3.1. The impact of PCN-treatment on plasma [3H]-digoxin concentrations following oral and IV administration
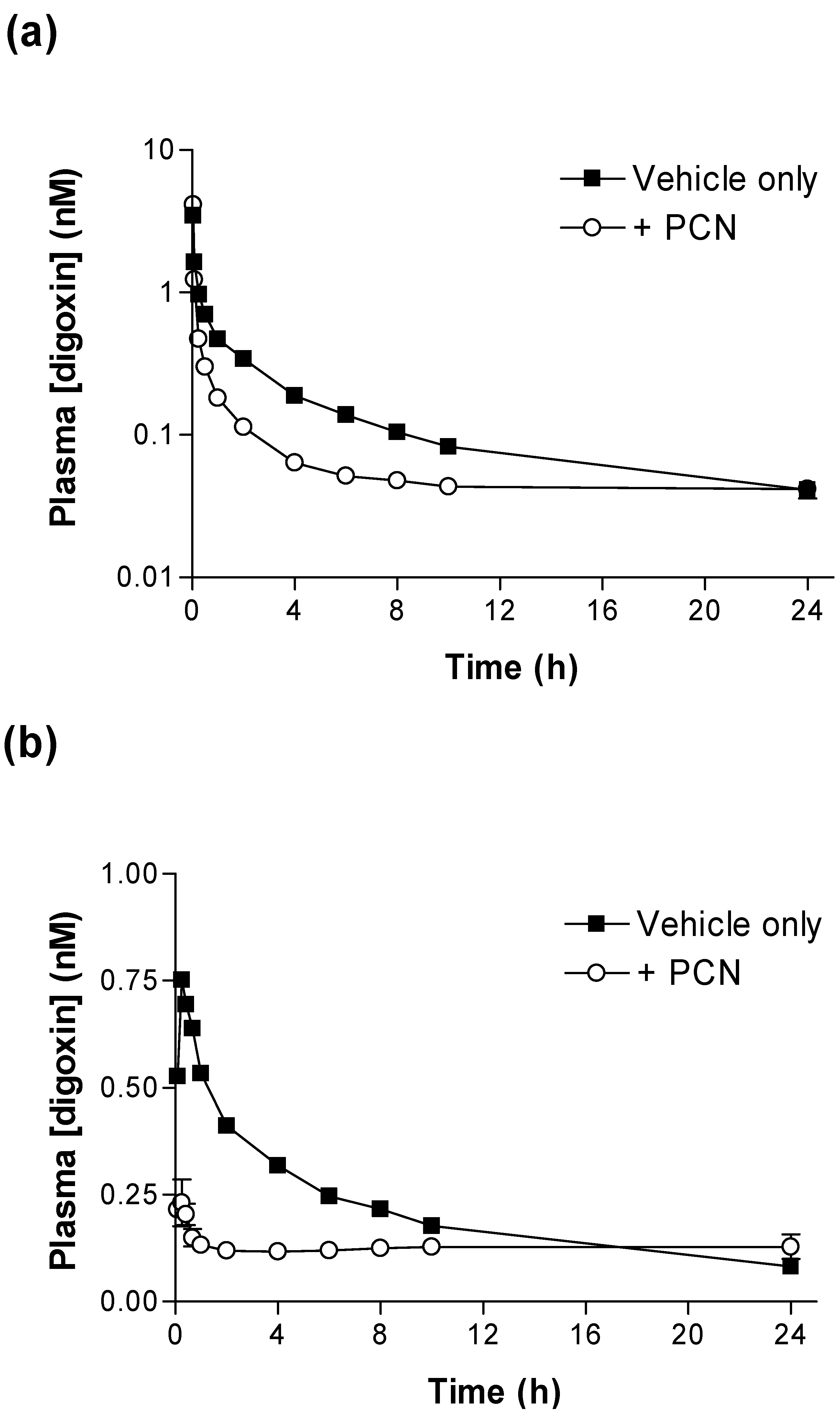
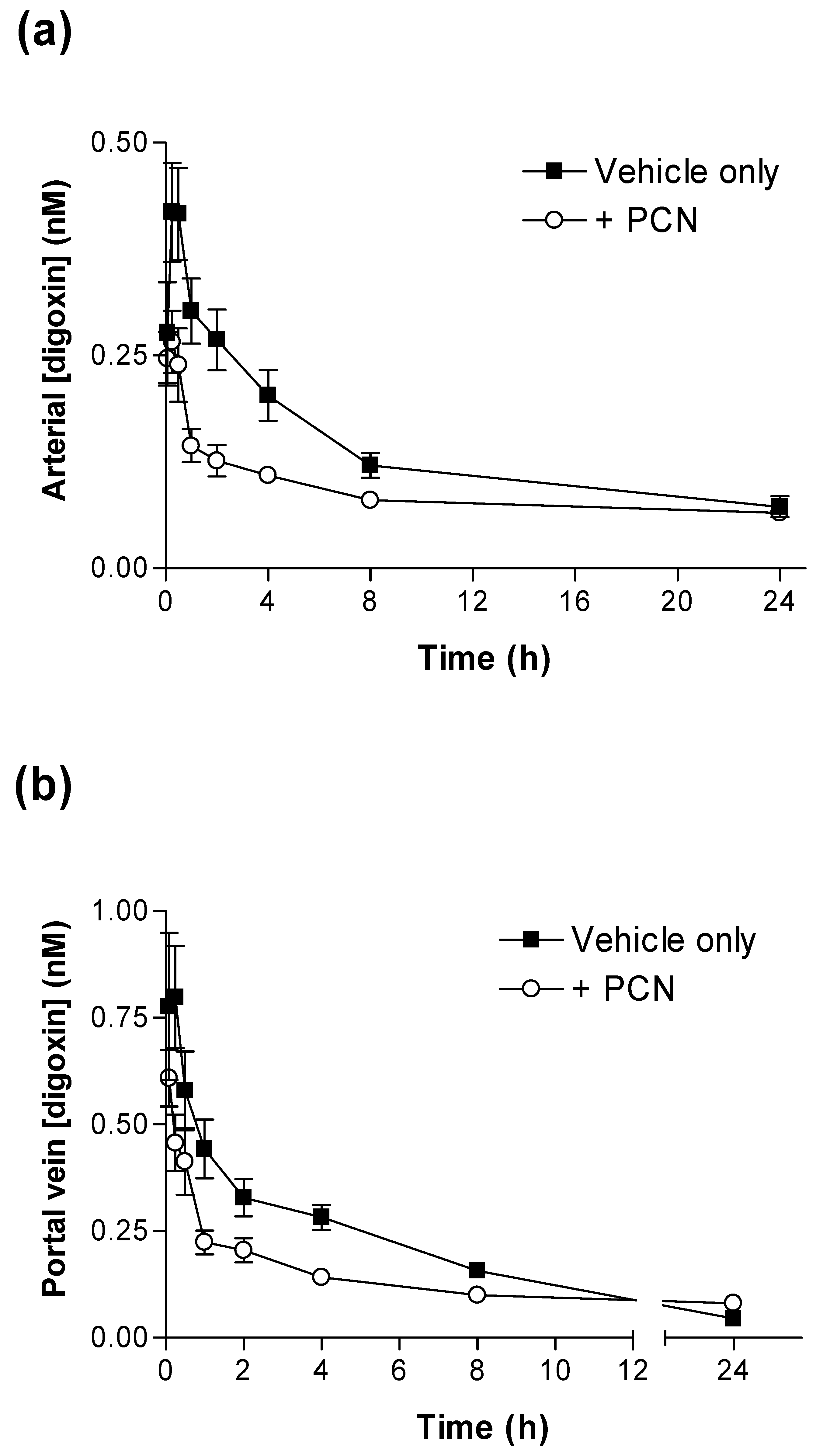
Figure 1 shows that PCN pre-treatment substantially alters the pharmacokinetic plasma profiles of [
3H]-digoxin after intravenous (IV) (2.3 nmol.kg
-1) or oral (PO) (4.6 nmol.kg
-1) dosing. Following IV administration (
Figure 1a) the concentration of [
3H]-digoxin remaining in blood plasma was reduced at all time points in PCN-dosed rats in comparison to control animals. The IV and oral [
3H]-digoxin AUC
(0-24h) for control and PCN-treated animals are shown in
Table 1.
Table 1.
[3H]-digoxin arterial and portal vein AUC in control and PCN-treated rats. Significant difference as indicated by Student’s T-test *** p < 0.001, * p < 0.05.
Table 1.
[3H]-digoxin arterial and portal vein AUC in control and PCN-treated rats. Significant difference as indicated by Student’s T-test *** p < 0.001, * p < 0.05.
| Arterial AUC (nmol.hr-1/L) | Portal AUC (nmol.hr-1/L) |
|---|
| Dosing | Control | PCN | Control | PCN |
|---|
| IV | 3.55 ± 0.09 | 1.90 ± 0.03*** | - | - |
