
Application of Modelling and Simulation Approaches to Predict Pharmacokinetics of Therapeutic Monoclonal Antibodies in Pediatric Population

Abstract
:1. Introduction
2. Methodology
2.1. Comparison of Modelling and Simulation Methodologies Used in Pediatric mAb Development
2.2. Screening Age-Dependency of Physiological Parameters for Pediatric PBPK Model Development
2.3. Analysis of Pediatric PBPK Modelling Studies
3. Results and Discussion
3.1. Classical Modelling Approach for Pediatric Dosing Regimen

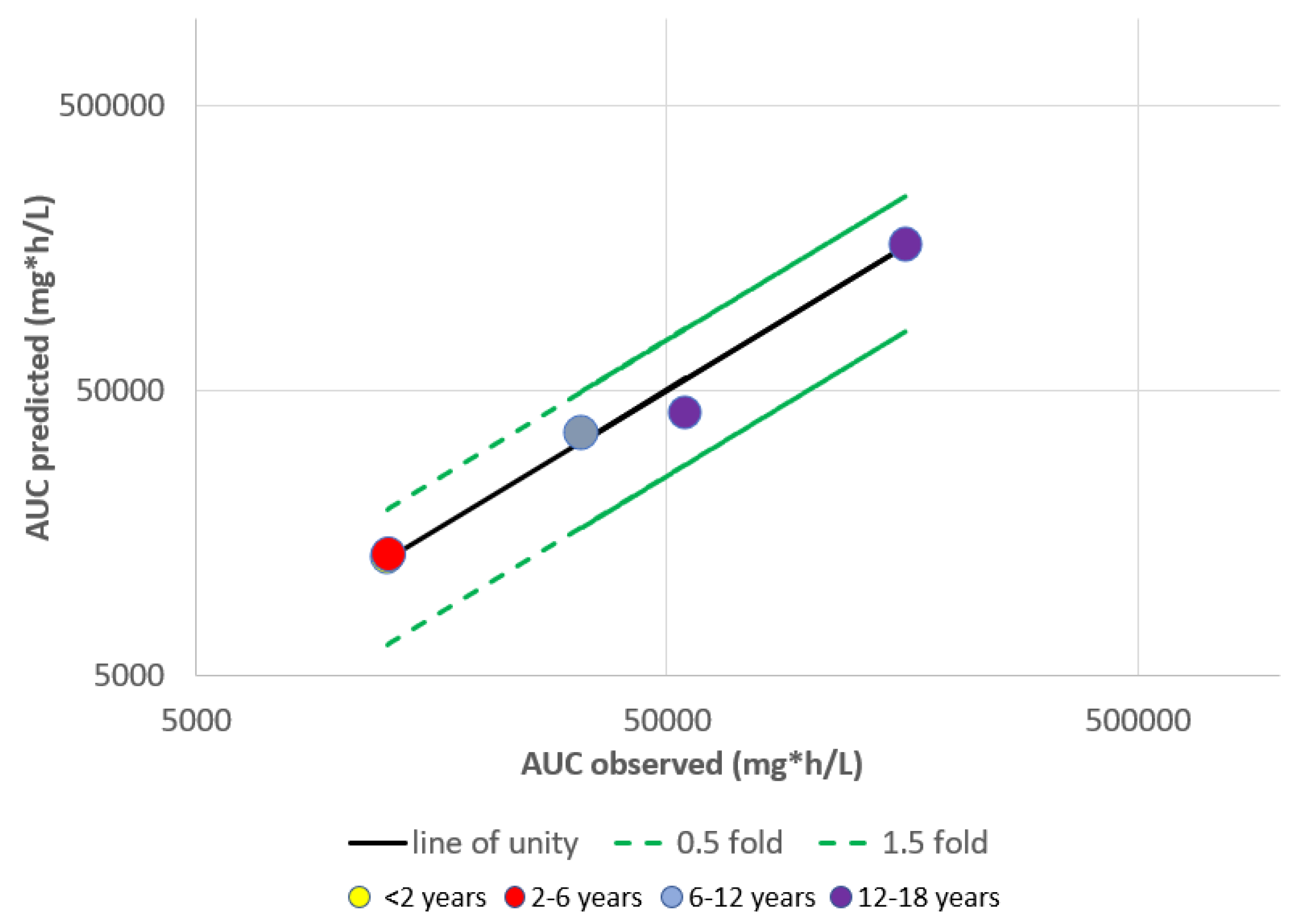{kind=link}
{kind=link}
| Methods | Allometric Scaling | Pop-PK | PBPK |
|---|---|---|---|
| Characteristics | Empirically derived function, predicting individual PK parameters (e.g., CL and V) based on demographic information (e.g., BW) (e.g., k = 0.75 for CL, 1.0 for V) [8] | Prediction based on retrospective analysis of pooled clinical data with the allometric and maturation function incorporated. Can predict within dose range studied or other doses and age range [31]. | Based on understanding complex physiological processes to mechanistically predict PK based on the interplay between drug-specific characteristics [35]. |
| Main applications | Extrapolate clinical PK information from adults to pediatric patients, typically combined with pop-PK to support the design of pediatric clinical studies [31] | Statistically driven analysis of pooled PK data from different clinical studies. Covariate analysis (age, gender, weight) can explain sources of PK variability. [31] | Leverage mechanistic mathematical models that recapitulate the physiology of humans, from neonates to adults, to assess the impact of ontogeny on mAb PK [36,37] |
| Strengths | Simple and quick with minimal resources required [8] | Analyse sparse data (typical for pediatric studies), and identify covariates that affect PK. Can integrate complex customised allometric and maturation functions [38]. | Can be used for predictions with limited clinical data. Accounts for the ontogeny of physiological processes in pediatrics, especially younger infants, and its impact on mAb PK [39,40]. |
| Gaps | Only captures body-size related information. Limited representation of complex physiological process such as TMDD or FcRn recycling. [31] Promising for mAbs with linear PK, which is affected by few well-understood parameters. However, less scientifically vigorous compared to pop-PK and PBPK [24]. | Knowledge about the appropriate allometric and maturation functions required. Predictions limited to scaling of selected parameters within the population and doses studied [38]. | Heavily reliant on biological understanding of ontogeny considering the physiological processes in pediatrics for initial model development. Reliability of data largely hinges on underlying ontogeny data [40,41]. |
