost of the high fidelity backup can very expensive.
- (3)
- Currently, transferring the data from the NGS platform to a file system where the data is analyzed is carried out by either copying it into a large storage device then shipping it to the destination or by transmitting it over the Internet, which is bound by the network bandwidth. The network speeds are too slow to allow the transfer of terabytes of NGS data routinely over the Internet. With the advent of Internet 2 [98], there is significant optimism in the market; however, the production of new NGS datasets outpaces the growth in network throughput.
6.2. CPU/Memory
6.3. Cloud Computing
- (1)
- Infrastructure as a service (IaaS): In this model, the service provider offers the computing infrastructure that includes computational, storage and network resources as a service. Amazon EC2, Google Compute Engine and Microsoft’s Azure cloud services are the examples for this model type. Users, however, should be aware of significant costs associated in development and adaptation of tools, moving data to and from the environment. The Cloud does not provide end to end solutions; it does provide computers and network hardware with some job scheduling and system deployment facilities, most of which are usually not attuned to bioinformatics and big data I/O heavy processes.
- (2)
- Platform as a service (PaaS): The provider gives the freedom to the users to run their applications on the cloud using the provided computing platforms, which typically includes operating system, programming language execution environment, database, web servers, etc. The service provider hides the implementation details from the users. For this model, Amazon’s EC2 and Windows Azure can still be considered as primary examples as they provide both IaaS and PaaS services to users.Theoretically, all the de novo genome assembly methods designed to work in a parallel fashion on a HPC, can also be utilized to work on a cloud computing environment, when the underlying cloud platform or the infrastructure is set-up as a HPC. However, efficient methods are needed to distribute the computation across multiple nodes in a HPC or cloud computing environment. For this purpose, methods, such Contrail and CloudBrush, are specifically designed to use Hadoop, an open source implementation of the MapReduce that was developed by Google to simplify large data processing needs by scaling computation across many computers [106,107,108].The challenge in this approach is the limited efficiency in the generic environments supported by cloud providers. Small to medium size compute units available are usually affordable, however, the de novo assembly, being memory intensive requires larger memory, and more CPU configurations which costs significantly more. Extremely I/O and memory heavy processes (such as assemblers) encounter additional difficulties in moving data from one compute unit to another for parallel execution. Message Passing Interface (MPI) [109], shared memory or other message communication paradigms can be very challenging in cloud environments working through generic network configurations which are optimized for running small internet stores, but are not optimized for heavy tasks with significant reliance on message passing.
- (3)
- Software as a service (SaaS): The provider supplies all the software and databases to the user as a service, which eliminates the need to install and maintain the software. To mention a few: Illumina’s BaseSpace service with storage, read mapping, variant calling and de novo genome assembly services (backed by AWS) [110]; DNANexus’s cloud service (also backed by AWS) with tools for ChiP-seq, RNA-seq, read mapping, and variant detection [111]; High-performance Integrated Virtual Environment (HIVE) with storage and various tools including reference based or de novo assembly services hosted on enterprise, appliance or cloud deployments [112]; Galaxy providing spectrum of miscellaneous tools for mapping and de novo assembly through cloud or datacenters can be considered as major examples [113]. Another commercial example of SaaS is Life Technologies’ LifeScope cloud computing service, which provides one core of a 2.4-GHz Xeon processor with a 4 GB memory for $0.17 per core hour. There are also open source cloud solutions such as BioKepler, GenomeSpace, and Cloud BioLinux, which are accessible through Amazon EC2 cloud, as well as downloadable versions [113,114,115].
7. Quality Metrics and Parameters for Assembled Genome
8. Perspectives and Remaining Challenges
- (1)
- Quality metrics for personal genome assembly assessment. Currently, there is no “gold standard” for personal genome assessment. Many parameters need to be considered in genome assembly assessment, including completeness, continuity, accuracy, etc. There have been many scoring metrics developed for genome assembly assessment [73,74,75]. However, not all of them have been directly applied for human genome assessment.
- (2)
- Best practice of personal genome assembly workflow. As stated in Section 4 (Current solutions for de novo assembly and post-assembly), there is no single “one-size-fits-all” pipeline for de novo genome assembly. However, this conclusion was made based on studies of using simulated data or a single chromosome of the human genome [72,75]. Therefore, a comprehensive study needs to be performed on human genome assembly with real sequencing data. The best practice guidelines for a personal genome assembly pipeline starting from study design and ending with bioinformatics data analysis will be derived from such a study and will allow understanding of the relationships among various parameters such as NGS platforms, read length, sequence coverage and assembly process.
- (3)
- Personalized genome annotation. In order to use a personal genome as reference to uncover genetic variations in diseased tissues, each individual genome needs to be well annotated with various biological features. A comprehensive bioinformatics process needs to be established to perform multiple tasks such as comparative genome analysis with the public reference genome (to identify SNPs/SVs and create cross-reference), structure analysis of gene/transcripts on the genome, identification of functional genomic loci, and calculation of sequence conservation score at each base position. This basic information may facilitate the understanding of biological effects for variants discovered in diseased tissue from the same individual.
