Enhanced Cellular Delivery and Biocompatibility of a Small Layered Double Hydroxide–Liposome Composite System


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
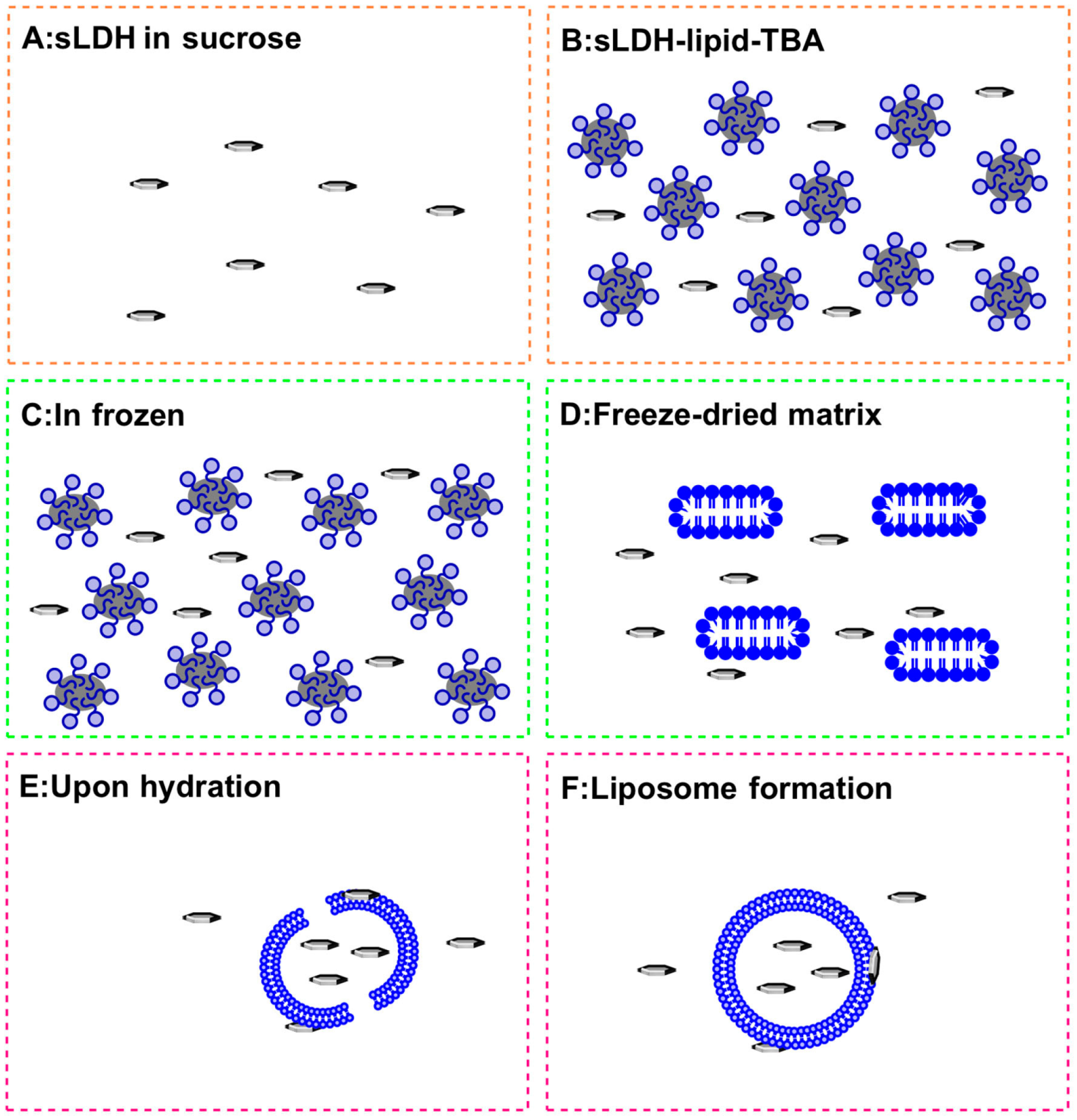
