1. Introduction
A key for successful gene therapy is whether or not therapeutic genes can be effectively delivered and internalized into targeted organs and cells. Clinical trials for gene therapy against several intractable and lethal diseases have actively progressed in the world [
1]. However, the approach for gene therapy by systemic administration has some obstacles, including degradation by endonuclease in blood and non-targeted distribution, subsequently leading to poor therapeutic effects and adverse effects, such as the interferon response. Therefore, the approach of local administration, which enables the direct delivery of therapeutic genes into target organs, has been examined [
2].
Pulmonary administration is a powerful tool for achieving effective pulmonary gene therapy against several lung diseases, such as cystic fibrosis, α1-antitrypsin deficiency, and lung cancer, which are also the target of our pulmonary gene delivery system, due to direct and noninvasive delivery into deep lung areas through the respiratory tract [
3]. Furthermore, viral and non-viral vectors have been applied to pulmonary gene delivery to realize higher gene transfection efficiencies [
4]. On the other hand, the development of inhalable aerosol systems for pulmonary gene delivery is critical for clinical use. At present, three aerosol inhalation systems are available for clinical application: nebulizers, pressurized metered-dose inhalers (pMDIs), and dry powder inhalers (DPIs) [
5]. A nebulizer formulation containing plasmid DNA (pDNA) encoding the therapeutic gene and a cationic liposome, as a vector, was used in a phase I clinical trial of gene therapy against cystic fibrosis [
6,
7].
Among these systems, DPIs have attracted much attention due to their low cost, the portable device, no propellant, and ease of handling [
8]. Although reports on the development of DPIs for pulmonary gene therapy are still limited, some powder production techniques, including spray drying (SD), supercritical fluid precipitation, and lyophilization, might be applicable for a specific formulation or condition [
9,
10,
11]. However, the destabilization of the gene or delivery system caused by several stresses, including heating, freezing, spraying, and shear force, must be considered during powder production [
9,
12,
13,
14,
15]. Moreover, the morphology and particle size of prepared powders are critical factors affecting their inhalability. In general, aerodynamic particles of 1~5 μm are considered suitable for delivery by inhalation [
8]. On the other hand, particles in this range of geometric size generate a strong adhesion force, resulting in poor dispersibility. To overcome the dilemma between aerodynamic and geometric particle sizes in the development of DPIs, recently, some low-density dry powders have been produced, facilitating an increase in the geometric particle size with a suitable aerodynamic particle size for inhalation [
8].
Spray freeze drying (SFD) is a recent and simple technique to produce porous dry powders with a low density [
16]. Maa
et al. revealed that a dry powder produced by SFD could exhibit superior inhalation characteristics compared to that by SD irrespective of having similar geometric particle sizes [
17]. In addition, SFD can guarantee the high-level recovery of produced dry powders, even if the initial amount of the formulation is small, which is important for the study of dry gene powders on a laboratory scale since the employed genes are relatively expensive. From these fine characteristics, we have introduced SFD into powder production for gene DPIs. In our previous study, it was demonstrated that dry gene powder could be stably prepared by SFD without the loss of pDNA integrity, and that the powder exhibited a gene expressing effect in the lungs of mice following pulmonary administration [
18]. For clinical application, however, a higher gene transfection efficiency in the lungs is necessary.
The choice of suitable vectors for gene DPIs is a critical element determining gene transfection efficiency in the lungs following inhalation. Biodegradable polycations are attractive non-viral vectors for effective gene therapy because of their quick endosomal escape potential and high tolerability in the body [
19]. In our previous reports, chitosan, a natural biodegradable polycation, was selected as a vector for inhalable dry gene powder prepared by SFD [
18]. Chitosan has been reported to show high tolerability in the body [
20]. Unfortunately, on the other hand, it has been reported that the transfection efficiency of chitosan is low compared to that of polyethyleneimine (PEI), a non-biodegradable polycation [
21]. As novel biodegradable polycations for higher gene transfection efficiency, we have synthesized poly(aspartamide) derivatives with an ethylenediamine unit as a side chain (poly{
N-[
N-(2-aminoethyl)-2-aminoethyl]aspartamide} (PAsp(DET)) and their block copolymers with poly(ethylene)glycol (PEG-PAsp(DET)) [
22,
23]. These polycations have superior transfection efficiencies with minimal cytotoxicity compared to PEI, probably due to effective endosomal escape promoted by a marked shift of protonation between the physiological pH and endosomal acidic pH [
24]. In particular, PEG-polycation block copolymers including PEG-PAsp(DET) can form a core-shell type of polyplex micelle composed of (1) the inner complex core constructed from electrostatic interaction between a negatively-charged gene and positively-charged segment in the polymers, and (2) the hydrophilic outer shell constructed from the PEG segment in the polymers, allowing it to escape the degradation of genes by endonuclease and aggregation caused by the interaction with blood components [
25,
26]. So far, PEG-PAsp(DET) polyplex micelles have successfully achieved the effective
in vivo transfection of therapeutic genes into some target sites, such as: (1) vascular lesions via intraarterial administration [
27], (2) a bone defect area in skull bone by regulated release from a calcium phosphate cement scaffold locally implanted [
28], and (3) the lung via intratracheal administration [
29]. These results prompted us to apply these polycations as vectors for effective gene DPIs with higher gene transfection efficiencies.
Thus, in this study, inhalable dry gene powders including PAsp(DET) and PEG-PAsp(DET) were produced by SFD. To improve the inhalable potential of dry powders prepared by SFD,
l-leucine was added as a dispersibility enhancer in the formulation. The integrity of pDNA, physicochemical properties, and gene transfection efficiencies of the polyplexes after powder production were evaluated, respectively. Following the pulmonary administration of prepared dry powders to mice, the gene transfection efficiency and adverse effects in the lungs were compared to those of a chitosan-based dry powder developed in our previous study [
18].