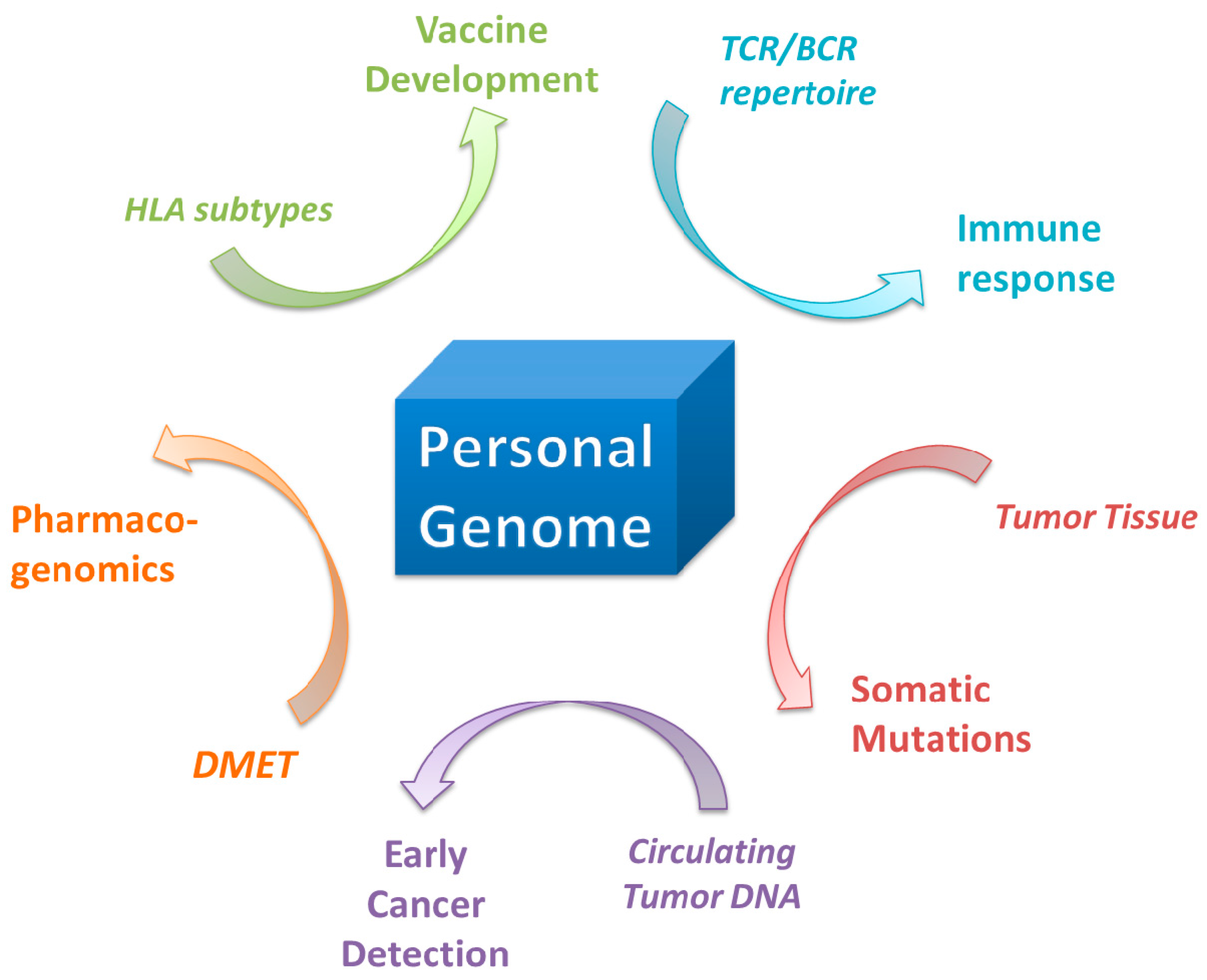
9. Application in Pharmaceuticals and Pharmacogenomics
10. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
Disclaimer
References
- The NIH Director. Available online: http://www.nih.gov/about/director/09172015-statement-PMI.htm (accessed on 12 April 2016).
- The Precision Medicine Initiative. Available online: https://www.whitehouse.gov/precision-medicine (accessed on 12 April 2016).