3.2. Physiologically Based Pharmacokinetic Modelling—An Emerging Alternative?
3.3. Ontogeny of Key Physiological Processes in Pediatric Monoclonal Antibody Disposition for Exploration in PBPK Studies
| Attributes | Small Molecules | mAbs |
|---|---|---|
| Molecular weight | <500 Da | 150 kDa |
| Target | Intracellular and surface targets | Membrane proteins or soluble proteins in circulation |
| Route of administration | Oral, intravascular (IV), subcutaneous (SC), intramuscular (IM) | Parenteral (intravascular (IV), subcutaneous (SC), intramuscular (IM)) |
| Posology | Short-acting: often dosed daily or multiple times a day | Long-acting with dosing intervals up to months |
| Absorption | Through passive diffusion and active transporters. Usually rapid after oral administration. | Mainly through lymphatic uptake due to their large molecular size. Slow after subcutaneous administration. |
| Distribution (Vd) | Volume of distribution high (0.1 to 1000 L/kg) | Volume of distribution is limited. Typically limited to plasma or interstitial fluid. |
| Metabolism/ | Typically eliminated by CYP, UGT, transporters, renal and biliary pathways. | Intracellular catabolism by lysosomal degradation after endocytosis |
| Elimination | Mainly via biliary and renal excretion | Mainly via target-mediated drug disposition (TMDD) but can be recycled via FcRn |
| Half-life (t1/2) | Short (<24 h) | Long (days or weeks) |
| Clearance (CL) | Mostly linear PK; non-linearity mainly due to the saturation of metabolic pathways | Non-linear clearance is observed at low dose levels due to TMDD; linear clearance observed at above saturable dose range |
| Immunogenicity | Typically not observed | Likely generation of antidrug antibodies (ADA) due to immunogenicity. ADA can form an immune complex with mAb, which can accelerate overall mAb clearance. |
| Drug–drug interaction | Expected and need to be investigated for CYP P450 and transporter interactions | Rarely observed with some exceptions (e.g., mAbs modulating cytokine pathway may interact with CYP3A4-mediated clearance of small molecule drugs) |
| Special population | Physiological parameters (e.g. body composition, organ size, metabolic enzyme and transporter activity, plasma protein levels) affecting ADME may differ based on demographic characteristics (age, sex, ethnicity) and on comorbidities (eg hepatic or renal impairment) | Age-dependent changes in Fc receptor for the paediatric population and pregnant population |