| Oral | 4.95 ± 0.38 | 2.67 ± 0.59* | 3.81 ± 0.09 | 2.75 ± 0.09* |
Significant reductions in IV and oral [
3H]-digoxin AUC
(0-24h) (p < 0.001 & p < 0.05, respectively) are evident for PCN-treated animals in comparison to controls (
Table 1). Following oral dosing in control animals, there was an initial rise in plasma [
3H]-digoxin between 5 minutes (0.53 ± 0.09 nM,
n = 4) and 15 minutes (0.75 ± 0.10 nM,
n = 4), indicating a period of rapid intestinal uptake into the systemic circulation (
Figure 1b). In the PCN-dosed animals the concentration of [
3H]-digoxin entering the circulation is greatly reduced (
Figure 1 b). This results in a reduced AUC
(0-24h) in the PCN-treated animals (p < 0.05
versus controls,
Table 1) and a reduced C
max from 0.77 ± 0.09 nM,
n = 4 in controls to 0.27 ± 0.02 nM,
n = 4 (p < 0.005).
3.2. Hepatic portal vein [3H]-digoxin activities and biliary [3H]-digoxin secretion
In order to assess the contribution of hepatic clearance and biliary excretion of [
3H]-digoxin/digoxin metabolites to the plasma profile following oral absorption, total [
3H] activities in hepatic portal vein, arterial plasma and in bile were determined after oral [
3H]-digoxin dosing (
Figure 2 and
Figure 3).
