
Venetoclax (ABT-199) Might Act as a Perpetrator in Pharmacokinetic Drug–Drug Interactions

Abstract
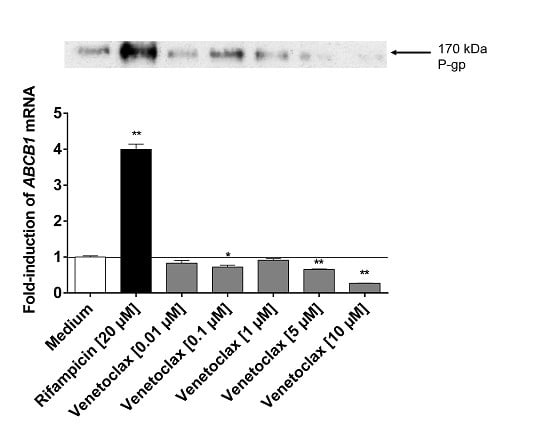
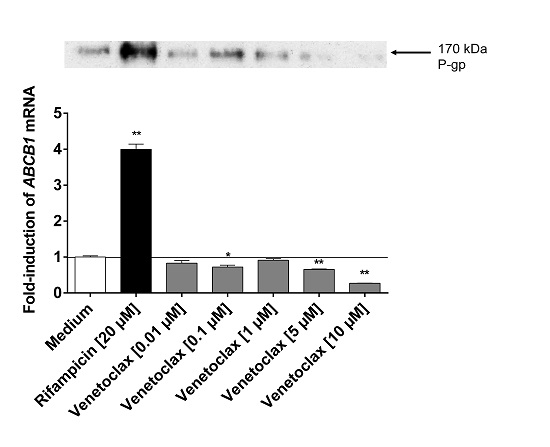
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
2.3. Cytotoxicity Assay
2.4. P-gp Inhibition Assay
2.5. BCRP Inhibition Assay
2.6. OATP Inhibition Assay
2.7. Inhibition of CYPs
2.8. Growth Inhibition (Proliferation) Assay
2.9. Induction Assay
2.10. Quantification of mRNA Expression by Real-Time RT-PCR
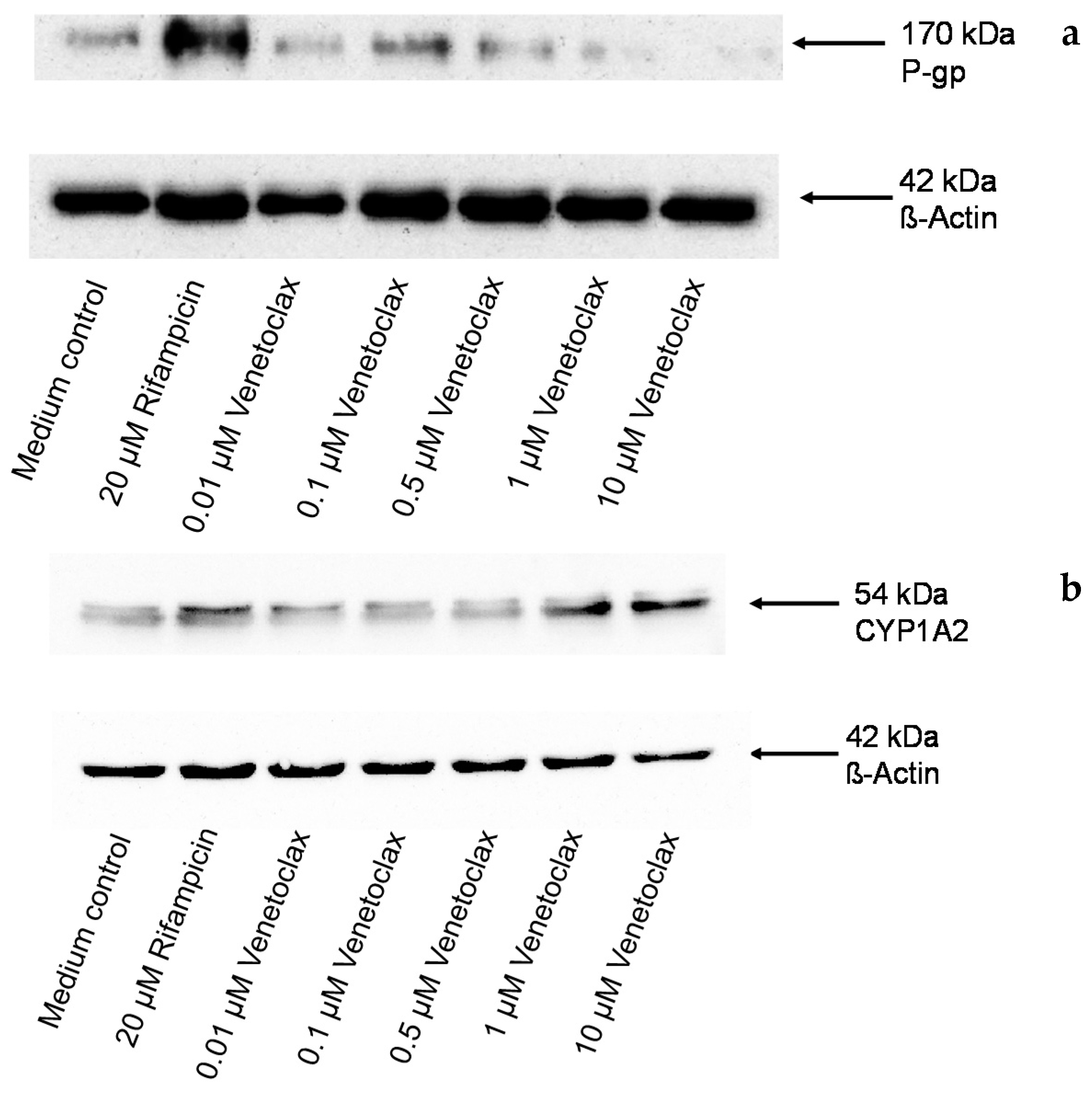
2.11. Western Blot Analysis of P-gp and CYP1A2 Protein Expression
2.12. AhR Reporter Gene Assay
2.13. Statistical Analysis
3. Results
3.1. Venetoclax Inhibits P-gp
3.2. Venetoclax Inhibits BCRP
3.3. Venetoclax Inhibits OATP1B1 and OATP1B3
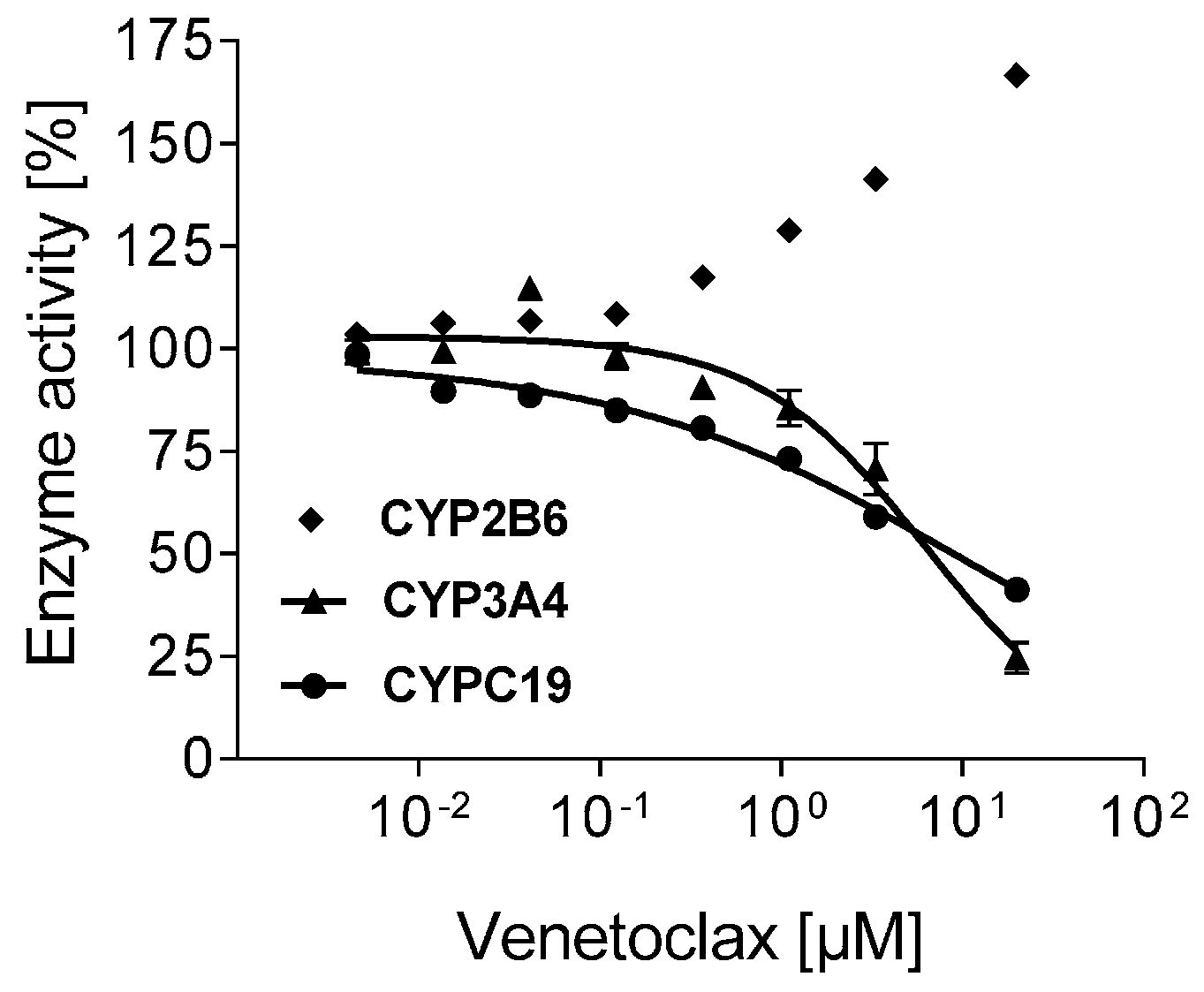
3.4. Inhibition of CYPs by Venetoclax
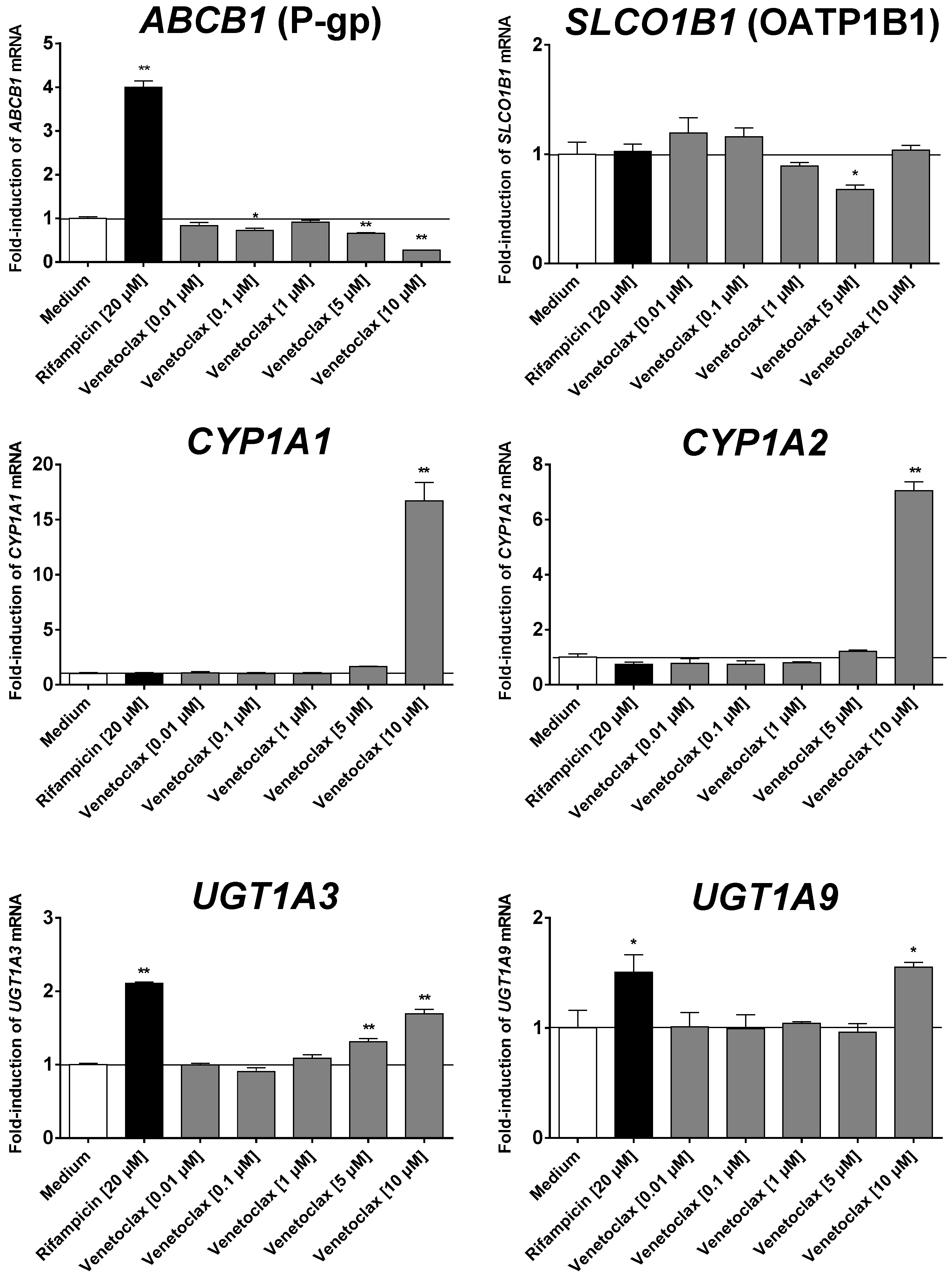
3.5. Influence of Venetoclax on the Expression of Drug Metabolising Enzymes and Drug Transporters
3.6. Venetoclax Does not Activate AhR
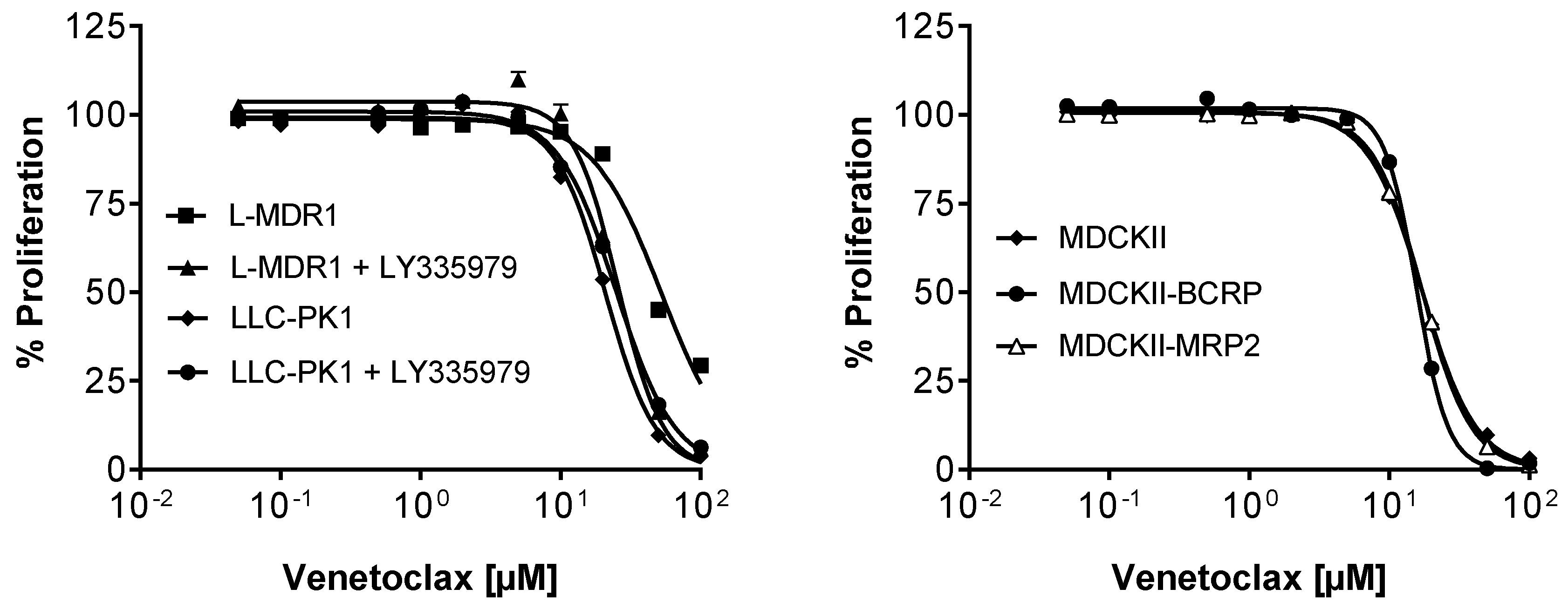
3.7. Efficacy in MDR Cell Lines
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, M.A.; Huang, D.; Roberts, A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin. Hematol. 2014, 51, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; Borst, P. Absence of the MDR1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Investig. 1995, 96, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Kool, M.; van Deemter, L.; Janssen, H.; Calafat, J.; Oomen, L.C.; Paulusma, C.C.; Oude Elferink, R.P.; Baas, F.; Schinkel, A.H.; et al. Drug export activity of the human canalicular multispecific organic anion transporter in polarized kidney MDCK cells expressing cMOAT (MRP2) cDNA. J. Clin. Investig. 