| Key Physiological Parameters | Age Group (Years) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Birth | 1 month | 2 months | 3 months | 6 months | 12 months | 18 months | <2 | 2–6 | 6–12 | 12–18 | Adults (>18) | Refs | |||||
| Extracellular fluid (ECF) volume % | 45 | 40 | 32 | 30 | 29 | 26 | 23 | 20 | 19 | 18 | 18 | 18 | [6] | ||||
| Plasma volume (mL/kg) | 40 | 45 | 45 | 50 | 50 | 50 | 55 | 55 | 50 | 46 | 43 | 43 | [5] | ||||
| Capillary density (capillaries/mm2) | 223 | 89.04 | 74.94 | 33.5 | 89.04 | 106.7 (18–40 years old) 171 (40–65 years old) | [41] | ||||||||||
| Leaky:Tight tissue mass ratio | 0.129 | 0.115 | 0.118 | 0.116 | 0.102 | 0.098 | [58,59] | ||||||||||
| Endogeneous IgG concentrations (μM) | 69.26 | 35.16 | 20.21 | 25.05 | 27.79 | 40.21 | 41.68 | 60.53 | 77 | 76.51 | [41] | ||||||
| Lymph Flow | Lymph flow data have not been quantified in pediatrics. However, research in the literature supports that the number of lymph nodes is less in pediatrics; thus, lymph flow is scaled allometrically with an exponent of 0.75 using 3.855 mL/h/kg as a reference value in adults. | [41] | |||||||||||||||
| FcRn p51 abundance * (pmol/mg protein) | 3.36 (3.07) * | 3.11 | 1.70 | 1.72 | 2.25 | [60] | |||||||||||
| FcRn B2M abundance * (pmol/mg protein) | 58.9 (40.722) * | 50.2 | 41.3 | 42.1 | 27.7 | [60] | |||||||||||
3.4. Perspectives on Existing Pediatric PBPK Models for Monoclonal Antibodies
4. Future Directions
5. PBPK Potential Role in Regulatory Submissions for Monoclonal Antibodies
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADA | Antidrug antibodies |
| AUC | Area Under Curve |
| BLA | Biologics license applications |
| BSA | Body surface area |
| BW | Body weight |
| CL | Clearance |
| Cmax | Peak (maximum) plasma concentration in plasma concentration curve |
| ECF | Extracellular fluid |
| EMA | European medicine agency |
| FcRn | Neonatal Fc receptor |
| IgG | Immunoglobulin G |
| IM | Intramuscular |
| IV | Intravascular |
| IVIVE | In vitro-in vivo extrapolation |
| mAbs | Monoclonal antibodies |
| PBPK | Physiologically based pharmacokinetic modelling |
| PSP | Paediatric study plan |
| popPK | population pharmacokinetics |
| PK | Pharmacokinetic |
| PREA | Paediatric research equity act |
| SC | Subcutaneous |
| t1/2 | Half life |
| TMDD | Target-mediated drug disposition |
| US FDA | U.S. Food and Drug Administration |
| V or Vd | Volume of distribution |
References
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Bamlanivimab EUA Letter of Authorization. Available online: https://www.fda.gov/media/143602/download (accessed on 30 November 2020).
- Food and Drug Administration. Casirivimab and Imdevimab EUA Letter of Authorization. Available online: https://www.fda.gov/media/143891/download (accessed on 30 November 2020).