Figure 2a confirms the initial observation that PCN pre-treatment produces a significant reduction in systemic arterial plasma [
3H] concentrations; the AUC
(0-24h) in controls was 3.16 ± 0.27 nmol.hr
-1/L (
n = 5), which decreased to 2.04 ± 0.15 nmol.hr
-1/L (
n = 6) in PCN-treated animals.
Figure 2b shows that in the same animals, PCN treatment also reduced portal vein plasma AUC
(0-24h). Portal vein plasma AUC
(0-24h) values are shown in
Table 1, indicating a significant reduction in PCN-treated plasma concentration as compared to controls (
n = 6&5 respectively; p < 0.02). However, portal vein C
max shows a non-significant reduction from 0.93 ± 0.17 nM,
n = 5 in controls to 0.62 ± 0.06 nM,
n = 6 in PCN treated animals (p < 0.1, n.s.). A significant hepatic clearance of [
3H]-digoxin is indicated by the difference in C
max between control animals for portal vein and arterial plasma samples (0.93 ± 0.17 nM, to 0.44 ± 0.05 nM,
n = 5 p < 0.05) and for PCN-treated animals (0.62 ± 0.06 nM, to 0.29 ± 0.04 nM,
n = 6 p < 0.001). This data is therefore consistent with both a reduced intestinal absorption of digoxin, but also a component of enhanced hepatic extraction in the whole animal response to PCN treatment. Whereas PCN treatment decreases portal vein AUC
(0-24h) by 28%, arterial AUC
(0-24h) is reduced by 35%. Hepatic extraction reduces arterial [
3H]-digoxin AUC
(0-24h) by 17% in control animals compared to 26% in PCN pre-treated rats.
Conventionally the presence of Pgp in the apical membrane of the intestine is thought to limit the epithelial absorption of Pgp substrates below the levels expected from the physicochemical properties of the compound alone (mainly lipophilicity). Increases in Pgp would therefore further limit intestinal absorptive permeability, so reducing circulating plasma levels. Hepatic portal vein blood digoxin concentrations are reduced at all but the initial time points after oral dosing, with a significant reduction in the AUC
(0-24h) as well as the C
max, suggesting that
in vivo a reduction in intestinal absorption must occur. Although digoxin is reported to be a substrate for rodent cyp3a4, studies have concluded that rat intestinal digoxin metabolism was negligible [
16], with the formation of digoxigenin bis-digitoxoside (Dg2) at <1% and levels of mono-digoxitoxoside (Dg1) and digoxigenin (DG0) even lower or undetectable [
12]. This metabolic profile was also found to be unaltered following PCN treatment [
12]. Accordingly it is reasonable to suggest that the reduced levels of digoxin in the hepatic portal vein are the result of an increased functional Pgp expression in the intestine.
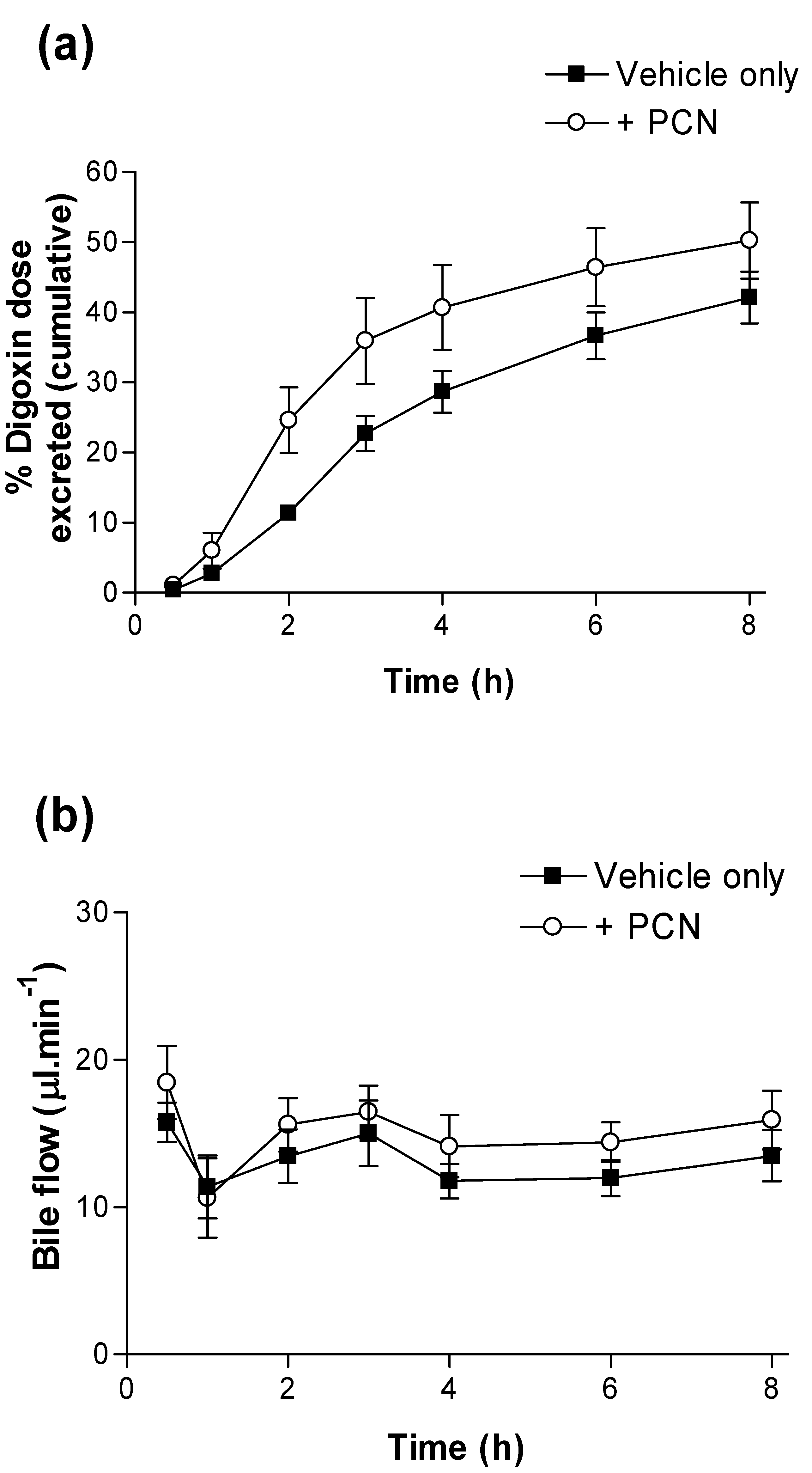
A hepatic component was confirmed by measurement of bile flow and cumulative [
3H]-digoxin secretion via bile (
Figure 3). Whereas bile flow is unaltered over the 8 hour time-course by PCN pre-treatment (
Figure 3b), [
3H]-digoxin excretion to bile is increased; at 2 hours 0.25 ± 0.05,
n = 6 of the [
3H]-digoxin dose is excreted into bile in the PCN-treated rats compared to controls, where just0.11 ± 0.01,
n = 6 (p < 0.05) of the dose is excreted. Furthermore, the calculated CL
bile at 8 h rose from 0.69 L/h/kg in controls to 1.39 L/h/kg in the PCN-treated rats, a 2-fold increase.