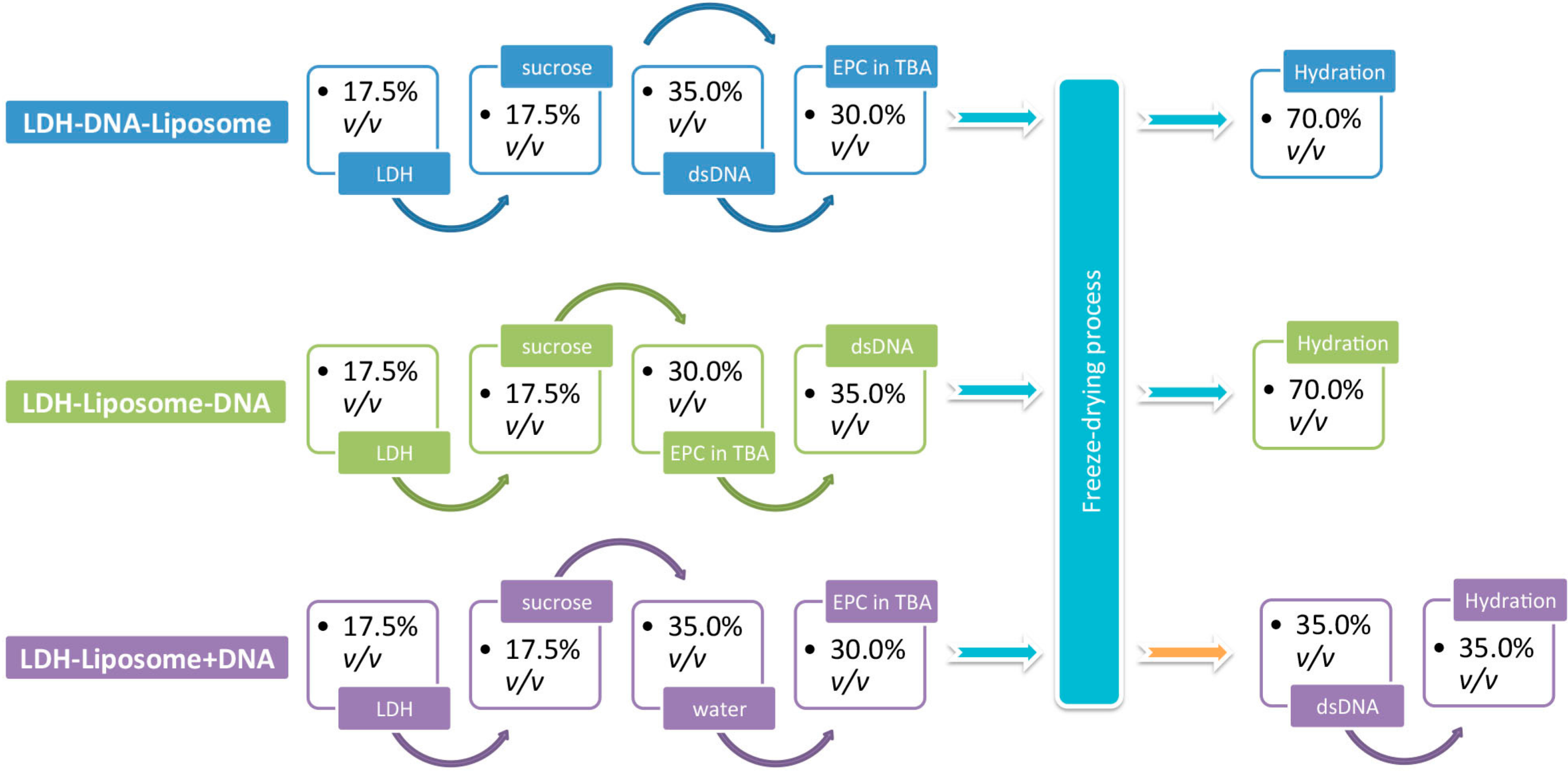
2.1. Preparation of LDH NPs and LDH–Liposome Composites
2.2. Incorporation of DNA into LDH–Liposome Composite
2.3. Suspension Stability Test