- Hoppu, K.; Anabwani, G.; Garcia-Bournissen, F.; Gazarian, M.; Kearns, G.L.; Nakamura, H.; Peterson, R.G.; Sri Ranganathan, S.; de Wildt, S.N. The status of paediatric medicines initiatives around the world––What has happened and what has not? Eur. J. Clin. Pharmacol. 2012, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Edginton, A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin. Drug Metab. Toxicol. 2018, 14, 585–599. [Google Scholar] [CrossRef]
- Friis-Hansen, B. Body water compartments in children: Changes during growth and related changes in body composition. Pediatrics 1961, 28, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Mulberg, A.E.; Murphy, M.D.; Dunne, J.; Mathis, L.L.; ProQuest. Pediatric Drug Development: Concepts and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Germovsek, E.; Cheng, M.; Giragossian, C. Allometric scaling of therapeutic monoclonal antibodies in preclinical and clinical settings. MAbs 2021, 13, 1964935. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.R.; Kern, S.E. The need for modeling and simulation to design clinical investigations in children. J. Clin. Pharmacol. 2010, 50 (Suppl. 9), 121S–129S. [Google Scholar] [CrossRef] [PubMed]
- Manolis, E.; Osman, T.E.; Herold, R.; Koenig, F.; Tomasi, P.; Vamvakas, S.; Saint Raymond, A. Role of modeling and simulation in pediatric investigation plans. Paediatr. Anaesth. 2011, 21, 214–221. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration (USFDA). General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products Guidance for Industry. Available online: https://www.fda.gov/media/90358/download (accessed on 30 November 2022).
- European Medicines Agency (EMA). Role of Pharmacokinetics in the Development of Medicinal Products in the Pediatric Population. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-role-pharmacokinetics-developmentmedicinal-products-paediatric-population_en.pdf (accessed on 25 September 2019).
- Mulugeta, Y.; Barrett, J.S.; Nelson, R.; Eshete, A.T.; Mushtaq, A.; Yao, L.; Glasgow, N.; Mulberg, A.E.; Gonzalez, D.; Green, D.; et al. Exposure Matching for Extrapolation of Efficacy in Pediatric Drug Development. J. Clin. Pharmacol. 2016, 56, 1326–1334. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Initial Pediatric Study Plans. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pediatric-study-plans-content-and-process-submitting-initial-pediatric-study-plans-and-amended (accessed on 5 January 2023).
- Ince, I.; Solodenko, J.; Frechen, S.; Dallmann, A.; Niederalt, C.; Schlender, J.; Burghaus, R.; Lippert, J.; Willmann, S. Predictive Pediatric Modeling and Simulation Using Ontogeny Information. J. Clin. Pharmacol. 2019, 59 (Suppl. 1), S95–S103. [Google Scholar] [CrossRef]
- Wu, Q.; Peters, S.A. A Retrospective Evaluation of Allometry, Population Pharmacokinetics, and Physiologically-Based Pharmacokinetics for Pediatric Dosing Using Clearance as a Surrogate. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 220–229. [Google Scholar] [CrossRef]
- Liu, T.G.P.; Gobburu, J.V. Allometry is a reasonable choice in pediatric drug development. J. Clin. Pharmacol. 2017, 57, 469–475. [Google Scholar] [CrossRef]
- Germovsek, E.; Barker, C.I.S.; Sharland, M.; Standing, J.F. Pharmacokinetic-Pharmacodynamic Modeling in Pediatric Drug Development, and the Importance of Standardized Scaling of Clearance. Clin. Pharmacol. 2019, 58, 39–52. [Google Scholar] [CrossRef]
- Xu, Y.; Langevin, B.A.; Zhou, H.; Xu, Z. Model-Aided Adults-to-Children Pharmacokinetic Extrapolation and Empirical Body Size-Based Dosing Exploration for Therapeutic Monoclonal Antibodies-Is Allometry a Reasonable Choice? J. Clin. Pharmacol. 2020, 60, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Xu, M.C.; Yao, Z. Evaluation of Weight Thresholds for Pediatric Patients to Use Adult Dosage of Therapeutic Monoclonal Antibodies. J. Clin. Pharmacol. 2019, 59, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Peyret, T.; Quartino, A.; Gosselin, N.H.; Gururangan, S.; Casanova, M.; Merks, J.H.; Massimino, M.; Grill, J.; Daw, N.C.; et al. Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Br. J. Clin. Pharmacol. 2016, 81, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, X.; Bajaj, G.; Barrett, J.S.; Meibohm, B.; Joshi, A.; Gupta, M. Challenges and considerations for development of therapeutic proteins in pediatric patients. J. Clin. Pharmacol. 2015, 55 (Suppl. 3), S103–S115. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Davis, H.M.; Zhou, H. Rational development and utilization of antibody-based therapeutic proteins in pediatrics. Pharmacol. Ther. 2013, 137, 225–247. [Google Scholar] [CrossRef]