2. Experimental Section
2.1. Materials
The pDNA encoding firefly luciferase with CAG promoter (pCAG-Luc) was provided by RIKEN Bioresource Center (Tsukuba, Japan). pCAG-Luc was amplified in the DH5 strain of Escherichia coli, and purified using an EndoFree Plasmid Giga Kit (Qiagen GmbH, Hilden, Germany). The concentration and purity of pCAG-Luc were determined by measuring UV absorption at 260 nm and the A260/A280 ratio, respectively. d(−)-mannitol (Wako Pure Chemical Industries Ltd., Osaka, Japan) and l-leucine (Sigma–Aldrich, St. Louis, MO, USA) were used as an excipient and a dispersibility enhancer for inhalable dry powder, respectively. Chitosan (Mw: 2000~5000, water-soluble, Wako Pure Chemical Industries Ltd.) was used as a control vector in this study. Fluorescein sodium salt (FLNa; Sigma–Aldrich) and indocyanine green (ICG; Sigma–Aldrich) were selected as fluorescent labels of dry powders for in vitro inhalation and in vivo gene transfection studies, respectively. Luciferin (Promega, Madison, WI, USA) was used as a substrate of luciferase. Isoflurane (Abbott Laboratories, Abbott Park, IL, USA) was used for inhalation anesthesia in the measurement of luminescence and fluorescence in mice with an in vivo imaging system (IVIS®; IVIS-SPECTRUM, Caliper Life Sciences, Hopkinton, MA, USA). Lipopolysaccharide (Sigma–Aldrich) was selected as a positive control for evaluating lung injury in vivo. The other reagents and solvents used were of analytical grade.
2.2. Synthesis and Characterization of PAsp(DET) and PEG-PAsp(DET)
PAsp(DET) and PEG-PAsp(DET) were prepared according to a ring-opening polymerization scheme, as previously reported [
22,
23]. Briefly, the polymerization of β-benzyl-
l-aspartate
N-carboxy-anhydride (Chuo Kaseihin Co., Inc., Tokyo, Japan) was started from
n-butylamine (Wako Pure Chemical Industries Ltd.) for PAsp(DET) and α-methoxy-ω-amino PEG (Mw: ~12,000, Nippon Oil and Fats, Tokyo, Japan) for PEG-PAsp(DET) to obtain poly(β-benzyl-
l-aspartate) (PBLA) and PEG-PBLA, respectively, followed by an aminolysis reaction to introduce diethylenetriamine (DET; Wako Pure Chemical Industries Ltd.) into the side chain of PBLA. From the
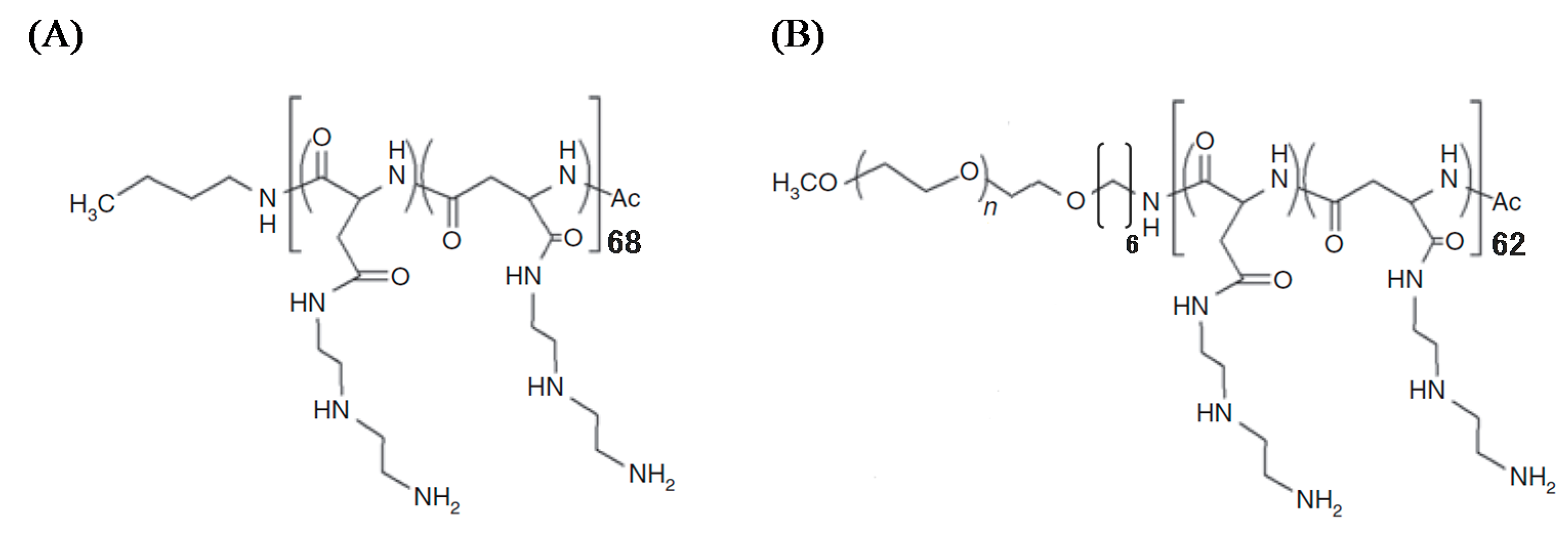
1H nuclear magnetic resonance spectrum, the polymerization degrees of PAsp(DET) and PAsp(DET) segments in PEG-PAsp(DET) were calculated to be 68 and 62, respectively, and the quantitative introduction of DET was confirmed (
Figure 1).
Figure 1.
Chemical structures of (A) PAsp(DET) and (B) PEG-PAsp(DET).
Figure 1.
Chemical structures of (A) PAsp(DET) and (B) PEG-PAsp(DET).