2.4. Agarose Gel Electrophoresis
2.5. Cell Viability
2.6. Cellular Uptake
3. Results and Discussion
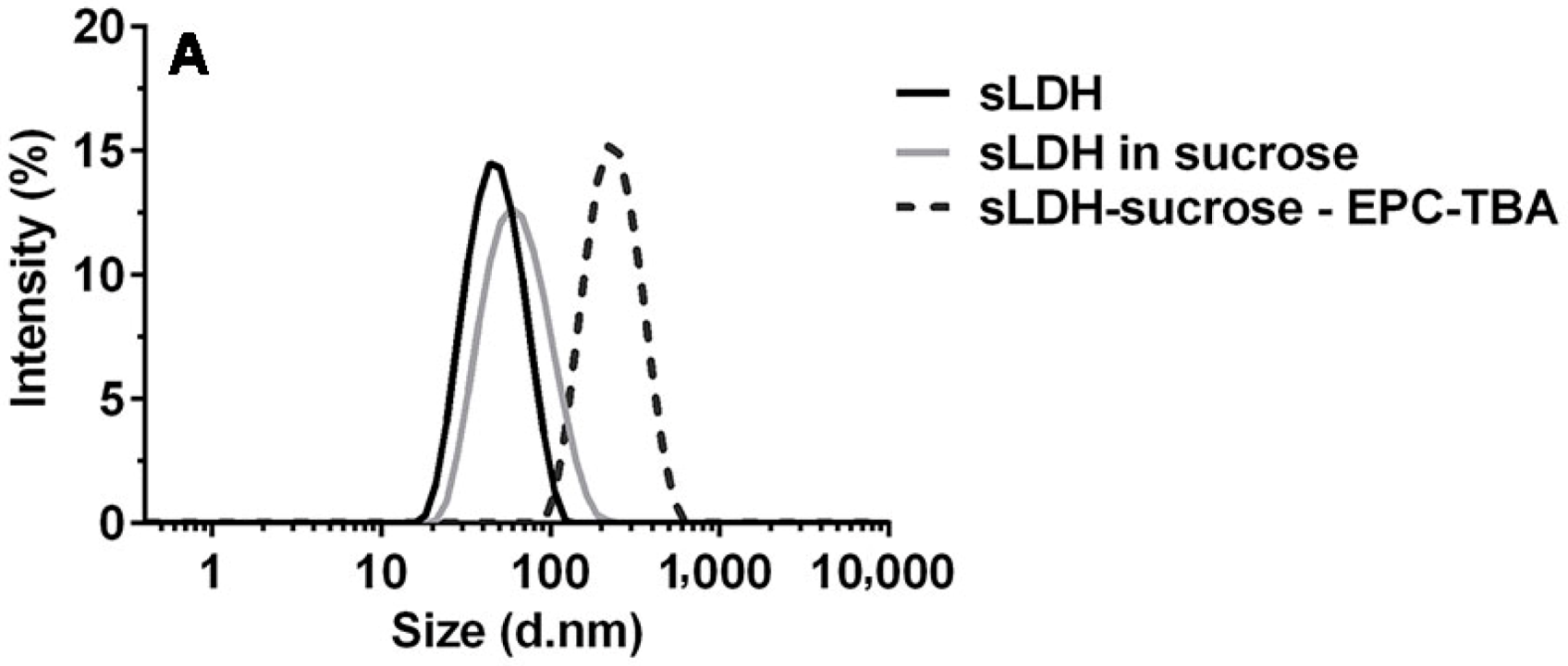
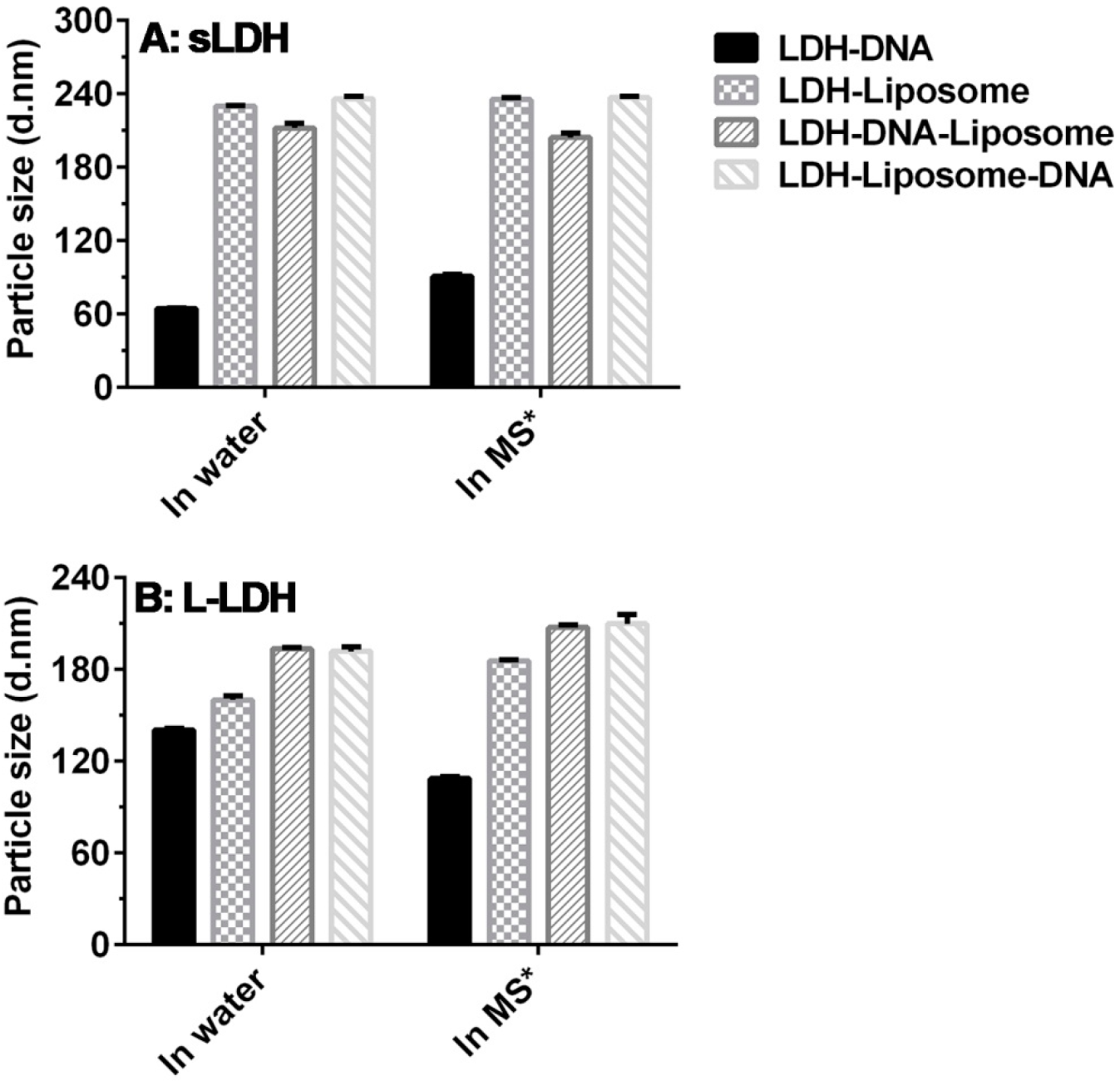
