Assessment of Thermal Transitions by Dynamic Mechanical Analysis (DMA) Using a Novel Disposable Powder Holder


Abstract
:
1. Introduction

2. Experimental Section
2.1. Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmaceutical Drugs/Additives | Glass Transition Temperature (ºC) Tg | Re-crystallization Temperature (ºC) | Melting Temperature (ºC) Tm |
|---|---|---|---|
| HPMC | 162 | ||
| PEO (MW 900,000 Da) | – | 65–70 | |
| Felodipine | ~43 | ~98 | 140–145 |
2.2. Instrumentation
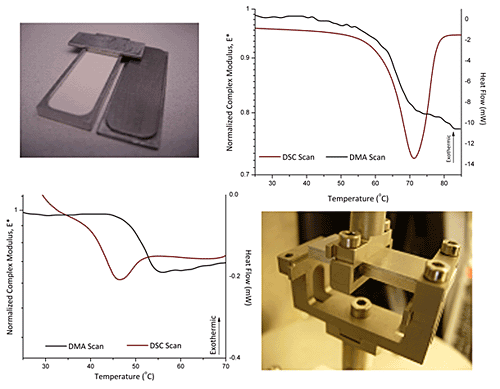
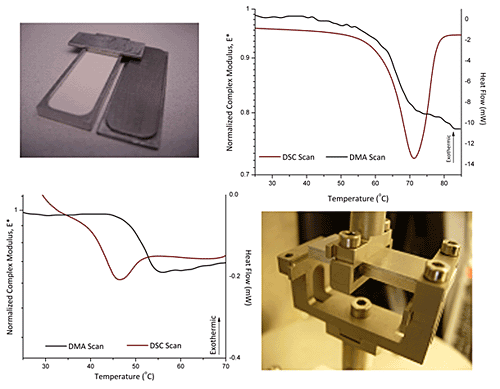
2.2.1. Powder sample holder
2.2.2. Dynamic mechanical analysis (DMA)
2.2.3. Differential Scanning Calorimetry (DSC) and Modulated Temperature DSC (MDSC)
3. Results and Discussion
3.1. Glass Transition Temperature of HPMC E4M

3.2. Melting of Polyethylene Oxide (PEO)


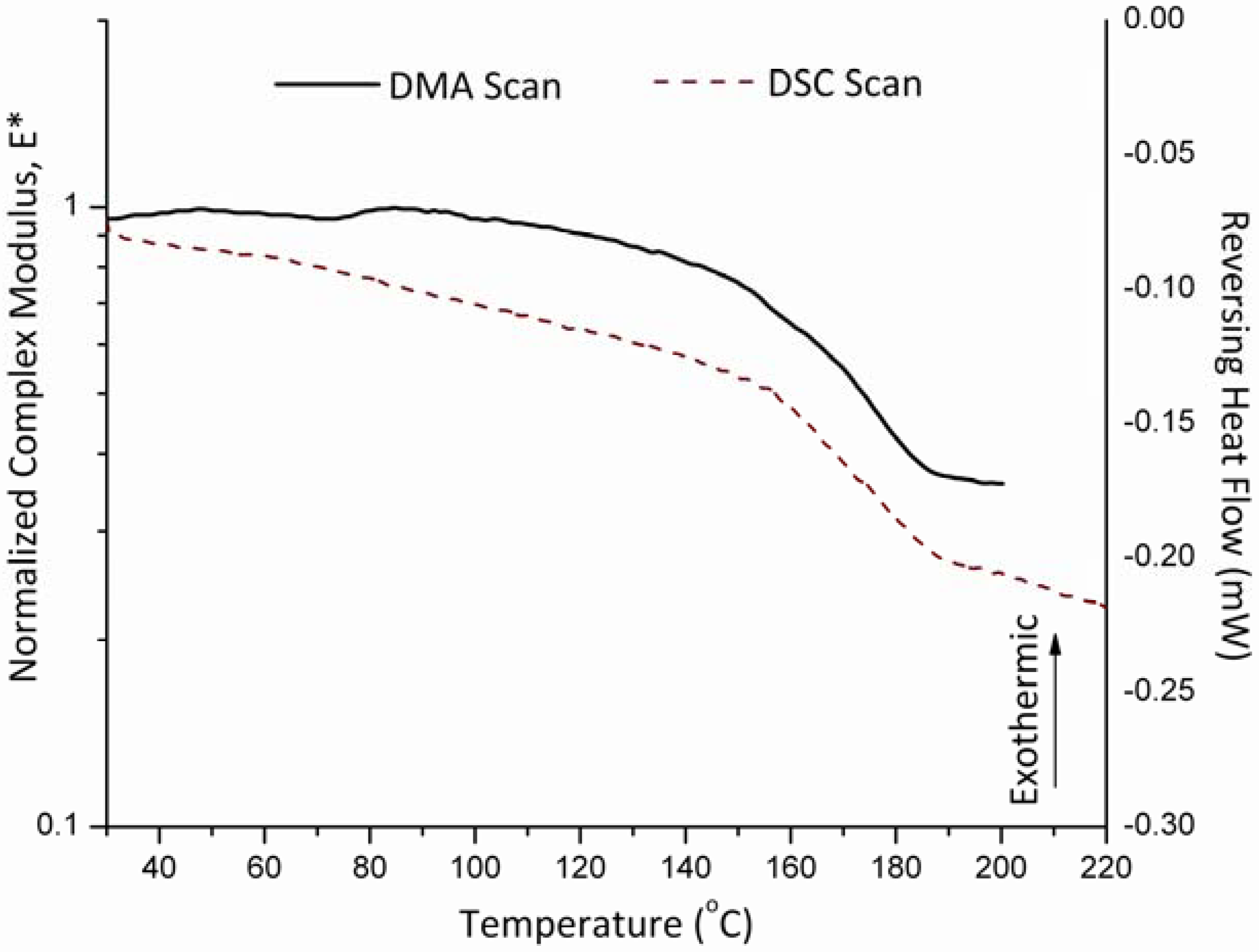
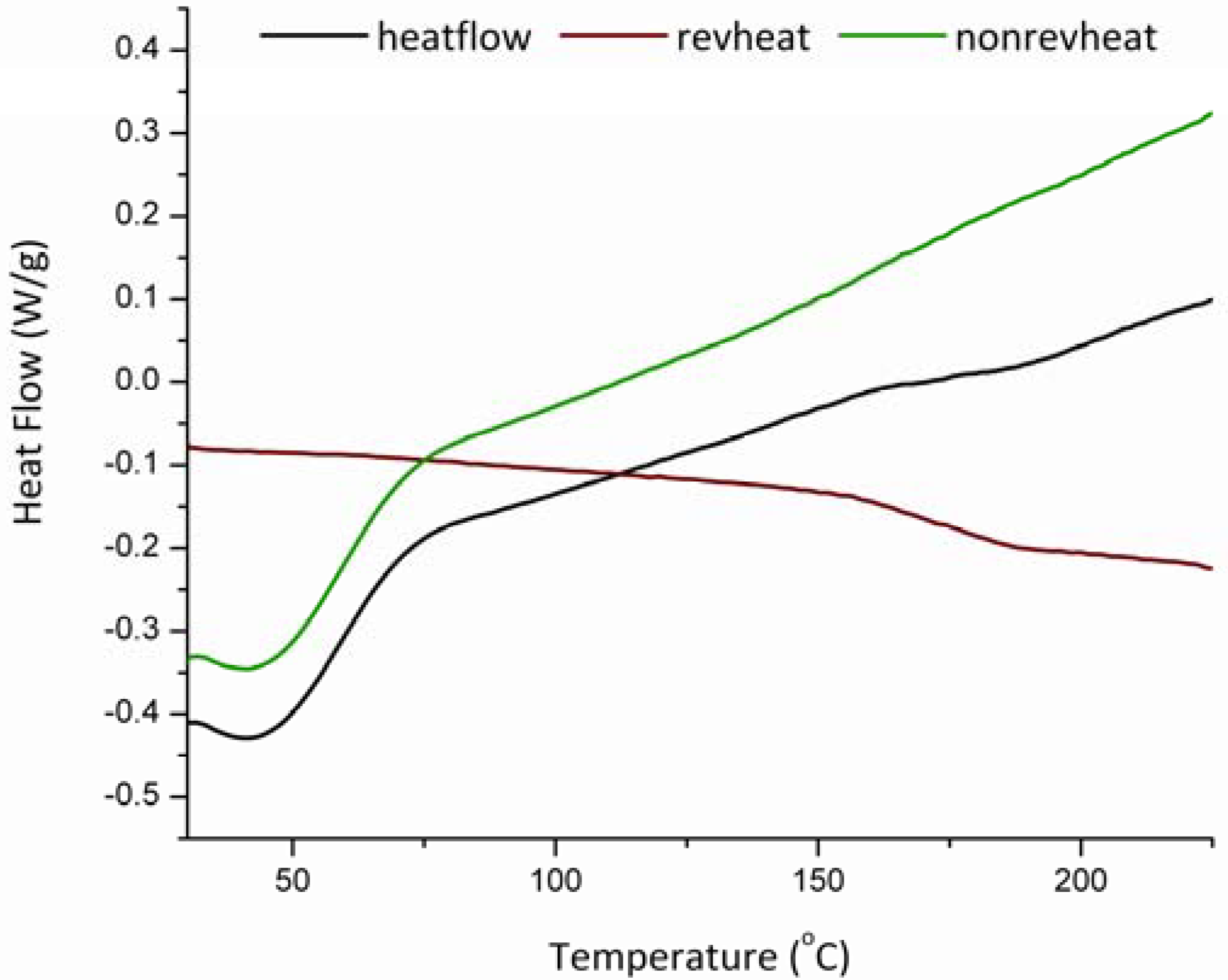
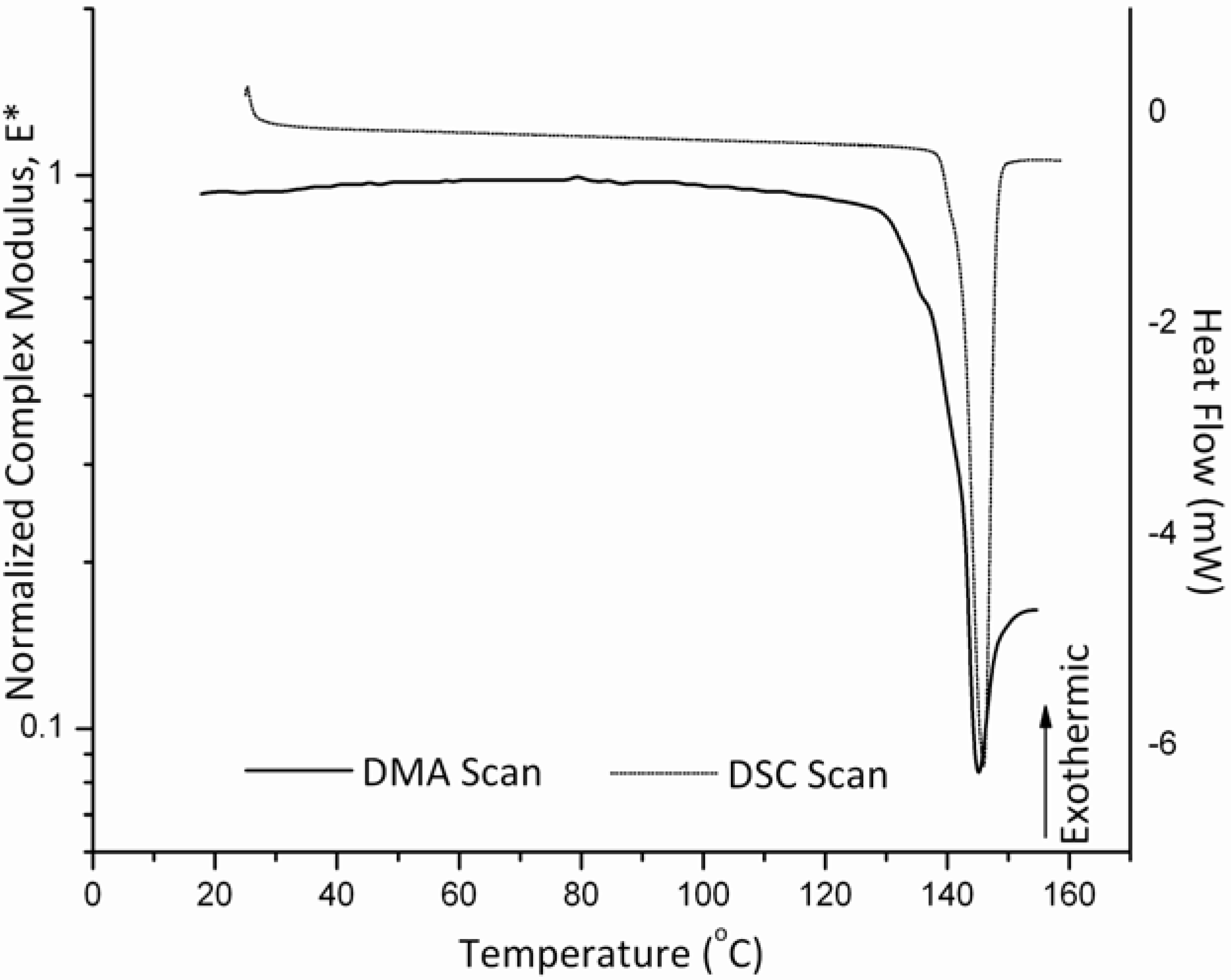
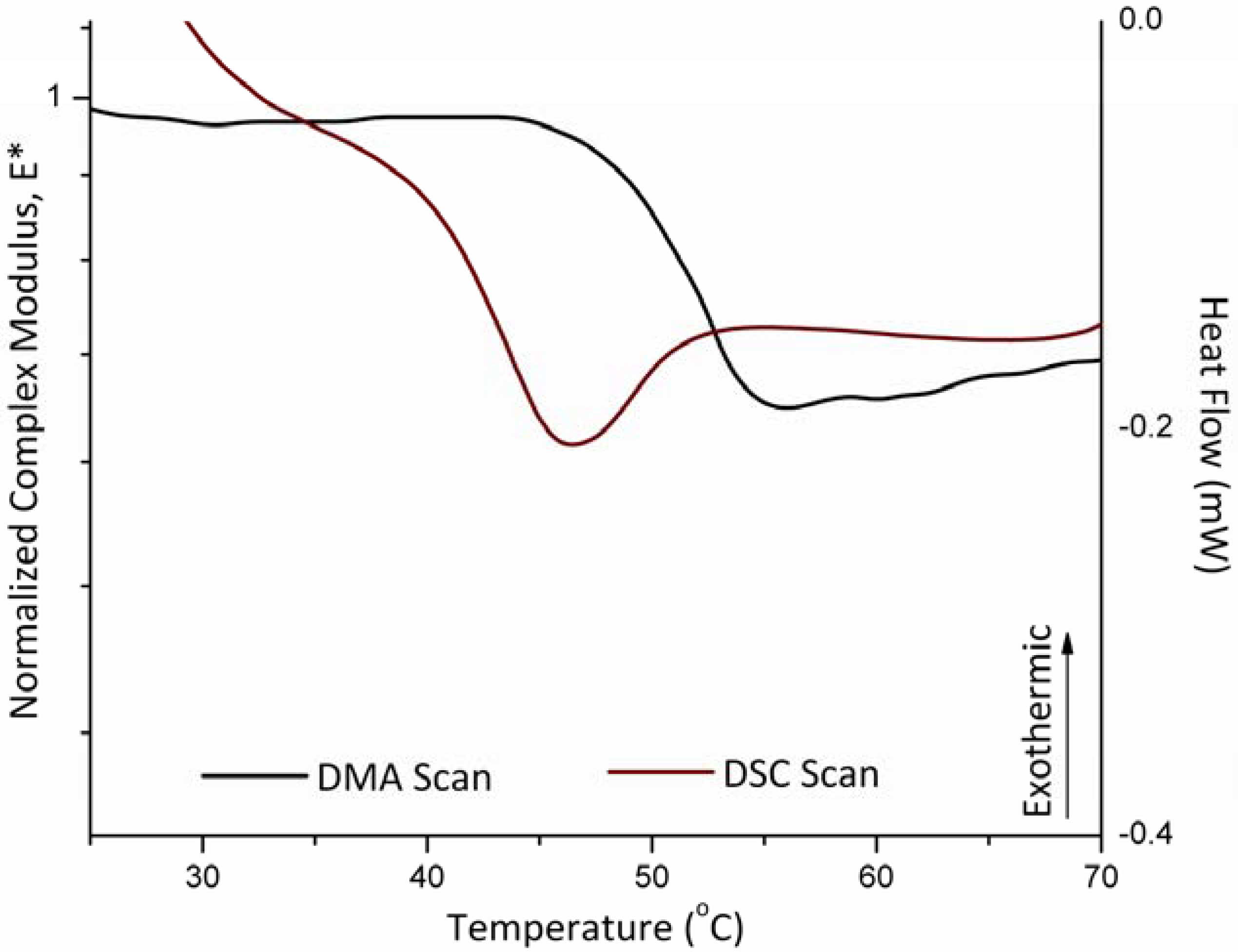