- Malik, P.R.V.; Edginton, A.N. Physiologically-Based Pharmacokinetic Modeling vs. Allometric Scaling for the Prediction of Infliximab Pharmacokinetics in Pediatric Patients. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 835–844. [Google Scholar] [CrossRef]
- Davda, J.P.; Dodds, M.G.; Gibbs, M.A.; Wisdom, W.; Gibbs, J. A model-based meta-analysis of monoclonal antibody pharmacokinetics to guide optimal first-in-human study design. MAbs 2014, 6, 1094–1102. [Google Scholar] [CrossRef]
- Vinks, A.A.; Emoto, C.; Fukuda, T. Modeling and simulation in pediatric drug therapy: Application of pharmacometrics to define the right dose for children. Clin. Pharmacol. Ther. 2015, 98, 298–308. [Google Scholar] [CrossRef]
- Anderson, B.J.; Allegaert, K.; Holford, N.H. Population clinical pharmacology of children: General principles. Eur. J. Pediatr. 2006, 165, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Krekels, E.H.; Neely, M.; Panoilia, E.; Tibboel, D.; Capparelli, E.; Danhof, M.; Mirochnick, M.; Knibbe, C.A. From pediatric covariate model to semiphysiological function for maturation: Part I-extrapolation of a covariate model from morphine to Zidovudine. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, e9. [Google Scholar] [CrossRef] [PubMed]
- De Cock, R.F.; Allegaert, K.; Sherwin, C.M.; Nielsen, E.I.; de Hoog, M.; van den Anker, J.N.; Danhof, M.; Knibbe, C.A. A neonatal amikacin covariate model can be used to predict ontogeny of other drugs eliminated through glomerular filtration in neonates. Pharm. Res. 2014, 31, 754–767. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e63. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Ke, A.B. Physiologically Based Pharmacokinetic Modeling and Allometric Scaling in Pediatric Drug Development: Where Do We Draw the Line? J. Clin. Pharmacol. 2021, 61 (Suppl. 1), S83–S93. [Google Scholar] [CrossRef] [PubMed]
- Edginton, A.N.; Schmitt, W.; Willmann, S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin. Pharmacol. 2006, 45, 1013–1034. [Google Scholar] [CrossRef] [PubMed]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental pharmacology--drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [CrossRef]
- Koren, G. Therapeutic drug monitoring principles in the neonate. National Academy of CLinical Biochemistry. Clin. Chem. 1997, 43, 222–227. [Google Scholar] [CrossRef]
- Templeton, I.E.; Jones, N.S.; Musib, L. Pediatric Dose Selection and Utility of PBPK in Determining Dose. AAPS J. 2018, 20, 31. [Google Scholar] [CrossRef]
- Wagner, C.; Zhao, P.; Pan, Y.; Hsu, V.; Grillo, J.; Huang, S.M.; Sinha, V. Application of Physiologically Based Pharmacokinetic (PBPK) Modeling to Support Dose Selection: Report of an FDA Public Workshop on PBPK. CPT Pharmacomet. Syst. Pharm. 2015, 4, 226–230. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Edginton, A.N. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacomet. Syst. Pharm. 2014, 3, e150. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, N.; Bhattaram, A.; Earp, J.C.; Florian, J.; Krudys, K.; Lee, J.E.; Lee, J.Y.; Liu, J.; Mulugeta, Y.; Yu, J.; et al. Role of Quantitative Clinical Pharmacology in Pediatric Approval and Labeling. Drug Metab. Dispos. 2016, 44, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Willmann, S.; Frei, M.; Sutter, G.; Coboeken, K.; Wendl, T.; Eissing, T.; Lippert, J.; Stass, H. Application of Physiologically-Based and Population Pharmacokinetic Modeling for Dose Finding and Confirmation During the Pediatric Development of Moxifloxacin. CPT Pharmacomet. Syst. Pharm. 2019, 8, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.L.; Jones, H.M. Opportunities and Challenges for PBPK Model of mAbs in Paediatrics and Pregnancy. AAPS J. 2022, 24, 72. [Google Scholar] [CrossRef]
- Pan, X.; Stader, F.; Abduljalil, K.; Gill, K.L.; Johnson, T.N.; Gardner, I.; Jamei, M. Development and Application of a Physiologically-Based Pharmacokinetic Model to Predict the Pharmacokinetics of Therapeutic Proteins from Full-term Neonates to Adolescents. AAPS J. 2020, 22, 76. [Google Scholar] [CrossRef]
- Bellanti, F.; Della Pasqua, O. Modelling and simulation as research tools in paediatric drug development. Eur. J. Clin. Pharmacol. 2011, 67 (Suppl. 1), 75–86. [Google Scholar] [CrossRef]
- Leong, R.; Vieira, M.L.; Zhao, P.; Mulugeta, Y.; Lee, C.S.; Huang, S.M.; Burckart, G.J. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 2012, 91, 926–931. [Google Scholar] [CrossRef]