Excretion into bile is significantly increased in the PCN treated animals at 2 hours. How is excretory transport into bile increased? Both Pgp and PXR are present in hepatocytes [
17] and induction of canalicular Pgp could represent a mechanism for increased digoxin transport. However, the liver-specific oatp1a4 (old nomenclature oatp2, gene Slco1A4, previously termed Slc21A5) is a high affinity digoxin transporter which impacts on digoxin disposition [
15,
16] . Oatp1a4 is present on the sinusoidal membrane of hepatocytes and numerous studies have reported PXR-mediated induction of oatp1a4 in response to xenobiotics, including PCN [
19,
20]. An increase in hepatic sinusoidal oatp1a4 expression, coupled to similar increases in canalicular Pgp levels would explain increased biliary clearance. Increased digoxin metabolism is also possible, as PCN is also known to induce cyp3a expression [
12] with hepatic metabolism by cyp3a comprising an estimated 60% of the dose [
21,
22,
23]. Potential metabolism must therefore be taken into account when assessing whole body digoxin disposition in rat. Our attempts at separating metabolites from total [
3H]-digoxin were unsuccessful. A similar increase in biliary secretion of ouabain, which is not metabolised, was previously observed in PCN pre-treated rats [
24].
Figure 1.
Time-dependence of [3H]-digoxin plasma concentrations in control and PCN treated rats following intravenous and oral administration (a) Arterial plasma concentrations of [3H]-digoxin in rats following IV administration at 2.3 nmol.kg-1 or (b) oral administration at 4.6 nmol.kg-1 for control (■) and PCN (○) dosed animals, treated for 72 hours prior to the experiment. Data are mean ± SEM of n = 3 (IV) and n = 4 (PO) animals.
Figure 1.
Time-dependence of [3H]-digoxin plasma concentrations in control and PCN treated rats following intravenous and oral administration (a) Arterial plasma concentrations of [3H]-digoxin in rats following IV administration at 2.3 nmol.kg-1 or (b) oral administration at 4.6 nmol.kg-1 for control (■) and PCN (○) dosed animals, treated for 72 hours prior to the experiment. Data are mean ± SEM of n = 3 (IV) and n = 4 (PO) animals.
Figure 2.
Time-dependence of [3H]-digoxin plasma concentrations in control and PCN treated rats following oral administration in (a) arterial plasma and (b) portal vein plasma for control (■) and PCN (○) dosed animals, treated for 72 hours prior to the experiment. Data are mean ± SEM of n = 5 control or 6 PCN-treated animals.
Figure 2.
Time-dependence of [3H]-digoxin plasma concentrations in control and PCN treated rats following oral administration in (a) arterial plasma and (b) portal vein plasma for control (■) and PCN (○) dosed animals, treated for 72 hours prior to the experiment. Data are mean ± SEM of n = 5 control or 6 PCN-treated animals.
Figure 3.
Excretion of digoxin into bile in control and PCN-dosed rats. (a) Cumulative dose (%) of [3H]-digoxin excreted into bile following oral administration (4.6 nmol.kg-1). Results indicate % dose in control (■) and PCN (○) dosed animals, treated for 72 hours prior to the experiment. Data are mean ± SEM of n = 5 (control) and n = 6 (PCN) animals. (b) shows the bile flow during 8 hours sampling.
Figure 3.