3.3. Glass Transition and Melting Temperatures of Crystalline Felodipine


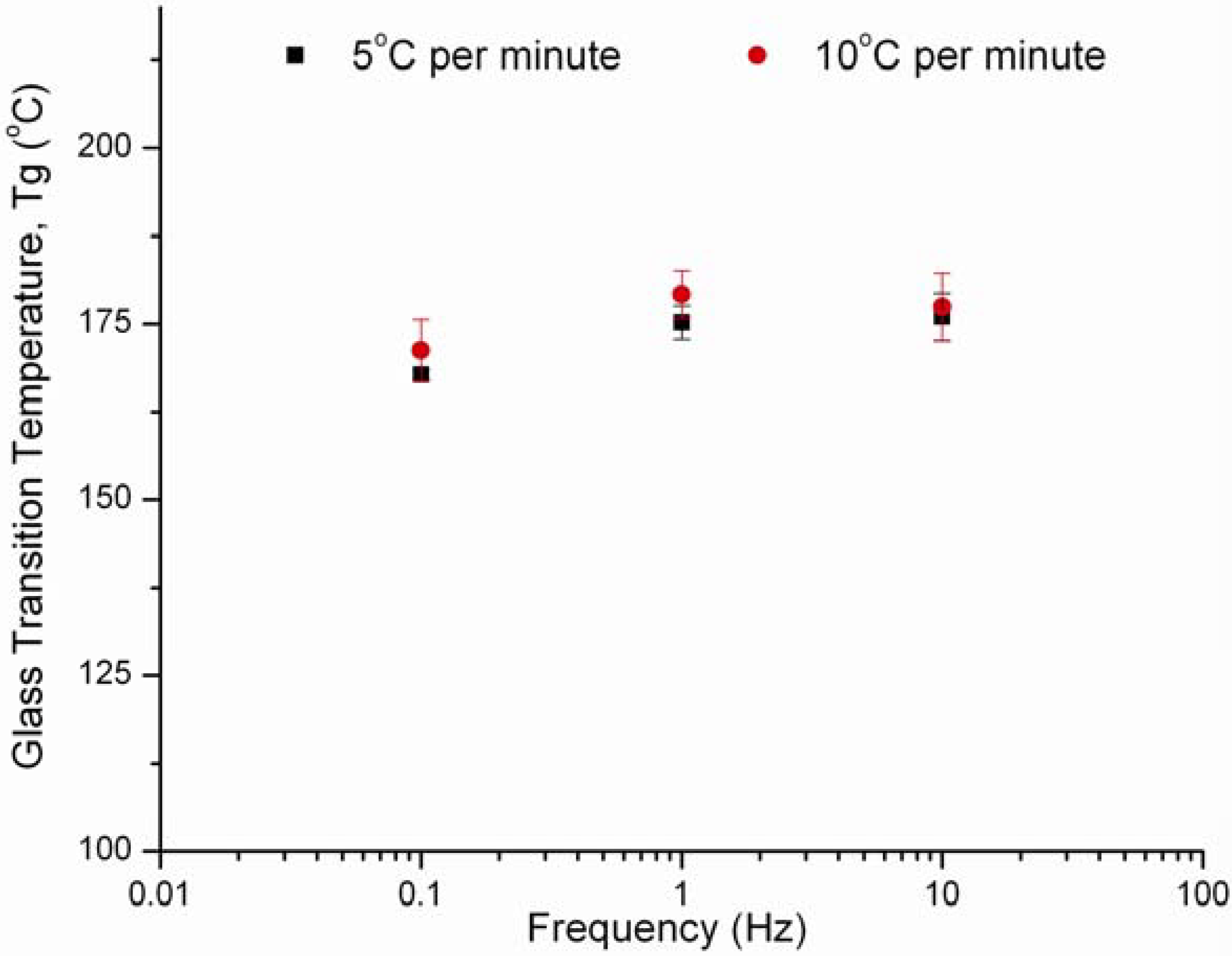
3.4. Effect of Frequency and Heating Rate on HPMC E4M Results
| Heating rate | Frequency Hz | Mean Tg (n = 3) (°C) | Standard Deviation (°C) | Percent Error (%) |
|---|---|---|---|---|
| 5 °C/min | 0.1 | 168a | 0.69 | 0.41 |
| 1.0 | 175a | 2.38 | 1.36 | |
| 10.0 | 176a | 3.37 | 1.92 | |
| 10 °C/min | 0.1 | 171a | 4.40 | 2.57 |
| 1.0 | 179a | 3.36 | 1.87 | |
| 10.0 | 177a | 4.83 | 2.72 |

4. Conclusions

Acknowledgements
References and Notes
- Angell, C.A. Formation of glasses from liquids and biopolymers. Science 1995, 267, 1924–1935. [Google Scholar]
- Brent, J.L.; Mulvaney, S.J.; Cohen, C.; Bartsch, J.A. Thermomechanical Glass Transition of Extruded Cereal Melts. J. Cereal Sci. 1997, 26, 301–312. [Google Scholar] [CrossRef]
- Brent, J.L.; Mulvaney, S.J.; Cohen, C.; Bartsch, J.A. Viscoelastic Properties of Extruded Cereal Melts. J. Cereal Sci. 1997, 26, 313–328. [Google Scholar] [CrossRef]