1998, 101, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Merino, G.; Wagenaar, E.; Bolscher, E.; Novotna, M.; Jonker, J.W.; Schinkel, A.H. Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J. Pharmacol. Exp. Ther. 2005, 312, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Wagenaar, E.; Mol, C.A.; van Deemter, L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Investig. 1996, 97, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- Boesch, D.; Gavériaux, C.; Jachez, B.; Pourtier-Manzanedo, A.; Bollinger, P.; Loor, F. In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res. 1991, 51, 4226–4233. [Google Scholar] [PubMed]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G156–G164. [Google Scholar] [PubMed]
- Weiss, J.; Theile, D.; Spalwisz, A.; Burhenne, J.; Riedel, K.D.; Haefeli, W.E. Influence of sildenafil and tadalafil on the enzyme- and transporter-inducing effects of bosentan and ambrisentan in LS180 cells. Biochem. Pharmacol. 2013, 85, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; Koster, A.S.; Beijnen, J.H.; Schellens, J.H.; Meijerman, I. Comparison of two immortalized human cell lines to study nuclear receptor-mediated CYP3A4 induction. Drug Metab. Dispos. 2008, 36, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mugundu, G.M.; Desai, P.B.; Thummel, K.E.; Unadkat, J.D. Intestinal human colon adenocarcinoma cell line LS180 is an excellent model to study pregnane X receptor; but not constitutive androstane receptor; mediated CYP3A4 and multidrug resistance transporter 1 induction: Studies with anti-human immunodeficiency virus protease inhibitors. Drug Metab. Dispos. 2008, 36, 1172–1180. [Google Scholar] [PubMed]