2.3. Preparation of Dry Powders by SFD
The procedure of powder production by SFD was described in our previous report [
18]. A schematic diagram is given in
Figure 2. The formulations are listed in
Table 1. The total mass of each formulation was adjusted to 50 mg. All formulation compositions for dry powders were dissolved in 2.5 mL of ultra-pure water. The solution was flowed at a rate of 5 mL/min, and atomized into liquid nitrogen at an air flow pressure of 150 kPa using a two-fluid nozzle (0.4 mm in inner diameter) for a spray dryer (SD-1000, TOKYO RIKAKIKAI Co., Ltd., Tokyo, Japan). We flowed water and air into the tubing connected with the nozzle before and after flowing the sample solution. The water flowed before the sample was to fix the flow rate and aid the wetting of the internal surfaces of the tubing and nozzle. The water flowed after the sample was to disperse the sample solutions left in the nozzle. The head of the nozzle was placed approximately 15 cm above the surface of liquid nitrogen. Following atomization, liquid nitrogen including frozen droplets was transferred to a freeze dryer (RLE-52, KYOWA VACUUM ENGINEERING Co., Tokyo, Japan) pre-cooled at a shelf temperature of −40 °C. After liquid nitrogen was evaporated, frozen droplets were dried at a pressure of less than 1 Pa, while the shelf temperature was gradually increased from −40 to 10 °C over a period of 24 h. The formulation names for prepared dry powders and solutions were abbreviated to “DP” and “SL” in each figure and table, respectively. The molecular ratio of amine in each polymer to phosphate in pDNA (N/P) was set to 4 or 8 for PAsp(DET), 40 or 80 for PEG-PAsp(DET), and 10 for chitosan, based on our previous studies [
18,
22,
24,
27,
29].
Figure 2.
Diagram of the SFD setup.
Figure 2.
Diagram of the SFD setup.
Table 1.
Composition of (A) several dry powders prepared by SFD and (B) solutions (dosage/body). N/P: molecular ratio of amine in polymer to phosphate in pDNA, pDNA: plasmid DNA, Leu: l-leucine, ICG: indocyanine green, FLNa: fluorescein sodium salt, Man: d(−)-mannitol.
(A) Dry powder
| Formulation name | N/P | pDNA (μg) | Polymer (μg) | Leu (μg) | ICG or FLNa (μg) | Man (μg) | Total amount (mg) |
|---|
| Man DP | – | – | – | – | 10 | 1490 | 1.5 |
| Man-Leu DP | – | – | – | 75 | 10 | 1415 | 1.5 |
| PAsp(DET) DP | 4
8 | 9
9 | 14.9
29.9 | 75
75 | 10
10 | 1391
1376 | 1.5
1.5 |
| PEG-PAsp(DET) DP | 40
80 | 9
9 | 255
510 | 75
75 | 10
10 | 1151
896 | 1.5
1.5 |
| Chitosan DP | 10 | 9 | 45 | 75 | 10 | 1361 | 1.5 |
(B) Solution
| Formulation name | N/P | pDNA (μg) | Polymer (μg) | Leu (μg) | ICG or FLNa (μg) | Man (μg) | Total amount (mg) | Water (μL) |
|---|
| PAsp(DET) SL | 4 | 9 | 14.9 | 75 | 10 | 1391 | 1.5 | 50 |
| PEG-PAsp(DET) SL | 40 | 9 | 255 | 75 | 10 | 1151 | 1.5 | 50 |
2.4. Morphology and Particle Size of Dry Powders
To examine the morphology of prepared dry powders, the particles were observed using a scanning electron microscope (SEM, Type JSM-6060, JEOL Ltd., Tokyo, Japan). Before the observation, prepared dry powders were dispersed by an appropriate apparatus using pulmonary administration to mice [
30]. The particle size distribution of the obtained powder was measured with the laser micron sizer LMS-30 (Seishin Enterprise Co., Ltd., Tokyo, Japan) based on laser diffraction.
2.5. Determination of pDNA Integrity in Dry Powders by Electrophoresis
Prepared dry powders were dissolved in ultra-pure water. We added 2 U of heparin sodium to enhance the dissociation of the complexes. Each sample solution containing 0.1 μg of pDNA was loaded on 0.6% agarose gel. Electrophoresis was carried out with a current of 100 V, 150 mA for 2 h in Tris–acetate–EDTA running buffer. A λ Hind III digest was used as a molecular mass marker, and naked pDNA and digested pDNA (by Hind III) were used as a control, respectively. After the electrophoresis, the gel was soaked in a 0.5 μg/mL ethidium bromide solution to detect pDNA with a fluorescent image analyzer (Typhoon FLA 9000; GE Healthcare Life Sciences, Tokyo, Japan).
2.6. Particle Size and Zeta Potential of Polyplexes
The particle size and zeta potential of the polyplexes formed between pDNA and PAsp(DET) or PEG-PAsp(DET) were assessed using an electrophoretic light scattering spectrophotometer (Zetasizer Nano ZS, Malvern Instruments, Worcestershire, UK) at 25 °C. Polyplex formation was performed in ultra-pure water, keeping it at room temperature for 30 min. Then, the final concentration of pDNA was adjusted to 20 μg/mL using 10 mM HEPES buffer at pH 7.4.
2.7. Cells
Murine colon adenocarcinoma (CT26) cells were kindly provided by Prof. Y. Takakura, Kyoto University. They were cultured in RPMI1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C in humidified air containing 5% CO2.