- Peleg, M. A Model of Mechanical Changes in Biomaterials at and around Their Glass Transition. Biotechnol. Progr. 1994, 10, 385–388. [Google Scholar] [CrossRef]
- Gidley, M.J.; Cooke, D.; Ward-Smith, S. Low moisture polysaccharide systems: Thermal and spectroscopic aspects. In The Glassy State in Foods; Blanshard, J.M.V., Lillford, P.J., Eds.; Nottingham University Press: Leicestershire, UK, 1993; pp. 303–316. [Google Scholar]
- Roos, Y.H.; Karel, M. Amorphous state and delayed ice formation in sucrose solutions. Int. J. Food Sci. Technol. 1993, 26, 553–566. [Google Scholar] [CrossRef]
- Coleman, N.J.; Craig, D.Q.M. Modulated temperature differential scanning calorimetry: A novel approach to pharmaceutical thermal analysis. Int. J. Pharm. 1996, 135, 13–29. [Google Scholar] [CrossRef]
- Hill, V.L.; Craig, D.Q.M.; Feely, L.C. Characterisation of spray-dried lactose using modulated differential scanning calorimetry. Int. J. Pharm. 1998, 161, 95–107. [Google Scholar] [CrossRef]
- O'Neill, M.J. The analysis of a temperature-controlled scanning calorimeter. Anal. Chem. 1964, 36, 1238–1245. [Google Scholar] [CrossRef]
- van der Plaats, G. A theoretical evaluation of a heat-flow differential scanning calorimeter. Thermochim. Acta 1984, 72, 77–82. [Google Scholar] [CrossRef]
- McPhillips, H.; Craig, D.Q.M.; Royall, P.G.; Hill, V.L. Characterisation of the glass transition of HPMC using modulated temperature differential scanning calorimetry. Int. J. Pharm. 1999, 180, 83–90. [Google Scholar] [CrossRef]
- Reading, M.; Luget, A.; Wilson, R. Modulated differential scanning calorimetry. Thermochim. Acta 1994, 238, 295–307. [Google Scholar] [CrossRef]
- Kalichevsky, M.T.; Jaroszkiewicz, E.M.; Ablett, S.; Blanshard, J.M.V.; Lillford, P.J. The glass transition of amylopectin measured by DSC, DMTA and NMR. Carbohydr. Polym. 1992, 18, 77–88. [Google Scholar] [CrossRef]