- Heimbach, T.; Lin, W.; Hourcade-Potelleret, F.; Tian, X.; Combes, F.P.; Horvath, N.; He, H. Physiologically Based Pharmacokinetic Modeling to Supplement Nilotinib Pharmacokinetics and Confirm Dose Selection in Pediatric Patients. J. Pharm. Sci. 2019, 108, 2191–2198. [Google Scholar] [CrossRef]
- Temrikar, Z.H.; Suryawanshi, S.; Meibohm, B. Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr. Drugs 2020, 22, 199–216. [Google Scholar] [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef]
- Rossing, N.P.H.; Lassen, N.A. Albumin transcapillary escape rate as an approach to microvascular physiology in health and disease. In Plasma Protein Turnover; Springer: Berlin/Heidelberg, Germany, 1976; pp. 357–370. [Google Scholar]
- Robbie, G.J.; Zhao, L.; Mondick, J.; Losonsky, G.; Roskos, L.K. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob. Agents Chemother. 2012, 56, 4927–4936. [Google Scholar] [CrossRef] [PubMed]
- Ternant, D.; Paintaud, G.; Trachtman, H.; Gipson, D.S.; Joy, M.S. A possible influence of age on absorption and elimination of adalimumab in focal segmental glomerulosclerosis (FSGS). Eur. J. Clin. Pharmacol. 2016, 72, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Lerner, G.; Kale, A.S.; Warady, B.A.; Jabs, K.; Bunchman, T.E.; Heatherington, A.; Olson, K.; Messer-Mann, L.; Maroni, B.J. Pharmacokinetics of darbepoetin alfa in pediatric patients with chronic kidney disease. Pediatr. Nephrol. 2002, 17, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ji, P.; Li, Z.; Roy, P.; Sahajwalla, C.G. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J. Clin. Pharmacol. 2013, 53, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.; Kennedy, P.J.; Santos, H.A.; Barrias, C.; Sarmento, B. A comprehensive review of the neonatal Fc receptor and its application in drug delivery. Pharmacol. Ther. 2016, 161, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Cavagnaro, J.A. Preclinical safety evaluation of biotechnology-derived pharmaceuticals. Nat. Rev. Drug. Discov. 2002, 1, 469–475. [Google Scholar] [CrossRef]
- Han, T.H.; Zhao, B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab. Dispos. 2014, 42, 1914–1920. [Google Scholar] [CrossRef]
- Shi, S. Biologics: An update and challenge of their pharmacokinetics. Curr. Drug. Metab. 2014, 15, 271–290. [Google Scholar] [CrossRef]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharm. 2017, 6, 576–588. [Google Scholar] [CrossRef]
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Basic anatomical and physiological data for use in radiological protection: Reference values. A report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. Ann. ICRP 2002, 32, 5–265.
- Barber, J.; Al-Majdoub, Z.M.; Couto, N.; Howard, M.; Elmorsi, Y.; Scotcher, D.; Alizai, N.; de Wildt, S.; Stader, F.; Sepp, A.; et al. Toward Systems-Informed Models for Biologics Disposition: Covariates of the Abundance of the Neonatal Fc Receptor (FcRn) in Human Tissues and Implications for Pharmacokinetic Modelling. Eur. J. Pharm. Sci. 2023, 182, 106375. [Google Scholar] [CrossRef]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef]
- Pyzik, M.; Rath, T.; Lencer, W.I.; Baker, K.; Blumberg, R.S. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J. Immunol. 2015, 194, 4595–4603. [Google Scholar] [CrossRef]
- McCarthy, K.M.; Yoong, Y.; Simister, N.E. Bidirectional transcytosis of IgG by the rat neonatal Fc receptor expressed in a rat kidney cell line: A system to study protein transport across epithelia. J. Cell Sci. 2000, 113 Pt 7, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, T.S.; Detmer, S.A.; Martin, W.L.; Bjorkman, P.J. IgG transcytosis and recycling by FcRn expressed in MDCK cells reveals ligand-induced redistribution. EMBO J. 2002, 21, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Hardiansyah, D.; Ng, C.M. Effects of the FcRn developmental pharmacology on the pharmacokinetics of therapeutic monoclonal IgG antibody in pediatric subjects using minimal physiologically-based pharmacokinetic modelling. MAbs 2018, 10, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P. Report from the EMA workshop on qualification and reporting of physiologically based pharmacokinetic (PBPK) modeling and simulation. CPT Pharmacomet. Syst. Pharm. 2017, 6, 71–72. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the Qualification and Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-qualification-reporting-physiologically-based-pharmacokinetic-pbpk-modelling_en.pdf (accessed on 30 November 2022).