2.8. In Vitro Transfection Study by Polyplexes
CT26 cells were seeded on a 24-well microplate (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) at a concentration of 60,000 cells/600 μL/well. After incubation for 24 h, the growth medium in each well was replaced with OPTI-MEM® (Invitrogen Co., Carlsbad, CA, USA). Each polyplex was formed in ultra-pure water at room temperature for 30 min. Then, 60 μL of the polyplex solution was added to each well at an amount of 3 μg as pDNA. At 4 h after exposure, the OPTI-MEM® containing the polyplex in each well was replaced with new growth medium, followed by incubation for 44 h. After removal of the medium, lysis buffer (0.05 % Triton X-100, 2 mM EDTA, 0.1 M Tris, pH 7.8) was added to each well to lyse cells. Additionally, each lysate was treated with three cycles of freezing and thawing to fully lyse cells. The lysate was centrifuged at 13,000× g for 7 min at 4 °C to collect the supernatant. The luciferase activity and protein concentration in each supernatant were measured by the Picagene® luciferase assay (TOYO INK Co., Ltd., Tokyo, Japan) and Bradford protein assay, respectively.
2.9. In Vitro Inhalation Characteristics of Dry Powders
The
in vitro inhalation characteristics of the prepared dry powders were evaluated using an 8-stage Andersen cascade impactor (AN-200; SIBATA SCIENTIFIC TECHNOLOGY Ltd., Saitama, Japan). To prevent dry powder from bouncing off the plates and becoming re-entrained in the air stream, the metal plates for the stages were coated with a thin layer of glycerin. After setting a No. 2 hydroxypropyl methylcellulose (HPMC) hard capsule (Shionogi Qualicaps Co. Ltd., Nara, Japan) containing 4 mg of each dry powder in a dry powder inhaler (Jethaler
® dual type; Hitachi Automotive Systems, Ltd., Isesaki, Japan), inspiration at a flow rate of 28.3 L/min and for a flow time of 5 s was carried out using a vacuum pump. After the inspiration, dry powder deposited in each part (capsule, device, throat, stages, and filter) was washed out by 10 mL of phosphate-buffered saline (PBS) to measure the concentration of FLNa using a fluorescence microplate reader (GEMINI EM; Molecular Devices LLC., Sunnyvale, CA, USA). From the concentration, the amount of dry powder deposited in each part was calculated. The deposition of dry powder in each part was defined by the following equation:
For inhalation indexes, the output efficiency (OE) and fine particle fraction (FPF) were defined by the following equations, respectively:
The cut-off diameter in stage 3 was 4.7 μm. OE means the emission potential from the capsule and device, while FPF means the potential of delivery into the lung. The mass median aerodynamic diameter (MMAD) was determined from each deposition pattern, using special analysis software (AEROSOL particle density analysis system; SIBATA SCIENTIFIC TECHNOLOGY Ltd.).
2.10. Pulmonary Administration to Mice
All the animal experiments were approved by the Animal Care and Use Committee of Faculty of Pharmacy, Meijo University on 31 October 2011 (authorization number: Pharm-Exp-5) and conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals. Female ICR mice weighing approximately 20 g were anesthetized with pentobarbital (50 mg/kg, i.p.) and a board was secured to their backs during the experiments. The trachea was exposed and 2.5 cm of PE-60 polyethylene tubing (internal diameter: 0.76 mm; Becton, Dickinson and Company, Franklin Lakes, NJ, USA) was inserted to a depth of 1.0 cm through an incision. Prepared dry powders were administered through the trachea using an appropriate apparatus for mice [
30]. The powders (1.5 mg) were put in a disposable tip and dispersed in the trachea by releasing air (0.35 mL) compressed in a syringe by opening a three-way stopcock connecting the disposable tip and syringe. On the other hand, 50 µL of the solution dissolving 1.5 mg of a powder was similarly administered by a liquid aerosol apparatus for mice (MicroSprayer
®-Model IA-1C; Penn-Century Inc., Philadelphia, PA, USA).
2.11. Detection of Fluorescence and Luminescence in Vivo
Following the pulmonary administration of each formulation to mice, fluorescence and luminescence in mice were detected using IVIS
® as in our previous report, respectively [
18,
31]. The nominal pixel size and minimum detectable radiance of IVIS
® were 13.5 μm and 70 photons/s/cm
2/sr, respectively. To detect the fluorescence derived from ICG, an excitation filter of 745 nm and an emission filter of 820 nm were used, and the exposure time was set to 1 s. When the luminescence corresponding to luciferase activity was detected, luciferin as a substrate of luciferase was administered (150 mg/kg, i.p.) at 10 min before the detection point, and the exposure time was set to 1 min. During the measurement, the mice were anesthetized with isoflurane on a stage kept at 25 °C. To quantify the fluorescence and luminescence intensities in the lungs of mice, the region of interest (ROI) was adjusted to a rectangle with a width of 3 cm and height of 1 cm.
2.12. Histological Analysis of Excised Lung
For this evaluation, mice were selected with sufficient pulmonary gene expression in the in vivo gene transfection study described above. At 48 h following the pulmonary administration of each formulation to mice, cardiac perfusion from left the ventricle to right atrium with PBS was performed to wash out the blood in the lungs. Then, the lungs were fixed by perfusion with 4% paraformaldehyde in PBS. The fixed lungs were excised from the bodies and stored in 4% paraformaldehyde in PBS at 4 °C for 24 h. Subsequently, they were transferred into 30% sucrose in PBS and stored at 4 °C for 48 h to fully remove paraformaldehyde. Cryosections (10 μm) were cut from the lungs frozen in O.C.T. Compound (Sakura Finetek USA, Inc., Torrance, CA, USA) on dry ice, and stained by hematoxylin & eosin to observe them with a microscope (BIOREVO BZ-9000; KEYENCE, Osaka, Japan).
2.13. Statistical Analysis
Statistical comparisons were made using Aspin-Welch’s t-test for evaluation of the particle size, zeta potential, and in vitro transfection efficiencies of polyplexes formed before and after powder production, and a one-way ANOVA followed by Dunnett’s test for the other evaluations. Significant correlations between pulmonary delivery and gene expression in vivo were evaluated based on a test of no correlation. P < 0.05 was considered significant.