- Pereira, P.M.; Oliveira, J.C. Measurement of glass transition in native wheat flour by dynamic mechanical thermal analysis (DMTA). Int. J. Food Sci. Technol. 2000, 35, 183–192. [Google Scholar] [CrossRef]
- Tester, R.F.; Debon, S.J.J. Annealing of starch - a review. Int. J. Biol. Macromol. 2000, 27, 1–12. [Google Scholar] [CrossRef]
- BeMiller, J.N.; Whistler, R.L. Carbohydrates. In Food Chemistry, 3rd ed.; Fennema, O., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1996; pp. 157–224. [Google Scholar]
- Mahlin, D.; Wood, J.; Hawkins, N.; Mahey, J.; Royall, P.G. A novel powder sample holder for the determination of glass transition temperatures by DMA. Int. J. Pharm. 2009, 371, 120–125. [Google Scholar] [CrossRef]
- Abiad, M.G. Thermal properties of food and pharmaceutical powders. PhD dissertation, Purdue University, West Lafayette, IN, USA, 2009. [Google Scholar]
- Ahmed, H.; Buckton, G.; Rawlins, D.A. Crystallisation of partially amorphous griseofulvin in water vapour: determination of kinetic parameters using isothermal heat conduction microcalorimetry. Int. J. Pharm. 1998, 167, 139–145. [Google Scholar] [CrossRef]