- Collins, F.S.; Harold, V. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. Clinvar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Degner, J.F.; Marioni, J.C.; Pai, A.A.; Pickrell, J.K.; Nkadori, E.; Gilad, Y.; Pritchard, J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 2009, 25, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Ewing, A.D.; Houlahan, K.E.; Hu, Y.; Ellrott, K.; Caloian, C.; Yamaguchi, T.N.; Bare, J.C.; P’ng, C.; Waggott, D.; Sabelnykova, V.Y.; et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 2015, 12, 623–630. [Google Scholar] [CrossRef]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Kidd, J.M.; Sampas, N.; Antonacci, F.; Graves, T.; Fulton, R.; Hayden, H.S.; Alkan, C.; Malig, M.; Ventura, M.; Giannuzzi, G.; et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods 2010, 7, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Logan, D.W. Do you smell what I smell? Genetic variation in olfactory perception. Biochem. Soc. Trans. 2014, 42, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.K.; Woo, M.H.; McLeod, H.L. Genetic basis of drug metabolism. Am. J. Health Syst. Pharm. 2012, 59, 2061–2069. [Google Scholar]
- Zhou, S.F.; Liu, J.P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Arstila, T.P.; Casrouge, A.; Baron, V.; Even, J.; Kanellopoulos, J.; Kourilsky, P. A direct estimate of the human alphabeta t cell receptor diversity. Science 1999, 286, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Rasmussen, M. Beyond the reference genome. Nat. Biotechnol. 2015, 33, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Nature Genetics. Whole genome? Nat Genet. 2015, 47, 963. [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1305–1351. [Google Scholar] [CrossRef]
- DNA Sequencing Costs. Available online: http://www.genome.gov/sequencingcosts/ (accessed on 12 April 2016).
- Anonymous. Human genome at ten: The sequence explosion. Nature 2010, 464, 670–671. [Google Scholar]
- EBI Search. Available online: http://www.ebi.ac.uk/ebisearch/search.ebi?db=genome_assembly&t=assembly&sort=_relevance&page=1&f=TAXONOMY:9606 (accessed on 12 April 2016).
- Homo sapiens. Available online: http://www.ncbi.nlm.nih.gov/genome/genomes/51 (accessed on 12 April 2016).
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.E.; Fan, J.; Willour, V.; Pennacchio, L.A.; Warrington, J.A.; Hu, A.; Chapelle, A.; Lehesjoki, A.; Cox, D.R.; Myers, R.M. Construction of a 750-kb bacterial clone contig and restriction map in the region of human chromosome 21 containing the progressive myoclonus epilepsy gene. Genome Res. 1996, 6, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.C.; Boysen, C.; Wang, K.; Hood, L. Pairwise end sequencing: A unified approach to genomic mapping and sequencing. Genomics 1994, 26, 345–353. [Google Scholar] [CrossRef]
- Dear, P.H. Genome mapping. eLS 2005. [Google Scholar] [CrossRef]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.-J.; Makhijani, V.; Roth, G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Li, R.; Li, Y.; Tian, G.; Goodman, L.; Fan, W.; Zhang, J.; Li, J.; Zhang, J. The diploid genome sequence of an asian individual. Nature 2008, 456, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-I.; Ju, Y.S.; Park, H.; Kim, S.; Lee, S.; Yi, J.-H.; Mudge, J.; Miller, N.A.; Hong, D.; Bell, C.J. A highly annotated whole-genome sequence of a korean individual. Nature 2009, 460, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, H.; Luo, R.; Wu, H.; Zhu, H.; Li, R.; Cao, H.; Wu, B.; Huang, S.; Shao, H.; et al. Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. Nat. Biotechnol. 2011, 29, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [Green Version]
- Zook, J.M.; Catoe, D.; McDaniel, J.; Vang, L.; Spies, N.; Sidow, A.; Weng, Z.; Liu, Y.; Mason, C.; Alexander, N.; et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. bioRxiv 2015. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, R.R.; Heiner, C.; Kent, S.B.; Hood, L. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, H.; Gesteland, R. Capillary gel electrophoresis for rapid, high resolution DNA sequencing. Nucleic Acids Res. 1990, 18, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Luckey, J.A.; Drossman, H.; Kostichka, A.J.; Mead, D.A.; D’Cunha, J.; Norris, T.B.; Smith, L.M. High speed DNA sequencing by capillary electrophoresis. Nucleic Acids Res. 1990, 18, 4417–4421. [Google Scholar] [CrossRef] [PubMed]
- Drossman, H.; Luckey, J.; Kostichka, A.J.; D’Cunha, J.; Smith, L.M. High-speed separations of DNA sequencing reactions by capillary electrophoresis. Anal. Chem. 1990, 62, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Johannes, K.; Ptak, S.E.; Briggs, A.W.; Ronan, M.T.; Simons, J.F.; Du, L.; Egholm, M.; Rothberg, J.M.; Paunovic, M.; et al. Analysis of one million base pairs of neanderthal DNA. Nature 2006, 444, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Quinlan, A.; Peckham, H.E.; Makowsky, K.; Tao, W.; Woolf, B.; Shen, L.; Donahue, W.F.; Tusneem, N.; Stromberg, M.P.; et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008, 18, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Steinmann, K. Single molecule sequencing with a heliscope genetic analysis system. Curr. Protoc. Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Illumina. Available online: http://www.illumina.com/technology/next-generation-sequencing/solexa-technology.html (accessed on 12 April 2016).