3.1. sLDH–Liposome Composite Formation



3.2. Composite System Stability

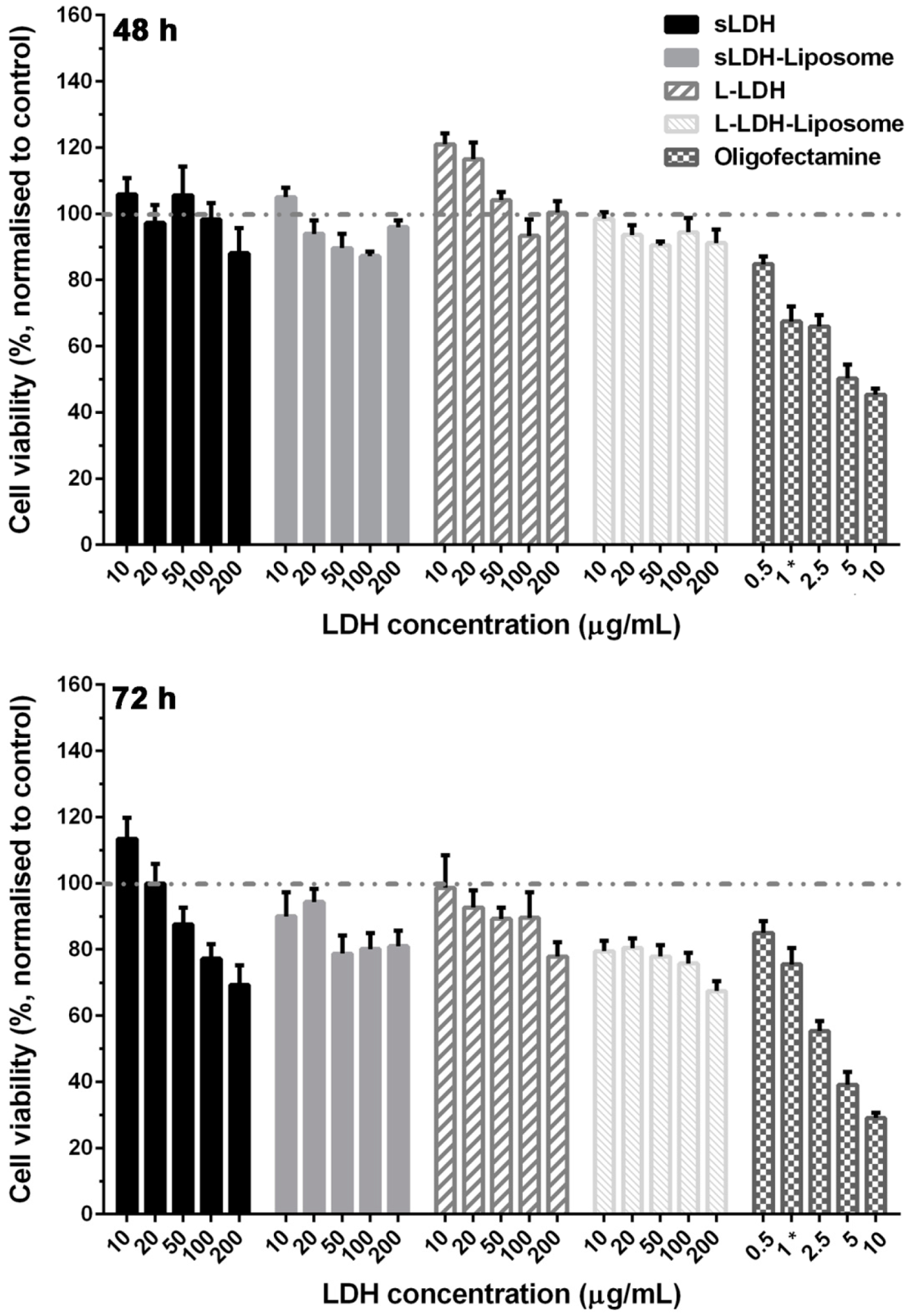
3.3. Cytotoxicity of LDH and LDH–Liposome NPs