Excretion of digoxin into bile in control and PCN-dosed rats. (a) Cumulative dose (%) of [3H]-digoxin excreted into bile following oral administration (4.6 nmol.kg-1). Results indicate % dose in control (■) and PCN (○) dosed animals, treated for 72 hours prior to the experiment. Data are mean ± SEM of n = 5 (control) and n = 6 (PCN) animals. (b) shows the bile flow during 8 hours sampling.
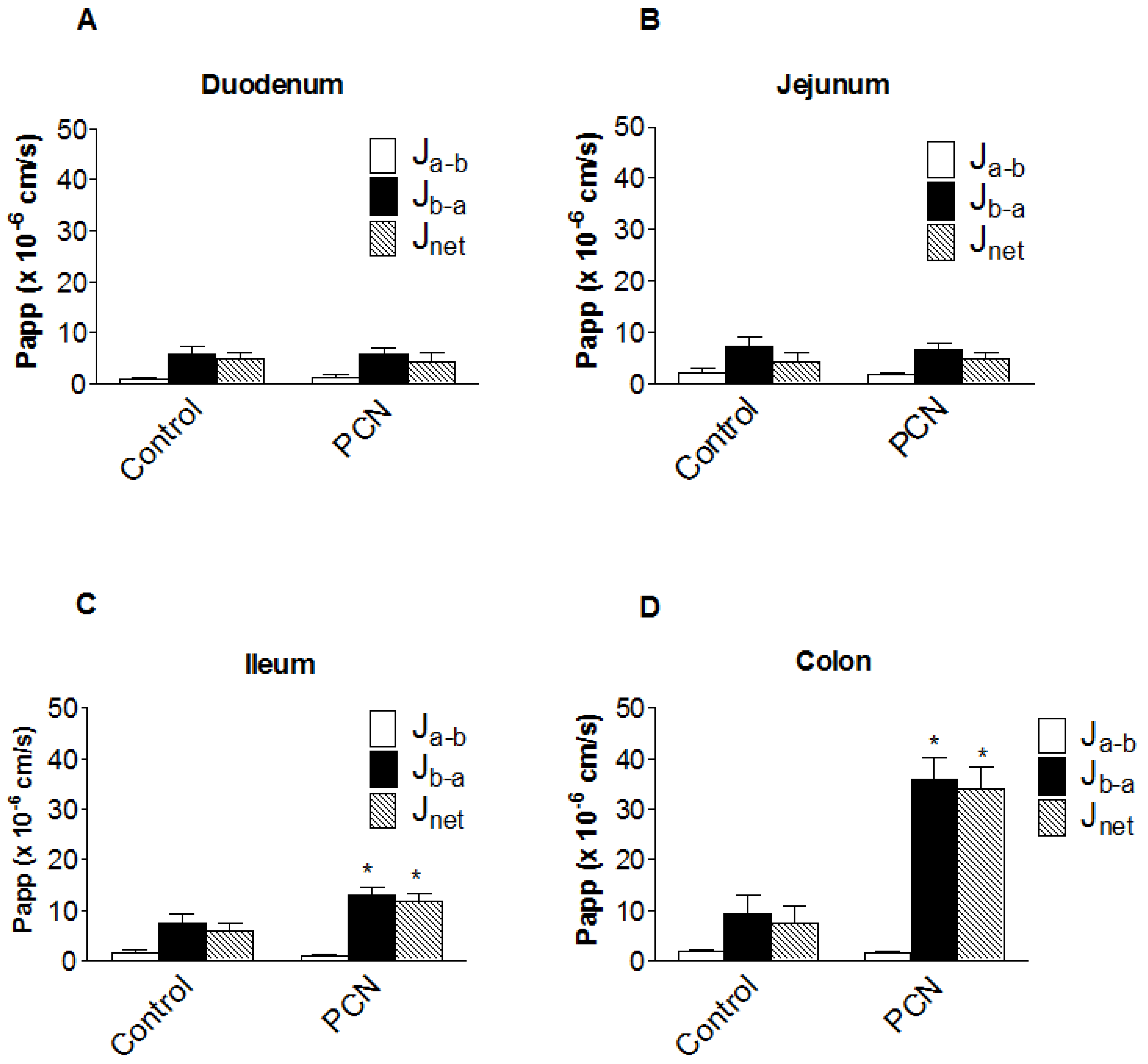
3.3. [3H]-Digoxin transport in excised intestinal sections
From the pharmacokinetic profiles, it is apparent that intestinal digoxin absorption is substantially altered following PCN treatment. In order to assess the impact of intestinal transport on the pharmacokinetic plasma profiles, transepithelial [
3H]-digoxin fluxes were measured across isolated intestinal sections, mounted in modified Ussing chambers (
Figure 4). [
3H]-Digoxin transport was determined to be greater in the basal-to-apical (P
b-a) than apical-to-basal (P
a-b) direction across all excised tissue sections (duodenum, jejunum, ileum and colon;
Figure 4). Under control conditions a net [
3H]-digoxin secretion (expressed as permeability P
net=J
net/C
digoxin) of 4.88 ± 1.36 cm.s
-1 (x 10
-6) (
n = 6) for duodenum, 4.20 ± 1.75 cm.s
-1 (x 10
-6) (
n = 12) for jejunum, 5.79 ± 1.60 cm.s
-1 (x 10
-6) (
n = 9) for ileum and 7.52 ± 3.33 cm.s
-1 (x 10
-6) (
n = 6) for the colon was observed.