- Eid, J.; Adrinan, F.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- SMRT Sequencing: Read Lengths. Available online: http://www.pacb.com/smrt-science/smrt-sequencing/read-lengths/ (accessed on 12 April 2016).
- Koren, S.; Harhay, G.; Smith, T.P.; Bono, J.L.; Harhay, D.M.; Mcvey, S.D.; Radune, D.; Bergman, N.H.; Phillippy, A.M. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 2013, 14, R101. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Phillippy, A. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015, 23C, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the oxford nanopore minion sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fiddes, I.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the minion nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Pathak, B.; Lofas, H.; Prasongkit, J.; Grigoriev, A.; Ahuja, R.; Scheicher, R.H. Double-functionalized nanopore-embedded gold electrodes for rapid DNA sequencing. Appl. Phys. Lett. 2012, 100, 154–159. [Google Scholar] [CrossRef]
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaisson, M.J.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L.; Sutton, G.G.; Jones, S.J.; Holt, R.A. Assembling millions of short DNA sequences using ssake. Bioinformatics 2007, 23, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Sharcgs, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing. Genome Res. 2007, 17, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Reinhardt, J.A.; Baltrus, D.A.; Hickenbotham, M.T.; Magrini, V.; Mardis, E.R.; Dangl, J.L.; Jones, C.D. Extending assembly of short DNA sequences to handle error. Bioinformatics 2007, 23, 2942–2944. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A.; et al. A whole-genome assembly of drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Batzoglou, S.; Jaffe, D.B.; Stanley, K.; Butler, J.; Gnerre, S.; Mauceli, E.; Berger, B.; Mesirov, J.P.; Lander, E.S. Arachne: A whole-genome shotgun assembler. Genome Res. 2002, 12, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.P. Generating a genome assembly with pcap. Curr. Protoc. Bioinform. 2005. [Google Scholar] [CrossRef]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Pevzner, P.A.; Tang, H.; Waterman, M.S. An eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA 2001, 98, 9748–9753. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; MacCallum, I.; Kleber, M.; Shlyakhter, I.A.; Belmonte, M.K.; Lander, E.S.; Nusbaum, C.; Jaffe, D.B. Allpaths: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008, 18, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. Abyss: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Yang, Y.; Tang, Y.; Shang, J.; Shen, B. A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLoS ONE 2011, 6, e17915. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B. Comparison of the two major classes of assembly algorithms: Overlap-layout-consensus and de-bruijn-graph. Brief. Funct. Genomics 2012, 11, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.; Bradnam, K.; St John, J.; Darling, A.; Lin, D.; Fass, J.; Yu, H.O.; Buffalo, V.; Zerbino, D.R.; Diekhans, M.; et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011, 21, 2224–2241. [Google Scholar] [CrossRef] [PubMed]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2013, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoc, T.; Pabinger, S.; Canzar, S.; Liu, X.; Su, Q.; Puiu, D.; Tallon, L.J.; Salzberg, S.L. Gage-b: An evaluation of genome assemblers for bacterial organisms. Bioinformatics 2013, 29, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Phillippy, A.M.; Zimin, A.; Puiu, D.; Magoc, T.; Koren, S.; Treangen, T.J.; Schatz, M.C.; Delcher, A.L.; Roberts, M.; et al. Gage: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012, 22, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Galardini, M.; Biondi, E.G.; Bazzicalupo, M.; Mengoni, A. Contiguator: A bacterial genomes finishing tool for structural insights on draft genomes. Sour. Code Biol. Med. 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- van Hijum, S.A.; Zomer, A.L.; Kuipers, O.P.; Kok, J. Projector 2: Contig mapping for efficient gap-closure of prokaryotic genome sequence assemblies. Nucleic Acids Res. 2005, 33, W560–W566. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.C.; Schuster, S.C.; Huson, D.H. Oslay: Optimal syntenic layout of unfinished assemblies. Bioinformatics 2007, 23, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Husemann, P.; Stoye, J. R2cat: Synteny plots and comparative assembly. Bioinformatics 2010, 26, 570–571. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Ye, L.; Gao, H.; Minx, P.; Warren, W.C.; Weinstock, G.M. Graph accordance of next-generation sequence assemblies. Bioinformatics 2012, 28, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Sanders, M.; Berriman, M.; Newbold, C. Iterative correction of reference nucleotides (icorn) using second generation sequencing technology. Bioinformatics 2010, 26, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.M.; Lorenzini, D.M.; Mendes, P.N.; Satake, T.S.; Sousa, G.R.; Campos, L.M.; Mazzoni, C.J.; Wagner, G.; Pires, P.F.; Grisard, E.C.; et al. Garsa: Genomic analysis resources for sequence annotation. Bioinformatics 2005, 21, 4302–4303. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.G.; Paixao, R.; Souza, R.C.; Costa, G.C.; Barrientos, F.J.; Santos, M.T.; Almeida, D.F.; Vasconcelos, A.T. A system for automated bacterial (genome) integrated annotation–sabia. Bioinformatics 2004, 20, 2832–2833. [Google Scholar] [CrossRef] [PubMed]