4. Discussion
A chitosan-based dry gene powder prepared by SFD in our previous report [
18] was not sufficient for human clinical application as an inhalant because of the relatively large particle size (approximately 20~40 μm). In this study, therefore, the atomizing pressure in the preparation procedure was increased from 20 to 150 kPa, consequently achieving the production of smaller-sized dry powders (
Figure 1 and
Table 2). Even in the powder production with more marked atomizing stress, the integrity of pDNA in PAsp(DET) DP and PEG-PAsp(DET) DP was demonstrated to be maintained (
Figure 4). Similar results were observed in some gene powder productions containing cationic vectors [
9,
15]. Thus, the polyplex formation between an anionic gene and a cationic vector in a sample solution for powder production must markedly contribute to avoiding the critical loss caused by some stresses in the procedure of SFD.
In our previous report, we used mouse lung metastasis models, which were prepared by the intravenous injection of CT-26 cells into mice, to examine the
in vivo efficacy of a chitosan-interferon-β gene complex powder for inhalation [
35]. In the present study, we thus selected CT26 cells in the
in vitro gene transfection study, expecting a future
in vivo efficacy study with the same mouse lung metastasis models. The decreased
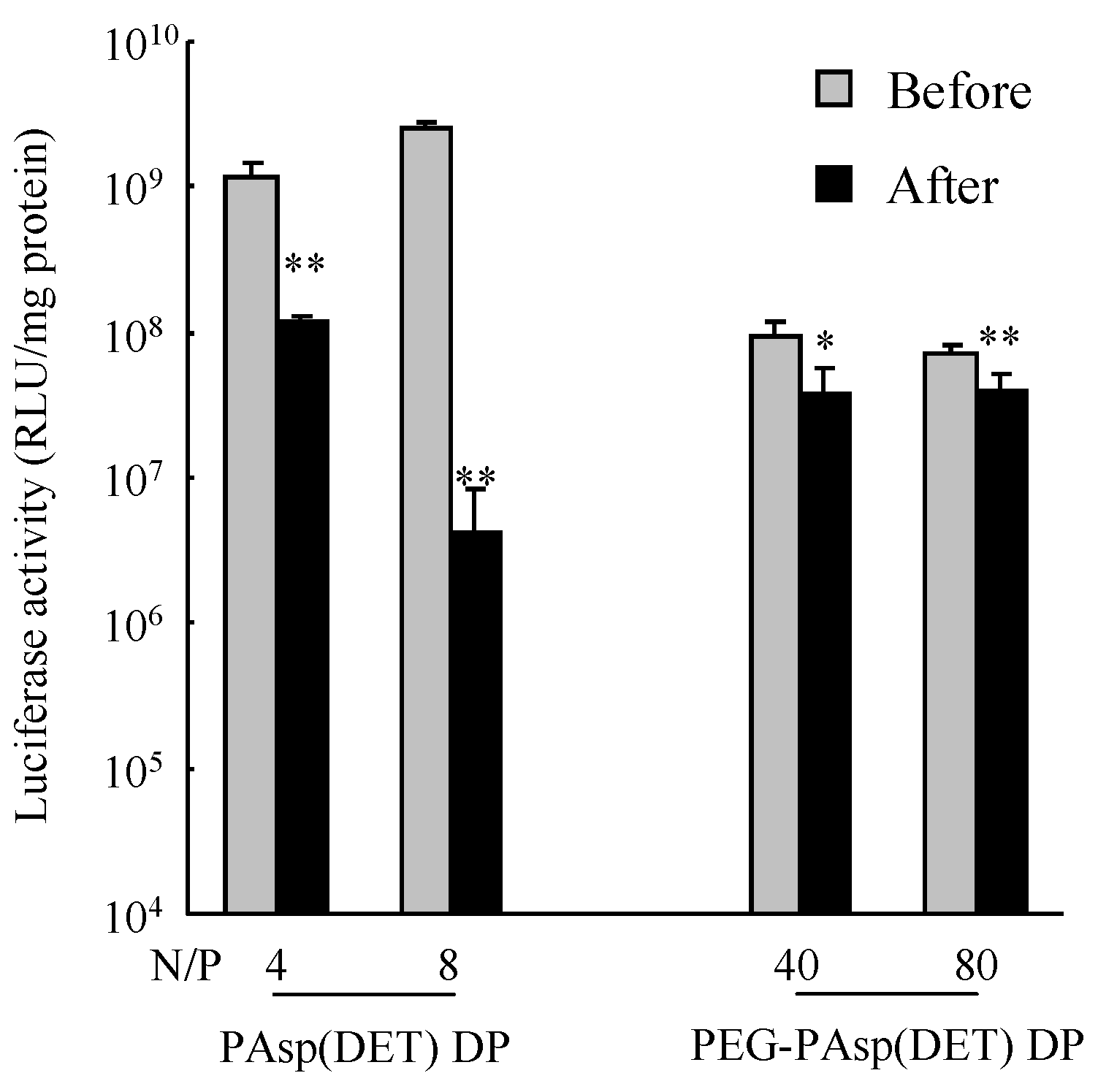
in vitro gene transfection efficiency of the PAsp(DET) polyplex after powder production (
Figure 6) was considered to be caused by the increased binding affinity of the DET segment with pDNA in the PAsp(DET) polyplex. Ramsy
et al. suggested that the binding affinity of a cationic poly(amino acid) vector with a gene is a more essential factor in determining the gene transfection efficiency of the polyplex than its particle size and surface charge [
36]. Moreover, Hahn
et al. reported that the binding affinity of PEI, a cationic vector, with pDNA was increased in a PEI polyplex reconstituted after powder production by lyophilization [
37]. The increased binding affinity interfered with the dissociation of pDNA from the PEI polyplex in the cytosol, consequently leading to a lower gene transfection efficiency. In this study, therefore, a similar phenomenon would occur in the PAsp(DET) polyplex reconstituted from PAsp(DET) DP. On the other hand, the maintained
in vitro gene transfection efficiency of the PEG-PAsp(DET) polyplex after powder production (
Figure 6) could be explained by the role of the PEG segment in PEG-PAsp(DET). In sample solution for powder production, PEG-PAsp(DET) formed the core-shell type of polyplex micelles, as mentioned above, which would maintain the environment of the inner complex core during powder production to maintain the binding affinity of the DET segment with pDNA in polyplex micelles. Armstrong
et al. reported that PEG modification of a cationic liposome could maintain the
in vitro gene transfection efficiency of the lipoplex reconstituted after powder production by lyophilization, partly supporting our hypothesis [
38].