The passive permeability of radiolabelled [14C]-mannitol was measured concurrently with [3H]-digoxin flux as a measure of the paracellular flux component and therefore the amount of ‘leak’ inherent within each section of intestine. Mannitol permeability was significantly different only in the colon (2.18 ± 0.40 cm.s-1, n = 20) compared to the other intestinal segments, e.g., the jejunum (6.02 ± 0.76 cm.s-1, n = 43). This difference correlates with a greater electrical resistance for colon compared to jejunum (97.9 ± 13.4 Ω.cm2, n = 12 compared to 28.6 ± 1.9 Ω.cm2, n = 43 p < 0.01).
There were no significant changes in PD or SCC in any section of the intestine following PCN-dosing. Also for the duodenum, jejunum and ileum, PCN treatment displayed no effect on trans-intestinal resistance recordings. The resistance across sections of colon excised from PCN-dosed animals (127.6 ± 5.9 Ω.cm-2, n = 9) were slightly increased compared to controls (97.9 ± 13.4 Ω.cm-2, n = 12) p < 0.05. In addition there were also no significant changes in mannitol permeability between controls and PCN-treated duodenal, jejunal and colonic tissue segments. Overall it is apparent that PCN-dosing of animals is without effect on either tissue integrity or ion transport of the four intestinal segments used in this study.
Figure 4a and 4b show that PCN pre-treatment does not significantly alter duodenal or jejunal net [
3H]-digoxin permeability. In the ileum however, there were significant increases in both P
b-a (7.43 ± 1.95 cm.s
-1 (x 10
-6),
n = 9 to 12.98 ± 1.51 cm.s
-1 (x 10
-6),
n = 7, p < 0.05) and P
net (5.79 ± 1.60 cm.s
-1 (x 10
-6),
n = 9 to 11.89 ± 1.48 cm.s
-1 (x 10
-6),
n = 7, p < 0.05) (
Figure 4c). This represents a 1.75-fold increase in P
b-a and a 2.05-fold increase in P
net, suggesting an increase in the functional activity of the secretory efflux transporter Pgp. A larger increase in the basal-to-apical and net permeability of [
3H]-digoxin was seen in the excised colon sections, with P
b-a significantly increasing (p < 0.01) from 9.25 ± 3.77 cm.s
-1 (x 10
-6) (
n = 6), to 35.93 ± 4.47 cm.s
-1 (x 10
-6) (
n = 4), a 3.9-fold increase (
Figure 4d). Net secretory flux also displayed the same pattern with a significant increase (p < 0.01) of 4.5-fold, from 7.52 ± 3.33 cm.s
-1 (x 10
-6) (
n = 6), to 34.11 ± 4.25 cm.s
-1 (x 10
-6) (
n = 4) (
Figure 4d). The larger increases in colonic [
3H]-digoxin secretion following PCN treatment suggest a greater capacity for increasing Pgp expression and activity in the distal GI tract.
Figure 4.
Segmental changes in intestinal bi-directional [3H]-digoxin flux in PCN-dosed rats. Transintestinal [3H]-digoxin permeability across excised sections of intestine mounted in modified Ussing chambers. Permeability was determined in both the apical-to-basal (Pa-b) and basal-to-apical (Pb-a) directions. Rats were dosed for 72 hours with an agarose vehicle control or PCN (35 mg.kg-1) prior to tissue excision. (a) duodenum, Data is mean ± SEM of n = 6 (Control) and n = 5 (PCN) tissue sections (b) jejunum, Data is mean ± SEM of n = 12 (Control) and n = 9 (PCN) tissue sections (c) ileum, Data is mean ± SEM of n = 9 (Control) and n = 7 (PCN) tissue sections (d) colon Data is mean ± SEM of n = 6 (Control) and n = 4 (PCN) tissue sections. * P < 0.05, significantly different from control values.
Figure 4.
Segmental changes in intestinal bi-directional [3H]-digoxin flux in PCN-dosed rats. Transintestinal [3H]-digoxin permeability across excised sections of intestine mounted in modified Ussing chambers. Permeability was determined in both the apical-to-basal (Pa-b) and basal-to-apical (Pb-a) directions. Rats were dosed for 72 hours with an agarose vehicle control or PCN (35 mg.kg-1) prior to tissue excision. (a) duodenum, Data is mean ± SEM of n = 6 (Control) and n = 5 (PCN) tissue sections (b) jejunum, Data is mean ± SEM of n = 12 (Control) and n = 9 (PCN) tissue sections (c) ileum, Data is mean ± SEM of n = 9 (Control) and n = 7 (PCN) tissue sections (d) colon Data is mean ± SEM of n = 6 (Control) and n = 4 (PCN) tissue sections. * P < 0.05, significantly different from control values.