- Correia, N.T.; Ramos, J.J.M.; Descamps, M.; Collins, G. Molecular Mobility and Fragility in Indomethacin: A Thermally Stimulated Depolarization Current Study. Pharm. Res. 2001, 18, 1767–1774. [Google Scholar] [CrossRef]
- Rowe, R.C.; Shesky, P.J.; Weller, P.J. Handbook of Pharmaceutical Excipients, 4th ed.; Pharmaceutical Press: London, UK, 2003. [Google Scholar]
- Trojak, A.; Kocevar, K.; Musevic, I.; Srcic, S. Investigation of the felodipine glassy state by atomic force microscopy. Int. J. Pharm. 2001, 218, 145–151. [Google Scholar] [CrossRef]
- Mao, C.; Chamarthy, S.P.; Byrn, S.R.; Pinal, R. Theoretical and experimental considerations on the enthalpic relaxation of organic glasses using differential scanning calorimetry. J. Phys. Chem. B 2010, 114, 269–279. [Google Scholar] [CrossRef]
- Yamamuro, O.; Tsukushi, I.; Lindqvist, A.; Takahara, S.; Ishikawa, M.; Matsuo, T. Calorimetric study of glassy and liquid toluene and ethylbenzene: Thermodynamic approach to spatial heterogeneity in glass-forming molecular liquids. J. Phys. Chem. 1998, 102, 1605–1609. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abiad, M.G.; Campanella, O.H.; Carvajal, M.T. Assessment of Thermal Transitions by Dynamic Mechanical Analysis (DMA) Using a Novel Disposable Powder Holder. Pharmaceutics 2010, 2, 78-90. https://doi.org/10.3390/pharmaceutics2020078
Abiad MG, Campanella OH, Carvajal MT. Assessment of Thermal Transitions by Dynamic Mechanical Analysis (DMA) Using a Novel Disposable Powder Holder. Pharmaceutics. 2010; 2(2):78-90. https://doi.org/10.3390/pharmaceutics2020078
Chicago/Turabian StyleAbiad, Mohamad G., Osvaldo H. Campanella, and M. Teresa Carvajal. 2010. "Assessment of Thermal Transitions by Dynamic Mechanical Analysis (DMA) Using a Novel Disposable Powder Holder" Pharmaceutics 2, no. 2: 78-90. https://doi.org/10.3390/pharmaceutics2020078