l-Leucine has been widely used as a dispersant reagent for inhalable dry powders prepared by SD [
39,
40]. Although the mechanism under enhanced dispersibility by
l-leucine has not been fully clarified,
l-leucine spontaneously covers the surface of droplets after aerosolization of sample solution due to a surfactant-like property and low solubility in water or other vehicles, which might contribute to lowering the cohesiveness and adhesiveness of prepared dry powders [
41]. In this study, therefore,
l-leucine was applied to powder production by SFD to further promote inhalation performance. The tap density of Man-Leu DP was 0.0094 g/mL, which was much lower than that of Man DP, 0.035 g/mL. The surface areas of these two particles measured by argon gas adsorption were 26.6 and 17.8 m
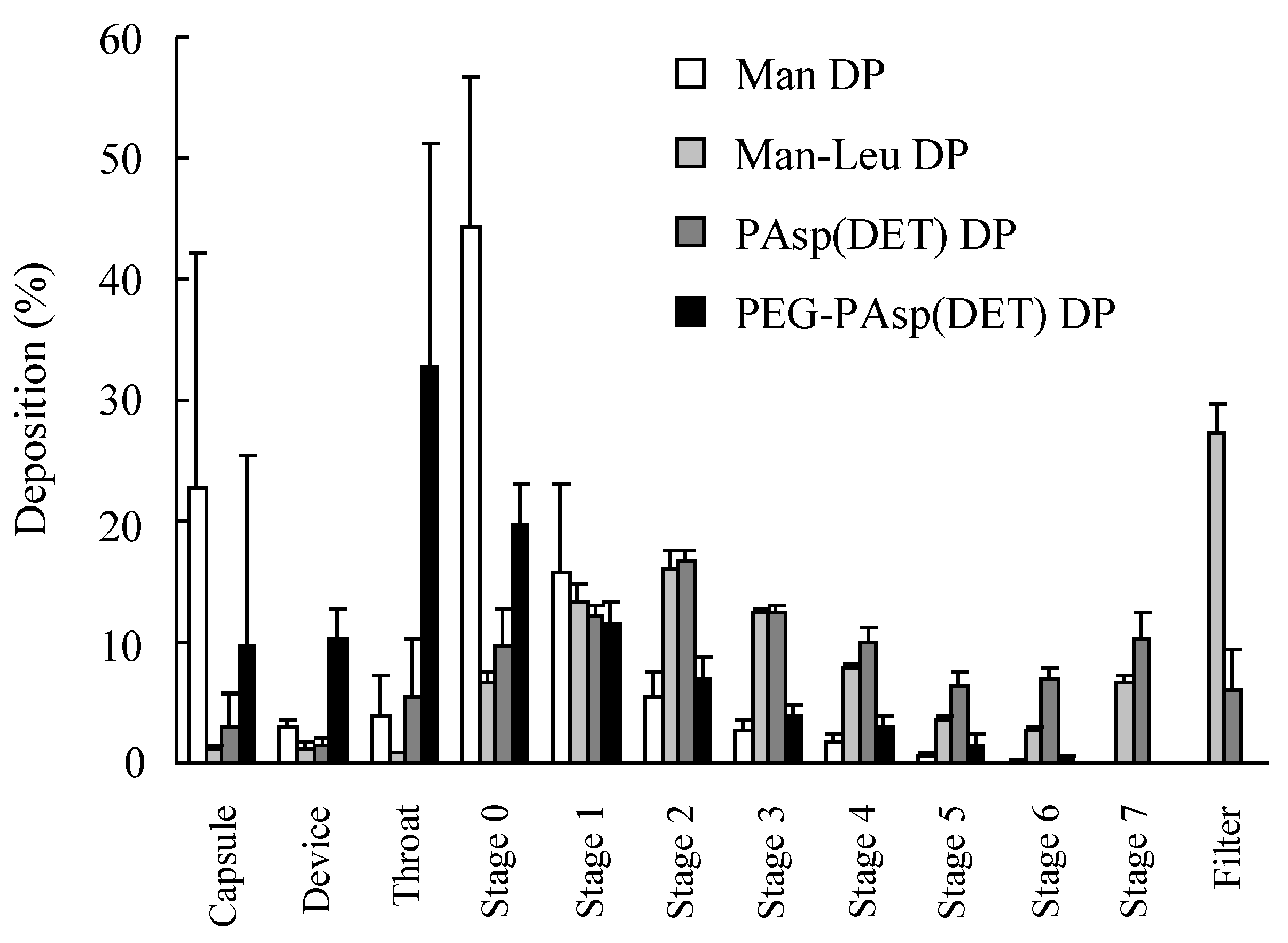
2, respectively, suggesting that these particles were porous. The water content measured with the Karl Fischer method was 0.89% and 0.86%, respectively. In the prepared dry gene powders, PAsp(DET) DP showed favorable inhalation performance similar to Man-Leu DP, while PEG-PAsp(DET) DP did not in spite of the addition of
l-leucine (
Figure 7 and
Table 3). This result could be easily explained by the difference of polymer occupation in the composition (PAsp(DET) DP; 1%, PEG-PAsp(DET); 17%). In our previous studies, it was clarified that PEG-PAsp(DET) needed larger amounts (
i.e., higher N/P) than PAsp(DET) to show effective gene expression, because the outer PEG shell would hamper the cellular uptake and intracellular process of the polyplex [
27]. Thus, a high N/P was similarly chosen for PEG-PAsp(DET) DP in this study. However, greater polymer occupation in PEG-PAsp(DET) DP would markedly affect its inhalation performance. This observation strongly indicated that we must select a vector for an effective inhalable dry gene powder in view of not only its gene transfection efficiency but also effective inhalability.