Pgp activity is conventionally thought to reduce the apparent absorptive permeability of substrates when expressed in model cell-lines and in intestinal tissues [
5,
25]. This is most evident following Pgp inhibition (e.g. by verapamil), with an increase in the absorptive permeability (P
a-b) of a concurrently measured substrate. Under basal conditions, [
3H]-digoxin P
a-b values were 1.05 ± 0.22 cm.s
-1 (x 10
-6) (
n = 6) for duodenum, 2.01 ± 0.80 cm.s
-1 (x 10
-6) (
n = 12) for jejunum, 1.64 ± 0.61 cm.s
-1 (x 10
-6) (
n = 9) for ileum and 1.73 ± 0.46 cm.s
-1 (x 10
-6) (
n = 6) for colon. Following PCN treatment, no significant changes in [
3H]-digoxin P
a-b were observed in any of the four sections (duodenum - 1.27 ± 0.51 cm.s
-1 (x 10
-6),
n = 5, jejunum - 1.74 ± 0.34 cm.s
-1 (x 10
-6),
n = 10, ileum - 1.08 ± 0.09 cm.s
-1 (x 10
-6),
n = 8, colon – 1.48 ± 0.42 cm.s
-1 (x 10
-6),
n = 5, P > 0.1). The lack of effect on P
a-b by the increased Pgp activity in colon and ileum mounted in Ussing chambers may indicate that
in vitro an additional pathway for absorption of [
3H]-digoxin is introduced by “edge effects” increasing “paracellular” diffusion above
in vivo levels so obscuring any reduction due to Pgp.
At the present time, there is little data on the regional expression of PXR in the intestine. PXR mRNA and protein expression is limited in proximal rat intestine relative to liver [
26]. Although PXR-mediated increases in Pgp expression in human and rat intestine have been reported [
12,
27], these studies do not address Pgp increases along the length of the gut. As Pgp expression is reported to increase aborally along the intestine [
9,
28,
29], Pgp-mediated digoxin efflux would be expected to be higher in the ileal and colonic segments. The present study has shown that for control rats there was only a limited increase in the magnitude of digoxin secretion (permeability) from duodenum to colon.
Direct measurements of absorptive digoxin permeability (P
a-b)
in vitro after PCN treatment in all intestinal segments do not show such a reduction in P
a-b, despite the increased net secretion of digoxin that is observed in ileum and colon. Experiments in rat brush-border intestinal vesicles have suggested that digoxin uptake is mediated by a proton-dependent carrier [
30] but an additional route
in vitro via a paracellular pathway in part arising from tissue damage may exist.
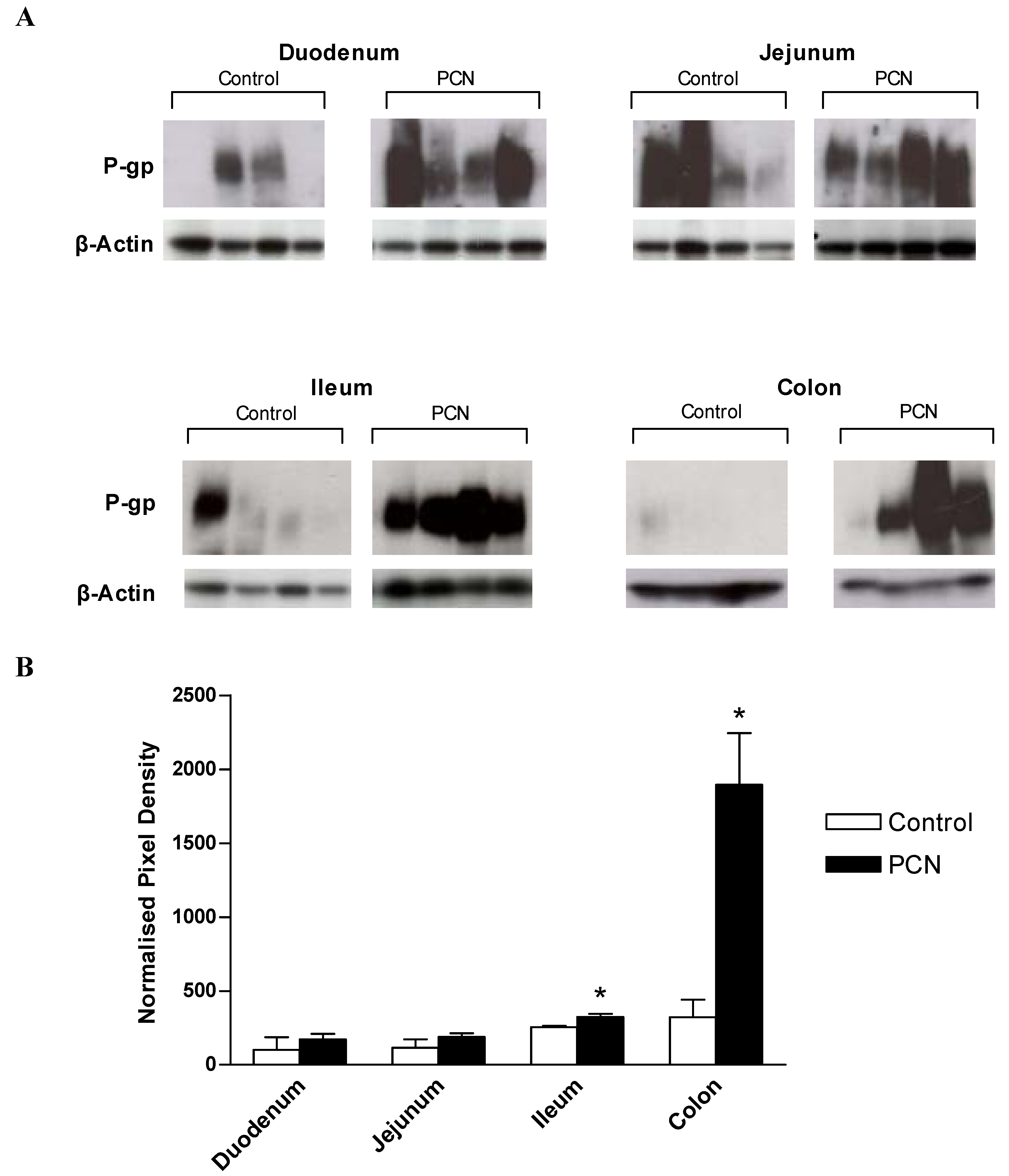
3.4. P-glycoprotein(Pgp) expression in rat intestinal tissue segments
Figure 5a shows the Western blot analysis for the expression of Pgp using the monoclonal antibody, C219. As Pgp expression levels were low in proximal intestine, chemiluminescent exposures of 5 minutes were required for duodenal and jejunal samples, whereas only a 10 second exposure was required to detect Pgp in PCN-treated ileum and colon samples. Prominent bands at approximately 170kD, representing the reported size for P-glycoprotein are evident for control samples; note that for ileal and colonic control samples, a 10 second exposure time is not sufficient to detect protein, however, longer exposure times would have led to saturation of the signal detected in the PCN-treated samples. In control samples of duodenum and jejunum there is marked variability between animals (
Figure 5a). C219-stained membranes were stripped and re-probed for the expression of the constitutively expressed house-keeping gene, β-actin. Densitometric analysis of digitised images allowed expression of Pgp to be normalised to β-actin expression (
Figure 5b). This data shows that whereas no significant change in the Pgp expression levels occurs in either duodenal or jejunal tissue despite the increase in mean values, both the ileum and colon display significant increases in Pgp protein levels following exposure to PCN (increases of 1.3-fold in the ileum and 2.9-fold in the colon; P < 0.05).