- Weiss, J.; Herzog, M.; Haefeli, W.E. Differential modulation of the expression of important drug metabolising enzymes and transporters by endothelin-1 receptor antagonists ambrisentan and bosentan in vitro. Eur. J. Pharmacol. 2011, 660, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Brandin, H.; Viitanen, E.; Myrberg, O.; Arvidsson, A.K. Effects of herbal medicinal products and food supplements on induction of CYP1A2; CYP3A4 and MDR1 in the human colon carcinoma cell line LS180. Phytother. Res. 2007, 21, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, D.; Nakamura, T.; Okamura, N.; Kokudai, M.; Inui, N.; Takeuchi, K.; Watanabe, H.; Hirai, M.; Okumura, K.; Sakaeda, T. Effects of acid and lactone forms of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on the induction of MDR1 expression and function in LS180 cells. Eur. J. Pharm. Sci. 2009, 37, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Harper, P.A.; Tang, B.K.; Okey, A.B. Regulation of cytochrome P450 enzymes by aryl hydrocarbon receptor in human cells: CYP1A2 expression in the LS180 colon carcinoma cell line after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin or 3-methylcholanthrene. Biochem. Pharmacol. 1998, 56, 599–612. [Google Scholar] [CrossRef]
- Harper, P.A.; Prokipcak, R.D.; Bush, L.E.; Golas, C.L.; Okey, A.B. Detection and characterization of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in the human colon adenocarcinoma cell line LS180. Arch. Biochem. Biophys. 1991, 290, 27–36. [Google Scholar] [CrossRef]
- Novotna, A.; Pavek, P.; Dvorak, Z. Novel stably transfected gene reporter human hepatoma cell line for assessment of aryl hydrocarbon receptor transcriptional activity: Construction and characterization. Environ. Sci. Technol. 2011, 45, 10133–10139. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, M.; Albermann, N.; Sauer, A.; Walter-Sack, I.; Haefel, W.E.; Weiss, J. In vitro and ex vivo evidence for modulation of P-glycoprotein activity by progestins. Biochem. Pharmacol. 2004, 68, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Rose, J.; Storch, C.H.; Ketabi-Kiyanvash, N.; Sauer, A.; Haefeli, W.E.; Efferth, T. Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J. Antimicrob. Chemother. 2007, 59, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Lindenmaier, H.; Haefeli, W.E.; Weiss, J. Interaction of the mitotic kinesin Eg5 inhibitor monastrol with P-glycoprotein. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 372, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Albermann, N.; Schmitz-Winnenthal, F.H.; Z’graggen, K.; Volk, C.; Hoffmann, M.M.; Haefeli, W.E.; Weiss, J. Expression of the drug transporters MDR1/ABCB1; MRP1/ABCC1; MRP2/ABCC2; BCRP/ABCG2; and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem. Pharmacol. 2005, 70, 949–958. [Google Scholar] [CrossRef] [PubMed]