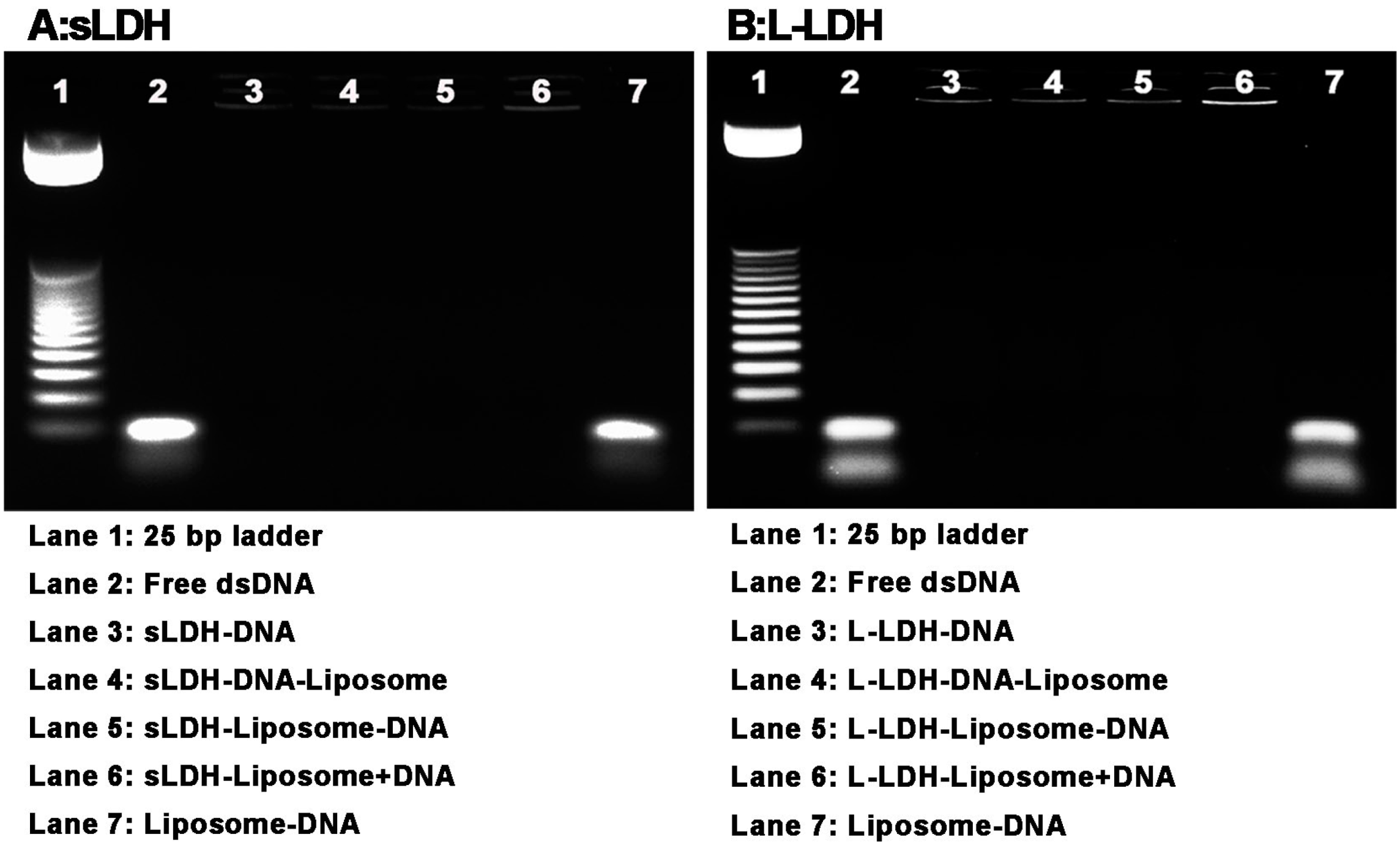
3.4. DNA Loading

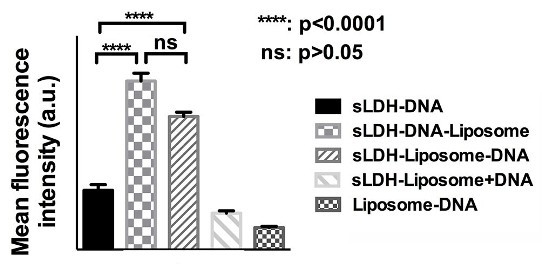
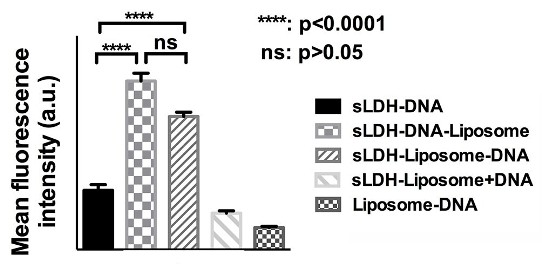
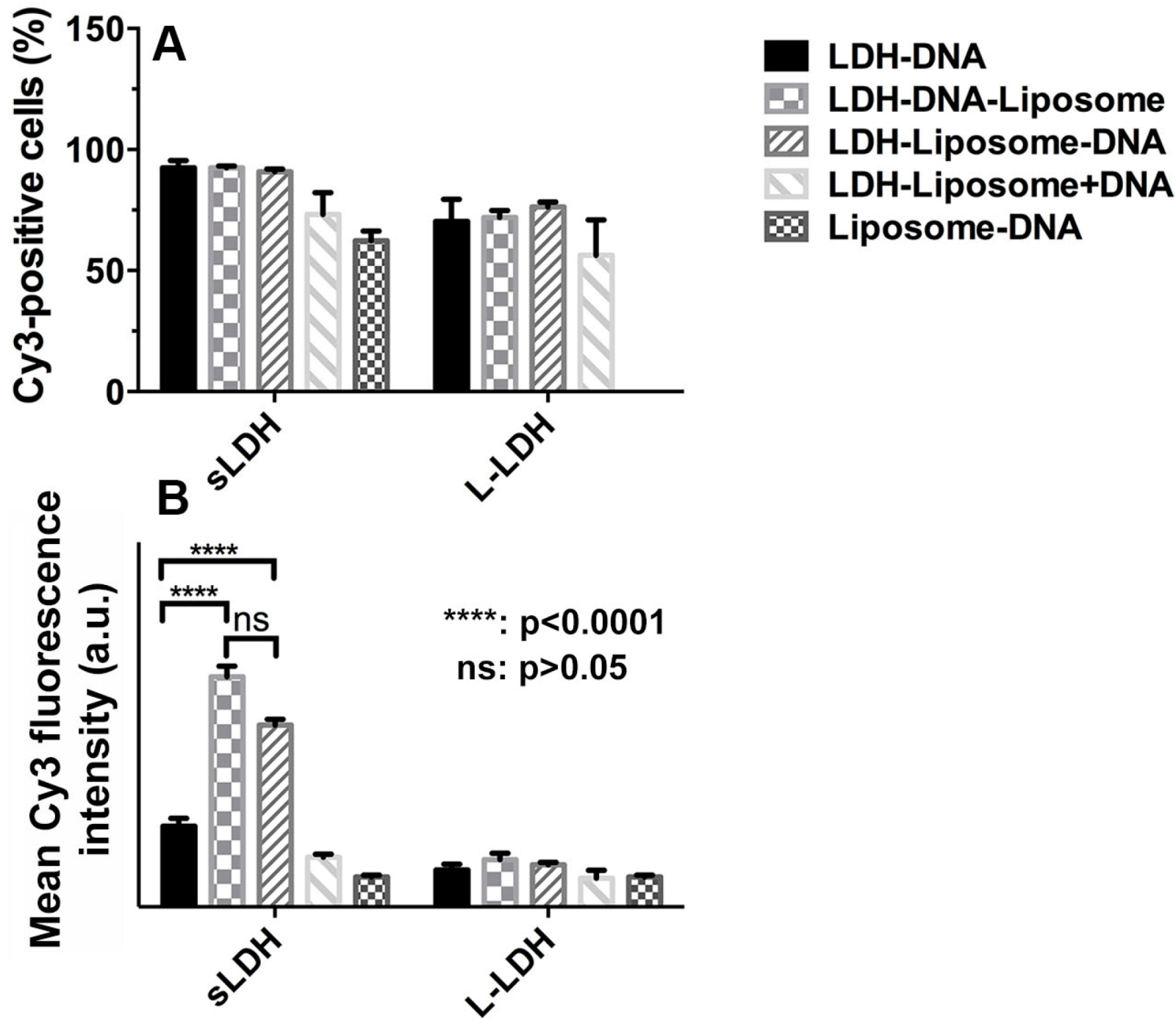
3.5. Cellular Delivery