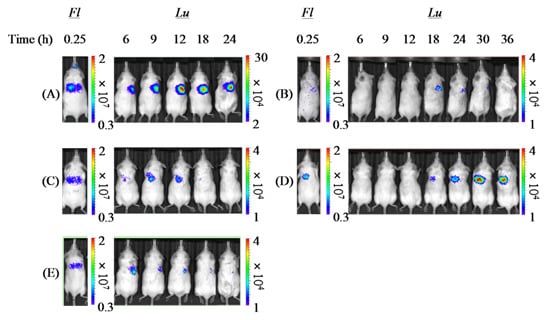
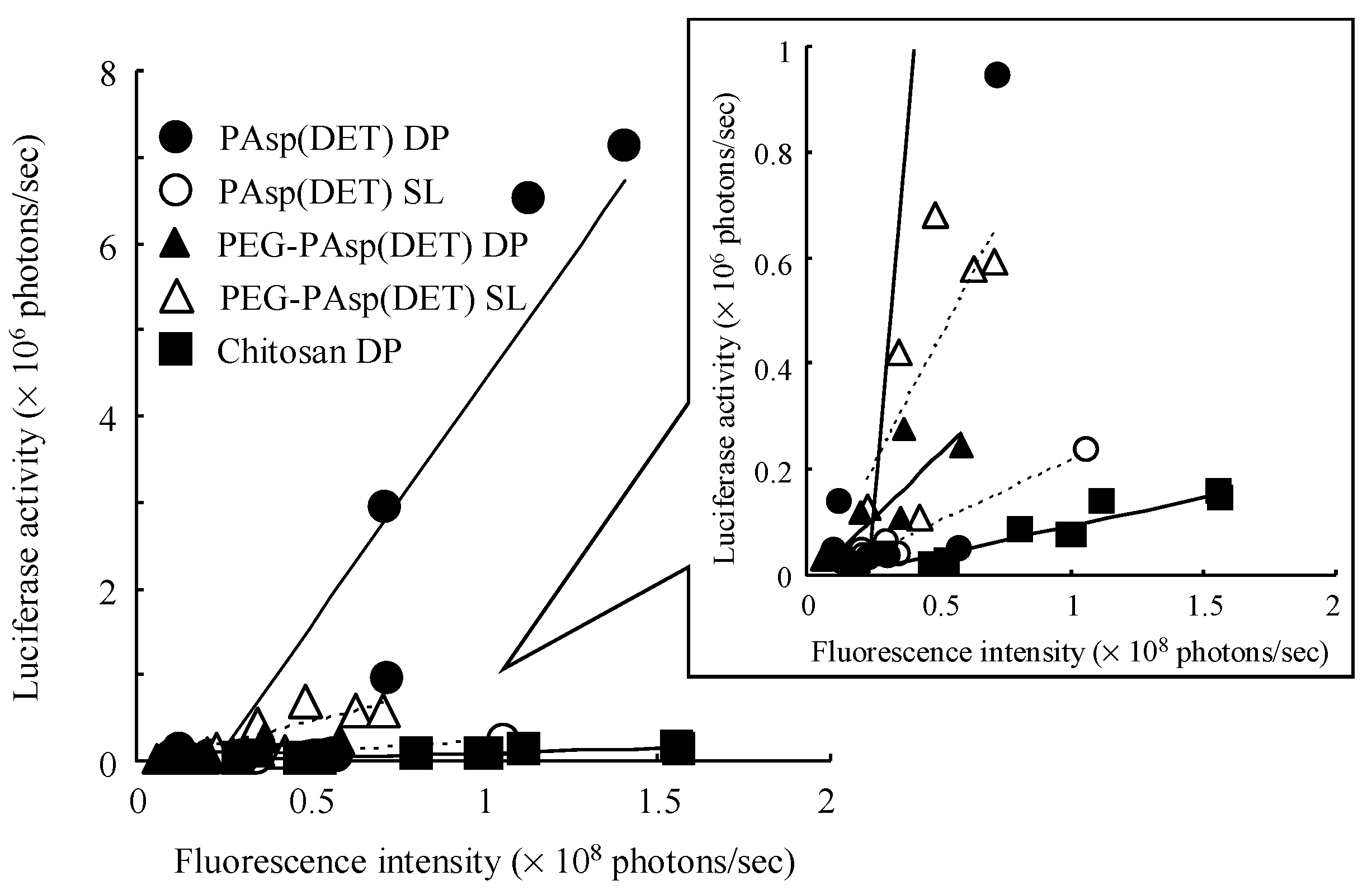
From the slope of the regression line gained from the correlation diagram between pulmonary delivery and gene expression by each formulation (
Table 4), it was demonstrated that both PAsp(DET) DP and PEG-PAsp(DET) DP had higher
in vivo pulmonary gene transfection efficiencies compared with Chitosan DP, which was in accordance with the higher
in vitro gene transfection potentials of these polymers themselves. In the
in vitro gene transfection study (before powder production), as shown in
Figure 6, both PAsp(DET) and PEG-PAsp(DET) polyplexes showed superior gene-expressing effects (1~10 × 10
8 RLU/mg protein), while the chitosan polyplex did not (<1 × 10
4 RLU/mg protein, data not shown). These results suggest that a vector with a higher gene transfection potential should be selected for developing an effective dry gene powder for inhalation.