- König, S.J.; Herzog, M.; Theile, D.; Zembruski, N.; Haefeli, W.E.; Weiss, J. Impact of drug transporters for the cellular resistance towards saquinavir and darunavir. J. Antimicrob. Chemother. 2010, 65, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, Z.; Vrzal, R.; Henklova, P.; Jancova, P.; Anzenbacherova, E.; Maurel, P.; Svecova, L.; Pavek, P.; Ehrmann, J.; Havlik, R.; et al. JNK inhibitor SP600125 is a partial agonist of human aryl hydrocarbon receptor and induces CYP1A1 and CYP1A2 genes in primary human hepatocytes. Biochem. Pharmacol. 2008, 5, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Ayed-Boussema, I.; Pascussi, J.M.; Maurel, P.; Bacha, H.; Hassen, W. Zearalenone activates pregnane X receptor; constitutive androstane receptor and aryl hydrocarbon receptor and corresponding phase I target genes mRNA in primary cultures of human hepatocytes. Environ. Toxicol. Pharmacol. 2011, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.; Haefeli, W.E. Evaluation of inhibitory potencies for compounds inhibiting P-glycoprotein but without maximum effects: f2 values. Drug Metab. Dispos. 2006, 34, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Sauer, A.; Divac, N.; Herzog, M.; Schwedhelm, E.; Böger, R.H.; Haefeli, W.E.; Benndorf, R.A. Interaction of angiotensin receptor type 1 blockers with ATP-binding cassette transporters. Biopharm. Drug Dispos. 2010, 31, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Theile, D.; Dvorak, Z.; Haefeli, W.E. Interaction potential of the multitargeted receptor tyrosine kinase inhibitor dovitinib with drug transporters and drug metabolizing enzymes assessed in vitro. Pharmaceutics 2014, 6, 632–650. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Raza, A.; Carter, T.H.; Claxton, D.; Erba, H.; DeAngelo, D.J.; Tallman, M.S.; Goard, C.; Borthakur, G. A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS ONE 2014, 9, e108694. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, T.W.; McNeil, M.J.; Kamal, A.H.; Currow, D.C.; Abernethy, A.P. Polypharmacy in patients with advanced cancer and the role of medication discontinuation. Lancet Oncol. 2015, 16, e333–e341. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.J.; Busman, T.; McKeegan, E.; Salem, A.; Zhu, M.; Ricker, J.L.; Blum, W.; et al. A phase 2 study of venetoclax (ABT-199/GDC-0199) in patients with acute myelogenous leukemia (AML). Blood 2014, 124, 118. [Google Scholar]
- Zhang, L.; Zhang, Y.D.; Strong, J.M.; Reynolds, K.S.; Huang, S.M. A regulatory viewpoint on transporter-based drug interactions. Xenobiotica 2008, 38, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Läpple, F.; von Richter, O.; Fromm, M.F.; Richter, T.; Thon, K.P.; Wisser, H.; Griese, E.U.; Eichelbaum, M.; Kivistö, K.T. Differential expression and function of CYP2C isoforms in human intestine and liver. Pharmacogenetics 2003, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Okey, A.B.; Riddick, D.S.; Harper, P.A. The Ah receptor: Mediator of the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related compounds. Toxicol. Lett. 1994, 70, 1–22. [Google Scholar] [CrossRef]
- Lankisch, T.O.; Gillman, T.C.; Erichsen, T.J.; Ehmer, U.; Kalthoff, S.; Freiberg, N.; Munzel, P.A.; Manns, M.P.; Strassburg, C.P. Aryl hydrocarbon receptor-mediated regulation of the human estrogen and bile acid UDP-glucuronosyltransferase 1A3 gene. Arch. Toxicol. 