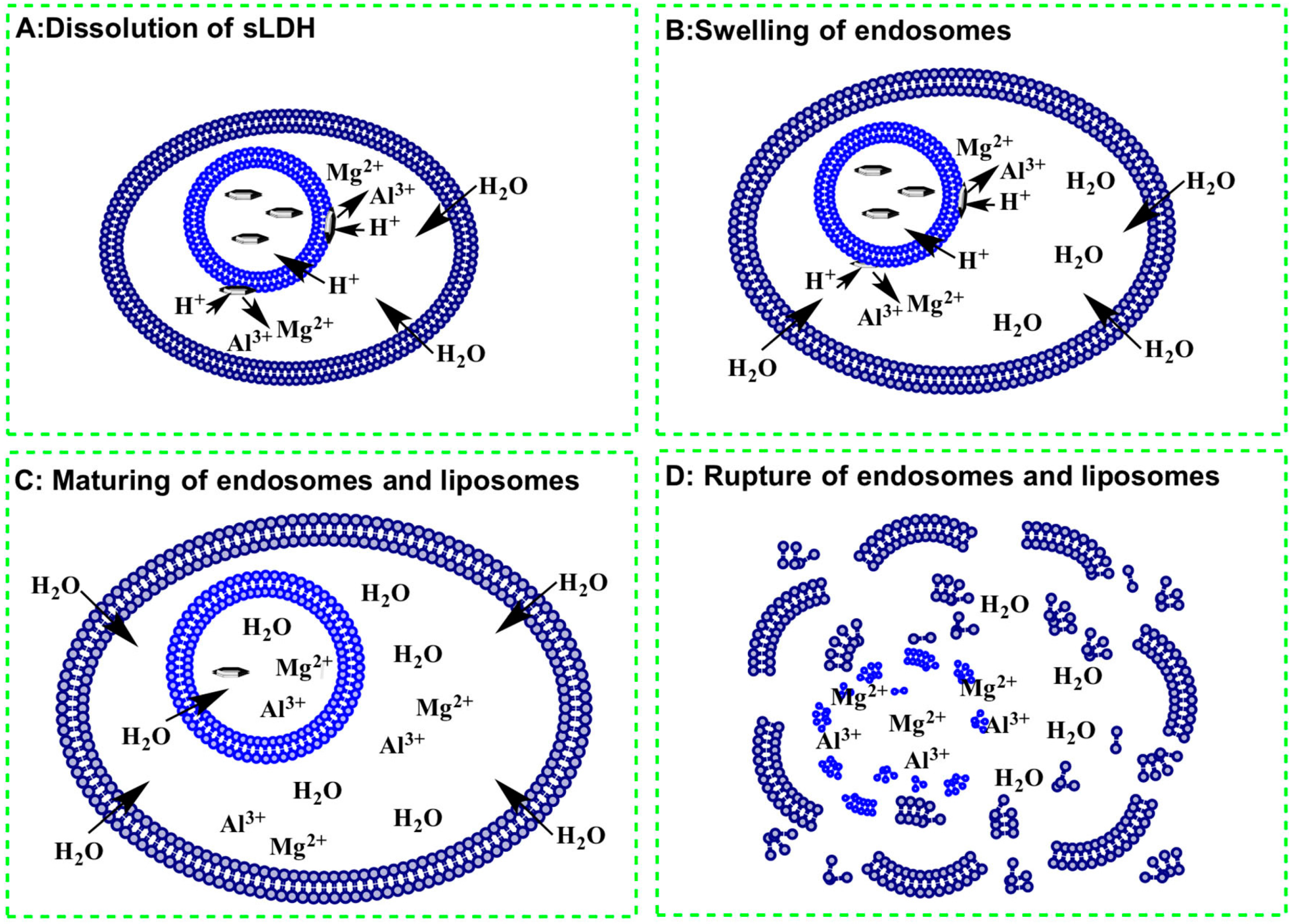
3.6. Why does the sLDH–Liposome Composite Enhance Cellular Delivery?

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mulligan, R.C. The basic science of gene therapy. Science 1993, 260, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Giacca, M. Gene Therapy; Springer: New York, NY, USA, 2010. [Google Scholar]
- Panno, J. Gene Therapy: Treatments and Cures for Genetic Diseases; Facts On File: New York, NY, USA, 2011. [Google Scholar]
- Verma, I.M.; Somia, N. Gene therapy—Promises, problems and prospects. Nature 1997, 389, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Schatzlein, A.G.; Uchegbu, I.F. Gene delivery with synthetic (non viral) carriers. Int. J. Pharm. 2001, 229, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Huang, L. Nanoparticles escaping RES and endosome: Challenges for siRNA delivery for cancer therapy. J. Nanomater. 2011. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Middaugh, C.R. Barriers to nonviral gene delivery. J. Pharm. Sci. 2003, 92, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Ruponen, M.; Honkakoski, P.; Ronkko, S.; Pelkonen, J.; Tammi, M.; Urtti, A. Extracellular and intracellular barriers in non-viral gene delivery. J. Control. Release 2003, 93, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, S.M.; Carrado, K.A.; Dutta, P.K. Handbook of Layered Materials; Marcel Dekker, Inc: New York, NY, USA, 2004; p. 646. [Google Scholar]
- Xu, Z.P.; Stevenson, G.S.; Lu, C.-Q.; Lu, G.Q.; Bartlett, P.F.; Gray, P.P. Stable suspension of layered double hydroxide nanoparticles in aqueous solution. J. Am. Chem. Soc. 2006, 128, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Stevenson, G.; Lu, C.-Q.; Lu, G.Q. Dispersion and size control of layered double hydroxide nanoparticles in aqueous solutions. J. Phys. Chem. B 2006, 110, 16923–16929. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-M.; Hwang, S.-H.; Choy, J.-H. The effect of synthetic conditions on tailoring the size of hydrotalcite particles. Solid State Ion. 2002, 151, 285–291. [Google Scholar] [CrossRef]
- Ladewig, K. Nanoparticles with Application in the Delivery of Nucleic Acids to Mammalian Cells. Ph.D. Dissertation, The University of Queensland, Queensland, Australia, March 2009. [Google Scholar]
- Wong, Y. Layered Double Hydroxide (LDH) Nanoparticle-Based Nucleic Acid Delivery System. Ph.D. Dissertation, The University of Queensland, Queensland, Australia, July 2009. [Google Scholar]
- Li, L.; Gu, W.; Chen, J.; Chen, W.; Xu, Z.P. Co-delivery of siRNAs and anti-cancer drugs using layered double hydroxide nanoparticles. Biomaterials 2014, 35, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Ladewig, K.; Niebert, M.; Xu, Z.P.; Gray, P.P.; Lu, G.Q. Controlled preparation of layered double hydroxide nanoparticles and their application as gene delivery vehicles. Appl. Clay Sci. 2010, 48, 280–289. [Google Scholar] [CrossRef]
- Ladewig, K.; Niebert, M.; Xu, Z.P.; Gray, P.P.; Lu, G.Q.M. Efficient siRNA delivery to mammalian cells using layered double hydroxide nanoparticles. Biomaterials 2010, 31, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.; Markham, K.; Xu, Z.P.; Chen, M.; Lu, G.Q.; Bartlett, P.F.; Cooper, H.M. Efficient delivery of siRNA to cortical neurons using layered double hydroxide nanoparticles. Biomaterials 2010, 31, 8770–8779. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.-H.; Park, M.; Oh, J.-M. Gene and Drug Delivery System with Soluble Inorganic Carriers; Humana Press: Towowa, NJ, USA, 2008; pp. 349–367. [Google Scholar]
- Choy, J.-H.; Kwak, S.-Y.; Jeong, Y.-J.; Park, J.-S. Inorganic layered double hydroxides as nonviral vectors. Angew. Chem. Int. Ed. 2000, 39, 4041–4045. [Google Scholar] [CrossRef]
- Oh, J.-M.; Biswick, T.T.; Choy, J.-H. Layered nanomaterials for green materials. J. Mater. Chem. 2009, 19, 2553–2563. [Google Scholar] [CrossRef]