- Swain, M.T.; Tsai, I.J.; Assefa, S.A.; Newbold, C.; Berriman, M.; Otto, T.D. A post-assembly genome-improvement toolkit (pagit) to obtain annotated genomes from contigs. Nat. Protoc. 2012, 7, 1260–1284. [Google Scholar] [CrossRef] [PubMed]
- Assefa, S.; Keane, T.M.; Otto, T.D.; Newbold, C.; Berriman, M. Abacas: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics 2009, 25, 1968–1969. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.J.; Otto, T.D.; Berriman, M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010, 11, R41. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Dillon, G.P.; Degrave, W.S.; Berriman, M. Ratt: Rapid annotation transfer tool. Nucleic Acids Res. 2011, 39, e57. [Google Scholar] [CrossRef] [PubMed]
- Wences, A.H.; Schatz, M. Metassembler: Merging and optimizing de novo genome assemblies. Genome Biol. 2015, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef] [PubMed]
- Mapleson, D.; Drou, N.; Swarbreck, D. Rampart: A workflow management system for de novo genome assembly. Bioinformatics 2015, 31, 1824–1826. [Google Scholar] [CrossRef] [PubMed]
- FALCON: Experimental PacBio diploid assembler. Available online: https://github.Com/pacificbiosciences/falcon (accessed on 12 April 2016).
- Pendleton, M.; Sebra, R.; Pang, A.W.C.; Ummat, A.; Franzen, O.; Rausch, T.; Stutz, A.M.; Stedman, W.; Anantharaman, T.; Hastie, A.; et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Meth. 2015, 12, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Utturkar, S.M.; Klingeman, D.M.; Land, M.L.; Schadt, C.W.; Doktycz, M.J.; Pelletier, D.A.; Brown, S.D. Evaluation and validation of de novo and hybrid assembly techniques to derive high-quality genome sequences. Bioinformatics 2014, 30, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Lai, Z.; Lane, T.; Nageswara-Rao, M.; Okada, M.; Jasieniuk, M.; O’Geen, H.; Kim, R.W.; Sammons, R.D.; Rieseberg, L.H. De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms. Plant Physiol. 2014, 166, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, A.H.; Derrington, I.M.; Ross, B.C.; Brinkerhoff, H.; Adey, A.; Nova, I.C.; Craig, J.M.; Langford, K.W.; Samson, J.M.; Daza, R. Decoding long nanopore sequencing reads of natural DNA. Nat. Biotechnol. 2014, 32, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Monjo, T.; Hoang, P.H.; Yoshimura, J.; Yurino, H.; Mitsui, J.; Ishiura, H.; Takahashi, Y.; Ichikawa, Y.; Goto, J. Rapid detection of expanded short tandem repeats in personal genomics using hybrid sequencing. Bioinformatics 2014, 30, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Internet2: Uninhibited Performance. Available online: http://www.Internet2.Edu/products-services/advanced-networking/ (accessed on 12 April 2016).
- Henson, J.; Tischler, G.; Ning, Z. Next-generation sequencing and large genome assemblies. Pharmacogenomics 2012, 13, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tarraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The european nucleotide archive. Nucleic Acids Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- CLCbio. Available online: http://www.clcbio.com/files/whitepapers/whitepaper-denovo-assembly-4.pdf (accessed on 12 April 2016).
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Amazon EC2. Available online: http://aws.Amazon.Com/ec2 (accessed on 12 April 2016).
- Azure. Available online: https://azure.Microsoft.Com/en-us/ (accessed on 12 April 2016).
- Google Cloud. Available online: https://cloud.Google.Com/ (accessed on 12 April 2016).
- Schatz, M. Assembly of Large Genomes Using Cloud Computing; Illumina Sequencing Panel: Toronto, ON, Canada, 2010. [Google Scholar]
- Chang, Y.-J.; Chen, C.-C.; Chen, C.-L.; Ho, J.-M. A de novo next generation genomic sequence assembler based on string graph and mapreduce cloud computing framework. BMC Genomics 2012, 13, S28. [Google Scholar] [PubMed]
- Dean, J.; Ghemawat, S. Mapreduce: Simplified data processing on large clusters. Commun. ACM 2008, 51, 107–113. [Google Scholar] [CrossRef]
- MPI Forum. MPI: A message-passing interface standard version 3.1. 2015. Available online: http://www.mpi-forum.org/docs/mpi-3.1/mpi31-report.pdf (accessed on 12 April 2016).
- BaseSpace. Available online: https://basespace.Illumina.Com/home/index (accessed on 12 April 2016).
- DNANexus. Available online: http://www.dnanexus.com (accessed on 12 April 2016).