2008, 82, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, S.; Ehmer, U.; Freiberg, N.; Manns, M.P.; Strassburg, C.P. Coffee induces expression of glucuronosyltransferases by the aryl hydrocarbon receptor and Nrf2 in liver and stomach. Gastroenterology 2010, 139, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer. 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Inhibited | Venetoclax [µM] | Control Compound | [µM] |
|---|---|---|---|
| P-gp | 30.0 ± 3.7 | Verapamil | 2.9 ± 0.8 [29] |
| BCRP | 19.6 ± 7.3 | FTC | 0.7 ± 0.3 [30] |
| OATP1B1 | 47.8 ± 10.1 | Rifampicin | 2.4 ± 0.9 [12] |
| OATP1B3 | 26.0 ± 9.6 | Rifampicin | 2.1 ± 1.0 [12] |
| CYP1A2 | no inhibition | ||
| CYP2B6 | activation | ||
| CYP2C19 | 14.21 ± 1.0 | Omeprazole | 0.8 ± 0.2 [31] |
| CYP2D6 | no inhibition | ||
| CYP3A4 | 7.2 ± 3.2 | Ketoconazole | 0.035 ± 0.015 [31] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, J.; Gajek, T.; Köhler, B.C.; Haefeli, W.E. Venetoclax (ABT-199) Might Act as a Perpetrator in Pharmacokinetic Drug–Drug Interactions. Pharmaceutics 2016, 8, 5. https://doi.org/10.3390/pharmaceutics8010005
Weiss J, Gajek T, Köhler BC, Haefeli WE. Venetoclax (ABT-199) Might Act as a Perpetrator in Pharmacokinetic Drug–Drug Interactions. Pharmaceutics. 2016; 8(1):5. https://doi.org/10.3390/pharmaceutics8010005
Chicago/Turabian StyleWeiss, Johanna, Thomas Gajek, Bruno Christian Köhler, and Walter Emil Haefeli. 2016. "Venetoclax (ABT-199) Might Act as a Perpetrator in Pharmacokinetic Drug–Drug Interactions" Pharmaceutics 8, no. 1: 5. https://doi.org/10.3390/pharmaceutics8010005