Western blot analysis for control rats is therefore similar to the permeability (secretion) data in that a marginal, but non-significant increase in Pgp protein expression is seen in the distal intestine. Since Pgp expression is dynamic and dependent on dietary and environmental factors, the lack of a marked aboral gradient in either protein expression or digoxin permeability may represent the strict diet of laboratory rats not exposed to a more challenging and variable “scavenger” diet.
Figure 5.
Western blot analysis of MDR1 and β-actin protein expression in excised sections of intestine from control and PCN-dosed rats (a) Samples of intestinal protein extracted from rat duodenum, jejunum, ileum (100 μg) and colon (50 μg) were separated by gel electrophoresis and probed for the presence of P-glycoprotein with the mouse-monoclonal antibody C219. Duodenal and jejunal blots were exposed for 5 minutes, ileal and colonic blots were exposed for 10 seconds. P-glycoprotein bands were visible at approximately 170 kDa. Blots were stripped and re-probed for the presence of β-actin with a cross-species polyclonal antibody (Abcam). β-actin bands were visible at approximately 37 kDa. (b) Densitometric analysis of P-glycoprotein blots indicating band pixel density relative to β-actin expression level. Data are mean ± SEM of n = 4 protein samples. * P < 0.05, significantly different from control values.
Figure 5.
Western blot analysis of MDR1 and β-actin protein expression in excised sections of intestine from control and PCN-dosed rats (a) Samples of intestinal protein extracted from rat duodenum, jejunum, ileum (100 μg) and colon (50 μg) were separated by gel electrophoresis and probed for the presence of P-glycoprotein with the mouse-monoclonal antibody C219. Duodenal and jejunal blots were exposed for 5 minutes, ileal and colonic blots were exposed for 10 seconds. P-glycoprotein bands were visible at approximately 170 kDa. Blots were stripped and re-probed for the presence of β-actin with a cross-species polyclonal antibody (Abcam). β-actin bands were visible at approximately 37 kDa. (b) Densitometric analysis of P-glycoprotein blots indicating band pixel density relative to β-actin expression level. Data are mean ± SEM of n = 4 protein samples. * P < 0.05, significantly different from control values.
Although there was a lack of variation in regional Pgp protein expression or function digoxin efflux in control animals, the present data now shows that Pgp induction potential increases aborally following 3 days PCN treatment. Net secretory digoxin transport is significantly increased in both ileal and colonic regions, with the greatest increase observed in the large intestine. This increase is matched to an increase in protein expression, with Western blot analysis indicating significant increases in Pgp protein relative to β-actin in both the ileum and colon, but with no change in the duodenum and jejunum. For the rat, secretory intestinal transport is likely limited to that mediated by Pgp [
2]. That Pgp is preferentially increased in the distal intestine also implies a functional linkage between PXR and Pgp expression restricted to the distal intestine. Transit time in the upper small intestine is rapid, which is in contrast to the colonic regions where transit time is prolonged to allow for microbial fermentation and faecal dehydration by water re-absorption [
31,
32]. The greater increase in Pgp expression may result from an increase in both exposure time and concentration of xenobiotics, in this case PCN. Likewise, due to the reduced transit rate and increased exposure to xenobiotics, it is possible that PXR expression may be higher in colon to allow a greater capacity for increasing the physiological barrier (Pgp and CYP3A4 expression).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