- Simonyan, V.; Mazumder, R. High-performance integrated virtual environment (hive) tools and applications for big data analysis. Genes 2014, 5, 957–981. [Google Scholar] [CrossRef] [PubMed]
- Goecks, J.; Nekrutenko, A.; Taylor, J. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [PubMed]
- Altintas, I. Distributed workflow-driven analysis of large-scale biological data using biokepler. In Proceedings of the ACM 2nd International Workshop on Petascal Data Analytics: Challenges and Opportunities, Sesttle, WA, USA, 12–18 November 2011.
- GenomeSpace. Available online: http://www.genomespace.org (accessed on 12 April 2016).
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. Quast: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Compass. Scripts to Compare a DNA Sequence Assembl to a Trusted Reference Sequence. Available online: https://github.com/jfass/compass (accessed on 12 April 2016).
- Hunt, M.; Kikuchi, T.; Sanders, M.; Newbold, C.; Berriman, M.; Otto, T.D. Reapr: A universal tool for genome assembly evaluation. Genome Biol. 2013, 14, R47. [Google Scholar] [CrossRef] [PubMed]
- Besenbacher, S.; Liu, S.; Izarzugaza, J.M.; Grove, J.; Belling, K.; Bork-Jensen, J.; Huang, S.; Als, T.D.; Li, S.; Yadav, R.; et al. Novel variation and de novo mutation rates in population-wide de novo assembled danish trios. Nat. Commun. 2015, 6, 5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilthey, A.; Cox, C.; Iqbal, Z.; Nelson, M.R.; McVean, G. Improved genome inference in the mhc using a population reference graph. Nat. Genet. 2015, 47, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wu, H.; Luo, R.; Huang, S.; Sun, Y.; Tong, X.; Xie, Y.; Liu, B.; Yang, H.; Zheng, H.; et al. De novo assembly of a haplotype-resolved human genome. Nat. Biotechnol. 2015, 33, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.J.; Phan, H.; Gerry, B.M.; Kuhn, A.; Hong, L.Z.; Yao, M.O.; Poon, P.S.; Unger, M.A.; Jones, R.C.; Quake, S.R.; et al. A microfluidic device for preparing next generation DNA sequencing libraries and for automating other laboratory protocols that require one or more column chromatography steps. PLoS ONE 2013, 8, e64084. [Google Scholar] [CrossRef] [PubMed]
- Markey, A.L.; Mohr, S.; Day, P.J. High-throughput droplet PCR. Methods 2010, 50, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Tewhey, R.; Warner, J.B.; Nakano, M.; Libby, B.; Medkova, M.; David, P.H.; Kotsopoulos, S.K.; Samuels, M.L.; Hutchison, J.B.; Larson, J.W.; et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2009, 27, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Leamon, J.H.; Link, D.R.; Egholm, M.; Rothberg, J.M. Overview: Methods and applications for droplet compartmentalization of biology. Nat. Methods 2006, 3, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Wang, J.; Potanina, A.; Quake, S.R. Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 2011, 29, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Parmar, N.; Bhella, G.K.; Robinson, I.K. A simple filtration technique for obtaining purified human chromosomes in suspension. Biotechniques 2014, 56, 257–261. [Google Scholar] [PubMed]
- Dolezel, J.; Vrana, J.; Safar, J.; Bartos, J.; Kubalakova, M.; Simkova, H. Chromosomes in the flow to simplify genome analysis. Funct. Integr. Genom. 2012, 12, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burge, C.B.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.B.; Karlin, S. Finding the genes in genomic DNA. Curr. Opin. Struct. Biol. 1998, 8, 346–354. [Google Scholar] [CrossRef]
- Parra, G.; Blanco, E.; Guigó, R. Geneid in drosophila. Genome Res. 2000, 10, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Schweikert, G.; Zien, A.; Zeller, G.; Behr, J.; Dieterich, C.; Ong, C.S.; Philips, P.; De Bona, F.; Hartmann, L.; Bohlen, A.; et al. Mgene: Accurate svm-based gene finding with an application to nematode genomes. Genome Res. 2009, 19, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Braker1: Unsupervised rna-seq-based genome annotation with genemark-et and augustus. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Stanke, M. Webaugustus—A web service for training augustus and predicting genes in eukaryotes. Nucleic Acids Res. 2013, 41, W123–W128. [Google Scholar] [CrossRef] [PubMed]
- Schweikert, G.; Behr, J.; Zien, A.; Zeller, G.; Ong, C.S.; Sonnenburg, S.; Ratsch, G. Mgene.Web: A web service for accurate computational gene finding. Nucleic Acids Res. 2009, 37, W312–W316. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Shailza, S.; Balwant Kumar, M.; Durlabh Kumar, S. Molecular drug targets and structure based drug design: A holistic approach. Bioinformation 2006, 1, 314–320. [Google Scholar]
- Seib, K.L.; Dougan, G.; Rappuoli, R. The key role of genomics in modern vaccine and drug design for emerging infectious diseases. PLoS Genet. 2009, 5, e1000612. [Google Scholar] [CrossRef] [PubMed]
- Green, E.D.; Guyer, M.S.; National Human Genome Research Institute. Charting a course for genomic medicine from base pairs to bedside. Nature 2011, 470, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. Available online: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/ucm284076.htm (accessed on 12 April 2016).