The much higher
in vivo gene transfection efficiency of PAsp(DET) DP than PAsp(DET) SL (
Table 4), which was consistent with our previous report about a chitosan-based dry gene powder prepared by supercritical CO
2 precipitation [
10], surprised us since it was opposite to the results of the
in vitro gene transfection study (
Figure 6). Although the reasons for this discrepancy between
in vivo and
in vitro remain unclear, one of them is considered to be the different concentration of the polyplex exposed on the cell surface. On reaching the lungs following pulmonary delivery
in vivo, PAsp(DET) DP was dissolved in a small volume of water on the lung epithelial surface to reconstitute a very high concentration of the PAsp(DET) polyplex. The high-concentration exposure might enhance the intracellular uptake of the PAsp(DET) polyplex, contributing to the higher
in vivo gene transfection efficiency of PAsp(DET) DP. On the other hand, the difference between
in vivo gene transfection efficiencies of PEG-PAsp(DET) DP and PEG-PAsp(DET) SL (
Table 4) were in good agreement with the results of the
in vitro gene transfection study (
Figure 6). It has been reported that PEG modification of lipoplexes and polyplexes interferes with the electrostatic interaction between positively charged complexes and the negatively charged cell membrane, resulting in limited intracellular uptake via endocytosis [
42,
43]. Possibly, the intracellular uptake of the PEG-PAsp(DET) polyplex might be restricted even at an extremely high concentration of polyplex formation on the lung epithelial surface, consequently leading to similar trends between
in vitro and
in vivo gene transfection efficiencies of PEG-PAsp(DET) DP and SL. In addition, the PEG segment in PEG-PAsp(DET) must also be related to the delayed gene expression of PEG-PAsp(DET) DP and SL (time of maximum gene expression: 6~12 h in PAsp(DET) DP and SL, 18~36 h in PEG-PAsp(DET) DP and SL), which was consistent with the results of the transfection study using multicellular tumor spheroids, as shown in a previous report [
44]. From the comparison between
in vitro and
in vivo gene transfection efficiencies, it was strongly indicated that the
in vivo gene transfection efficiency of an intact dry gene powder cannot be effectively predicted based on the
in vitro gene transfection efficiency of its dissolved form.
Although we did not record the emitted dose in this study, it was the case that some powders remained in the tip. Even if all the powder was emitted from the tip, the powder distribution in the lung would vary among animals. To overcome this problem, we employed the dual imaging technique [
18,
31]. PAsp(DET) DP showed the highest
in vivo gene transfection efficiency and higher FPF than PEG-PAsp(DET) DP. However, the
in vitro dispersion was conducted with 4 mg powder at 28.3 L/min from Jethaler
®. On the other hand, the
in vivo dispersion was conducted with 1.5 mg powder from a syringe device. The dispersion conditions and device design were different, and so the results cannot be directly compared. For clinical applications, an Andersen cascade impactor study with an appropriate device containing an optimal amount of powder is required.
Local toxic actions induced by some lipoplexes and polyplexes on the lung epithelial surface have limited clinical application as gene inhalation therapy [
45]. In human clinical trials, it was reported that the nebulization of a lipoplex containing GL67, a cationic lipid optimized for pulmonary gene therapy, caused some influenza-like symptoms (myalgia, headache, and a high body temperature) and mild respiratory symptoms (cough, wheeze, and a tight chest) [
6,
7]. Therefore, a guarantee of safety in the lungs is essential for the clinical application of gene formulations for inhalation. The present study indicated that PEG-PAsp(DET) DP had good tolerability but PAsp(DET) DP could cause more severe irritation in the lungs (
Figure 10). Similar trends were observed in our previous reports with their solution formulations [
29,
46], partly supporting the present results. The cationic surface charge is one of the most important factors related to the toxicities of lipoplexes and polyplexes, which can cause direct membrane damage and aggregation in the body. After the dissolution of PEG-PAsp(DET) DP on the lung epithelial surface, the PEG-PAsp(DET) polyplex would be reconstituted, in which the hydrophilic PEG shell covers the inner cationic core, greatly contributing to its favorable tolerability. Our preliminary study with a pulse oximeter (MouseOx
®, STARR Life Sciences, Oakmont, PA, USA) showed no changes in the oxygen saturation, number of breaths, and heartbeat rate of mice 24 and 48 h following the pulmonary administration of each formulation (9 μg as pDNA), suggesting no severe toxic effect of PAsp(DET) and PEG-PAsp(DET) polyplexes in solution and powder (data not shown). Unfortunately, information about the safety of PAsp(DET) DP and PEG-PAsp(DET) DP is limited in the present report, and comprehensive analysis including quantifications of some indicators (e.g., lactate dehydrogenase activity and inflammatory cytokine amounts in bronchoalveolar fluid) will be necessary to confirm their local and systemic safety more precisely. As a future plan to achieve a higher transfection efficiency in the lungs with minimized toxicity, we will try to apply the combination of PAsp(DET) and PEG-PAsp(DET) to the powder formulation for inhalation, whose efficiency was shown in our previous report with the solution formulation [
46].
In addition to the local toxicity of the gene complex, that of the carrier material is important. Lactose is commonly used as an additive for inhalable formulations; however, it is usual to use lactose as a carrier which is not expected to be delivered deep into the lungs. In the present study, mannitol was used as a carrier delivered deep into the lungs with the gene complex. Mannitol has been used as a carrier for SFD powders in published reports [
47,
48]. The
in vivo toxicity study showed that Man-Leu was not toxic (
Figure 10); however, the selection of a carrier is one of the important issues to be resolved in the future.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