- Ladewig, K.; Xu, Z.P.; Lu, G.Q. Layered double hydroxide nanoparticles in gene and drug delivery. Expert Opin. Drug Deliv. 2009, 6, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. Liposome Technology, Vol 2: Entrapment of Drugs and other Materials into Liposomes, 3rd ed.; Informa Healthcare: New York, NY, USA, 2007; Volume 2. [Google Scholar]
- Gabizon, A.; Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-D.; Huang, L. Stealth nanoparticles: High density but sheddable PEG is a key for tumor targeting. J. Control. Release 2010, 145, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Huang, L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J. Control. Release 2012, 158, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Qhattal, H.S.S.; Hye, T.; Alali, A.; Liu, X. Hyaluronan polymer length, grafting density, and surface poly(ethylene glycol) coating influence in vivo circulation and tumour targeting of hyaluronan-grafted liposomes. ACS Nano 2014, 8, 5423–5440. [Google Scholar] [CrossRef]
- Yuba, E.; Tajima, N.; Yoshizaki, Y.; Harada, A.; Hayashi, H.; Kono, K. Dextran derivative-based pH-sensitive liposomes for cancer immunotherapy. Biomaterials 2014, 35, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, Y.; Yuba, E.; Sakaguchi, N.; Koiwai, K.; Harada, A.; Kono, K. Potentiation of pH-sensitive polymer-modified liposomes with cationic lipid inclusion as antigen delivery carriers for cancer immunotherapy. Biomaterials 2014, 35, 8186–8196. [Google Scholar] [CrossRef] [PubMed]
- Raemdonck, K.; Braeckmans, K.; Demeester, J.; De Smedt, S.C. Merging the best of both worlds: Hybrid lipid-enveloped matrix nanocomposites in drug delivery. Chem. Soc. Rev. 2014, 43, 444–472. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Pharmacokinetics and Biodistribution of LCP Nanoparticles. Ph.D. Dissertation, The University of North Carolina at Chapel Hill, Ann Arbor, MI, USA, 2012. [Google Scholar]
- Zhang, Y. Systemic Delivery of Phosphorylated Nucleoside Analogues and siRNA via LCP Nanoparticles for Cancer Therapy. Ph.D. Dissertation, The University of North Carolina at Chapel Hill, Ann Arbor, MI, USA, 2013. [Google Scholar]
- Tseng, Y.-C. LCP Nanoparticle for Tumor and Lymph Node Metastasis Imaging. Ph.D. Dissertation, The University of North Carolina at Chapel Hill, Ann Arbor, MI, USA, 2013. [Google Scholar]
- Li, J.; Chen, Y.-C.; Tseng, Y.-C.; Mozumdar, S.; Huang, L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J. Control. Release 2010, 142, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Bégu, S.; Aubert-Pouessel, A.; Polexe, R.; Leitmanova, E.; Lerner, D.A.; Devoisselle, J.-M.; Tichit, D. New layered double hydroxides/phospholipid bilayer hybrid material with strong potential for sustained drug delivery system. Chem. Mater. 2009, 21, 2679–2687. [Google Scholar] [CrossRef]
- Huang, J.; Gou, G.; Xue, B.; Yan, Q.; Sun, Y.; Dong, L.-E. Preparation and characterization of “dextran-magnetic layered double hydroxide-fluorouracil” targeted liposomes. Int. J. Pharm. 2013, 450, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, Z.; Cui, S.; Lei, M.; Zeng, K.; Liao, Y.; Chu, W.; Deng, Y.; Zhao, C. Improvement of pharmacokinetic and antitumor activity of layered double hydroxide nanoparticles by coating with PEGylated phospholipid membrane. Int. J. Nanomed. 2014, 9, 4867–4878. [Google Scholar]