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome p450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Price, E.T.; Chang, C.W.; Li, Y.; Huang, Y.; Guo, L.W.; Guo, Y.; Kaput, J.; Shi, L.; Ning, B. Gene expression variability in human hepatic drug metabolizing enzymes and transporters. PLoS ONE 2013, 8, e60368. [Google Scholar] [CrossRef] [PubMed]
- Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: http://www.Fda.Gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.Htm (accessed on 12 April 2016).
- Lewis, D.F.; Watson, E.; Lake, B.G. Evolution of the cytochrome P450 superfamily: Sequence alignments and pharmacogenetics. Mutat. Res. 1998, 410, 245–270. [Google Scholar] [CrossRef]
- Londin, E.R.; Clark, P.; Sponziello, M.; Kricka, L.J.; Fortina, P.; Park, J.Y. Performance of exome sequencing for pharmacogenomics. Per. Med. 2014, 12, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yu, D.; Chen, J.; Cao, R.; Yang, J.; Wang, H.; Ji, X.; Ning, B.; Shi, T. Re-annotation of presumed noncoding disease/trait-associated genetic variants by integrative analyses. Sci. Rep. 2015, 5, 9453. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Fukushima, Y.; Yokota, S.; Fukami, T.; Takamiya, M.; Aoki, Y.; Yokoi, T.; Nakajima, M. Cyp2a7 pseudogene transcript affects cyp2a6 expression in human liver by acting as a decoy for mir-126. Drug Metab. Dispos. 2015, 43, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Hetherington, S.; Hughes, A.R.; Mosteller, M.; Shortino, D.; Baker, K.L.; Spreen, W.; Lai, E.; Davies, K.; Handley, A.; Dow, D.J.; et al. Genetic variations in HLA-b region and hypersensitivity reactions to abacavir. Lancet 2002, 359, 1121–1122. [Google Scholar] [CrossRef]
- McCormack, M.; Alfirevic, A.; Bourgeois, S.; Farrell, J.J.; Kasperavičiūtė, D.; Carrington, M.; Sills, G.J.; Marson, T.; Jia, X.; de Bakker, P.I.; et al. Hla-a*3101 and carbamazepine-induced hypersensitivity reactions in europeans. N. Engl. J. Med. 2011, 364, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.M.; Trowsdale, J. Major histocompatibility complex structure and function. Curr. Opin. Immunol. 1989, 2, 93–98. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genome Build # | Release Year | Total Genome Length | Total Non-N Bases | N50 | Number of Gaps | # of Scaffolds | # Unplaced Scaffolds |
|---|---|---|---|---|---|---|---|
| 35 | 2004 | 3,091,649,889 | 2,866,200,199 | 38,509,590 | 292 | 377 | 86 |
| 36 | 2006 | 3,104,054,490 | 2,881,649,121 | 38,509,590 | 292 | 367 | 88 |
| 37 | 2009 | 3,137,144,693 | 2,897,299,566 | 46,395,641 | 357 | 249 | 59 |
| 38 | 2013 | 3,209,286,105 | 3,049,316,098 | 67,794,873 | 875 | 473 | 169 |
| Platform | Mode | Read-Length | Reads Passing Filter per Run | Output | Run Time | Quality | Cost/Run | Instrument Price |
|---|---|---|---|---|---|---|---|---|
| Illumina HiSeq 2000/2500 | High-Output | 1 × 36–2 × 125 | 4 B | 128 GB–1 TB | 1–6 days | Q30 ≥ 80% | ~$29K | $740K |
| Rapid | 1 × 36–2 × 150 | 600 M | 18 GB–300 GB | 7–60 h | Q30 ≥ 75% | ~$8K | ||
| Illumina HiSeq X ten | X ten | 2×150 | 5.3–6 B | 1.6–1.8 TB | <3 days | Q30 ≥ 75% | ~$12K | $1M* |
| Roche 454 FLX system | Titanium XL+ | 700 | 1 M | 700 MB | 23 h | 99.997% | ~$6K | ~$500K |
| Life Technologies Ion Torrent | Proton I | 200 | 165 M | ~10 GB | 2–4 h | ~$1000 | $149K | |
| Proton II | 100 | 660 M | ~32 GB | 2–4 h | ||||
| Intelligent Biosystems (Qiagen) | MAX-Seq | 2 × 55 | 75 M/lane | 132 GB | 2.5 days | ~$1200 | ~$270K | |
| Mini-20 | 2 × 100 | 20 M/lane | 80 GB | ~$150–300/sample | $125K | |||
| PacBio RS | RS II | 10–15 KB | 50 K | 500 MB–1 GB | 4 h | >99.999% | ~$400 | ~$700K |
| Oxford Nanopore | miniON | >200 KB | no fixed run time (~1 bp per nanosecond) | ≤$900 | ~$1000 |
| Approaches | Commonly Used Tools | Notes |
|---|---|---|
| Assembly Approaches | ||
| de Bruijn graph | EULER, ALLPATHS, Velvet, ABySS, SOAPdenovo, etc. | For shorter reads (25–100 bp) assembly |
| Overlap-layout-consensus (OLC) | SSAKE, SHARCGS, VCAKE, Celera Assembler, Arachne, PCAP, HGAP, etc. | For longer reads (100–800 bp) and long reads assembly |
| Post-Assembly Approaches | ||