- Liu, X.I.; Dallmann, A.; Wang, Y.M.; Green, D.J.; Burnham, J.M.; Chiang, B.; Wu, P.; Sheng, M.; Lu, K.; van den Anker, J.N.; et al. Monoclonal Antibodies and Fc-Fusion Proteins for Pediatric Use: Dosing, Immunogenicity, and Modeling and Simulation in Data Submitted to the US Food and Drug Administration. J. Clin. Pharmacol. 2019, 59, 1130–1143. [Google Scholar] [CrossRef]
- Gill, K.L.; Machavaram, K.K.; Rose, R.H.; Chetty, M. Potential Sources of Inter-Subject Variability in Monoclonal Antibody Pharmacokinetics. Clin. Pharmacokinet. 2016, 55, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Avery, L.B.; Wade, J.; Wang, M.; Tam, A.; King, A.; Piche-Nicholas, N.; Kavosi, M.S.; Penn, S.; Cirelli, D.; Kurz, J.C.; et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs 2018, 10, 244–255. [Google Scholar] [CrossRef]
- Chung, S.; Nguyen, V.; Lin, Y.L.; Lafrance-Vanasse, J.; Scales, S.J.; Lin, K.; Deng, R.; Williams, K.; Sperinde, G.; Li, J.J.; et al. An in vitro FcRn- dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. MAbs 2019, 11, 942–955. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, Y.; Jusko, W.J. Across-Species Scaling of Monoclonal Antibody Pharmacokinetics Using a Minimal PBPK Model. Pharm. Res. 2015, 32, 3269–3281. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Iyer, S.; Fielder, P.J.; Davis, J.D.; Deng, R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: Comparative evaluation of prediction approaches in early drug development. Biopharm. Drug. Dispos. 2016, 37, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; McNeill, J.H. To scale or not to scale: The principles of dose extrapolation. Br. J. Pharmacol. 2009, 157, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Q.; Salinger, D.H.; Endres, C.J.; Gibbs, J.P.; Hsu, C.P.; Stouch, B.J.; Hurh, E.; Gibbs, M.A. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: Retrospective analysis of monkey as a single species for first-in-human prediction. Clin. Pharmacol. 2011, 50, 131–142. [Google Scholar] [CrossRef]
- Malik, P.R.V.; Temrikar, Z.H.; Chelle, P.; Edginton, A.N.; Meibohm, B. Pediatric Dose Selection for Therapeutic Proteins. J. Clin. Pharmacol. 2021, 61 (Suppl. 1), S193–S206. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, A.; Sharma, P.; Stepanov, O.; Reddy, V.P. Application of Modelling and Simulation Approaches to Predict Pharmacokinetics of Therapeutic Monoclonal Antibodies in Pediatric Population. Pharmaceutics 2023, 15, 1552. https://doi.org/10.3390/pharmaceutics15051552
Lim A, Sharma P, Stepanov O, Reddy VP. Application of Modelling and Simulation Approaches to Predict Pharmacokinetics of Therapeutic Monoclonal Antibodies in Pediatric Population. Pharmaceutics. 2023; 15(5):1552. https://doi.org/10.3390/pharmaceutics15051552
Chicago/Turabian StyleLim, Andrew, Pradeep Sharma, Oleg Stepanov, and Venkatesh Pilla Reddy. 2023. "Application of Modelling and Simulation Approaches to Predict Pharmacokinetics of Therapeutic Monoclonal Antibodies in Pediatric Population" Pharmaceutics 15, no. 5: 1552. https://doi.org/10.3390/pharmaceutics15051552