- Chen, M.; Cooper, H.M.; Zhou, J.Z.; Bartlett, P.F.; Xu, Z.P. Reduction in the size of layered double hydroxide nanoparticles enhances the efficiency of siRNA delivery. J. Colloid Interface Sci. 2013, 390, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Chen, M.; Rahman, S.; Parekh, H.S.; Cooper, H.M.; Xu, Z.P. Engineering small MgAl-layered double hydroxide nanoparticles for enhanced gene delivery. Appl. Clay Sci. 2014, 100, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Putral, L.N.; Liang, M.T.; Chang, H.I.; Davies, N.M.; McMillan, N.A.J. Development of a novel method for formulating stable siRNA-loaded lipid particles for in vivo use. Pharm. Res. 2009, 26, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, Y. A novel method for the preparation of liposomes: Freeze drying of monophase solutions. J. Pharm. Sci. 2004, 93, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, C.; Deng, Y.; Wang, Y.; Wang, W. Freeze-drying of liposomes using tertiary butyl alcohol/water cosolvent systems. Int. J. Pharm. 2006, 312, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. Liposome Technology, Vol 1: Liposome Preparation and Related Techniques, 3rd ed.; Informa Healthcare: New York, NY, USA, 2007; Volume 1. [Google Scholar]
- Kasraian, K.; DeLuca, P.P. Thermal analysis of the tertiary butyl alcohol-water system and its implications on freeze-drying. Pharm. Res. 1995, 12, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, S.V. Low concentration of extracellular hemoglobin affects shape of RBC in low ion strength sucrose solution. Bioelectrochemistry 2009, 75, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Crosasso, P.; Ceruti, M.; Brusa, P.; Arpicco, S.; Dosio, F.; Cattel, L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J. Control. Release 2000, 63, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Koo, H.; Na, J.H.; Lee, K.E.; Jeong, S.Y.; Choi, K.; Kim, S.H.; Kwon, I.C.; Kim, K. DNA amplification in neutral liposomes for safe and efficient gene delivery. ACS Nano 2014, 8, 4257–4267. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, P.; Maity, D.; Yong, C.X.; Chuang, K.H.; Ding, J.; Feng, S.S. Vitamin E (d-alpha-tocopheryl-co-poly(ethylene glycol) 1000 succinate) micelles-superparamagnetic iron oxide nanoparticles for enhanced thermotherapy and MRI. Biomaterials 2011, 32, 5663–5672. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Kim, S.Y.; Oh, J.M.; Choy, J.H. Drug-ceramic 2-dimensional nanoassemblies for drug delivery system in physiological condition. J. Am. Ceram. Soc. 2012, 95, 2758–2765. [Google Scholar] [CrossRef]
- Ogris, M.; Steinlein, P.; Carotta, S.; Brunner, S.; Wagner, E. DNA/polyethylenimine transfection particles: Influence of ligands, polymer size, and PEGylation on internalization and gene expression. AAPS J. 2001, 3, E21. [Google Scholar]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2008, 109, 259–302. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, C.R.; Wadsworth, S.C. Nucleic Acid Delivery Vehicle. U.S. Patent 6,287,857, 2001. [Google Scholar]
- Ye, G.F.; Gupta, A.; DeLuca, R.; Parang, K.; Bothun, G.D. Bilayer disruption and liposome restructuring by a homologous series of small Arg-rich synthetic peptides. Colloids Surf. B Biointerfaces 2010, 76, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Liang, E.; Rosenblatt, M.N.; Ajmani, P.S.; Hughes, J.A. Biodegradable pH-sensitive surfactants (BPS) in liposome-mediated nucleic acid cellular uptake and distribution. Eur. J. Pharm. Sci. 2000, 11, 199–205. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Parekh, H.S.; Xu, Z.P. Enhanced Cellular Delivery and Biocompatibility of a Small Layered Double Hydroxide–Liposome Composite System. Pharmaceutics 2014, 6, 584-598. https://doi.org/10.3390/pharmaceutics6040584
Dong H, Parekh HS, Xu ZP. Enhanced Cellular Delivery and Biocompatibility of a Small Layered Double Hydroxide–Liposome Composite System. Pharmaceutics. 2014; 6(4):584-598. https://doi.org/10.3390/pharmaceutics6040584
Chicago/Turabian StyleDong, Haiyan, Harendra S. Parekh, and Zhi Ping Xu. 2014. "Enhanced Cellular Delivery and Biocompatibility of a Small Layered Double Hydroxide–Liposome Composite System" Pharmaceutics 6, no. 4: 584-598. https://doi.org/10.3390/pharmaceutics6040584