| Contigs orientation and visualization | AlignGraph, ABACAS, CONTIGuator, Projector2, OSLay and r2cat, etc. | |
| Extending contigs and filling gaps | IMAGE, GAA program, Reconciliator, GAPFiller, Pilon etc. | |
| Reads error correction | ICORN, AutoEditor, REAPR etc. | |
| Unmapped reads Annotation | RATT, Ensembl, GARSA and SABIA, etc. | |
| Parameters | Notes | |
|---|---|---|
| Contig Statistics | ||
| Number of contigs | total number of assembled contigs | |
| Max length of contigs | the longest contig | |
| Min length of contigs | the shortest of contig | |
| Total length of contigs | sum of the length of all contigs | |
| Nx_plot | contig length for x% of the bases of assembled contigs, where 0 < x < 100 | |
| NGx_plot | contig length for x% of the bases of reference genome, where 0 < x < 100 | |
| NAx_plot | contig length for x% of the bases of assembled contigs after correction, where 0 < x < 100 | |
| NGAx_plot | contig length for x% of the bases of the reference genome after correction, where 0 < x < 100 | |
| Assembly Errors | ||
| Number of misassembles | total number of assembly errors, include miss-join, base error, false indel, etc. | |
| miss-join | number of miss-join | |
| base error | number of base error | |
| false indel | number of false indel | |
| Number of misassembled contigs (parsimony) | number of contigs with assembly errors | |
| Total length of misassembled contigs | sum of the length of misassembled contigs | |
| Unaligned cotigs | total number of contigs could not be mapped to the reference genome | |
| alternative human reference | could be mapped to alternative human reference genomes | |
| nonhuman primate genome references | could be mapped to nonhuman primate reference genomes | |
| Ambiguously mapped contigs | cotigs mapped to multiple location on the reference genome | |
| Fragment coverage distribution (FCD) | local assembly error detected by fragment coverage of assembled contigs by sequence reads | |
| Genome Coverage | ||
| Genome coverage fraction | percentage of the reference genome covered by assemblies | |
| Known gene complete coverage fraction | percentage of known gene covered completely by assemblies | |
| Known gene partial coverage fraction | percentage of known gene covered partially by assemblies | |
| Know exon complete coverage fraction | percentage of known exon covered completely by assemblies | |
| Know exon partial coverage fraction | percentage of known exon covered partially by assemblies | |
| Duplication ratio (multiplicity) | ratio of total length of aligned contigs vs. total covered the reference genome | |
| Alignable ratio (validity) | ratio of total aligned contigs vs. total assembled contigs | |
| GC content | percentage of GC content in assembled contigs | |
| Number of SNVs | total number of single nucleotide variation (SNV) detected in assembled contigs | |
| Number of SNPs | total number of single nucleotide polymorphism (SNP) detected in assembled contigs | |
| Number of small indels | total number of small indels detected in assembled contigs | |
| Number of inversion | total number of inversion detected in assembled contigs | |
| Number of translocation | total number of translocation detected in assembled contigs | |
| SNVs/100 kb | number of SNVs per 100 kb block | |
| SNPs/100 kb | number of SNPs per 100 kb block | |
| indels/100 kb | number of small indels per 100 kb block | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, W.; Wu, L.; Yavas, G.; Simonyan, V.; Ning, B.; Hong, H. Challenges, Solutions, and Quality Metrics of Personal Genome Assembly in Advancing Precision Medicine. Pharmaceutics 2016, 8, 15. https://doi.org/10.3390/pharmaceutics8020015
Xiao W, Wu L, Yavas G, Simonyan V, Ning B, Hong H. Challenges, Solutions, and Quality Metrics of Personal Genome Assembly in Advancing Precision Medicine. Pharmaceutics. 2016; 8(2):15. https://doi.org/10.3390/pharmaceutics8020015
Chicago/Turabian StyleXiao, Wenming, Leihong Wu, Gokhan Yavas, Vahan Simonyan, Baitang Ning, and Huixiao Hong. 2016. "Challenges, Solutions, and Quality Metrics of Personal Genome Assembly in Advancing Precision Medicine" Pharmaceutics 8, no. 2: 15. https://doi.org/10.3390/pharmaceutics8020015