
Application of Sucrose Acetate Isobutyrate in Development of Co-Amorphous Formulations of Tacrolimus for Bioavailability Enhancement

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
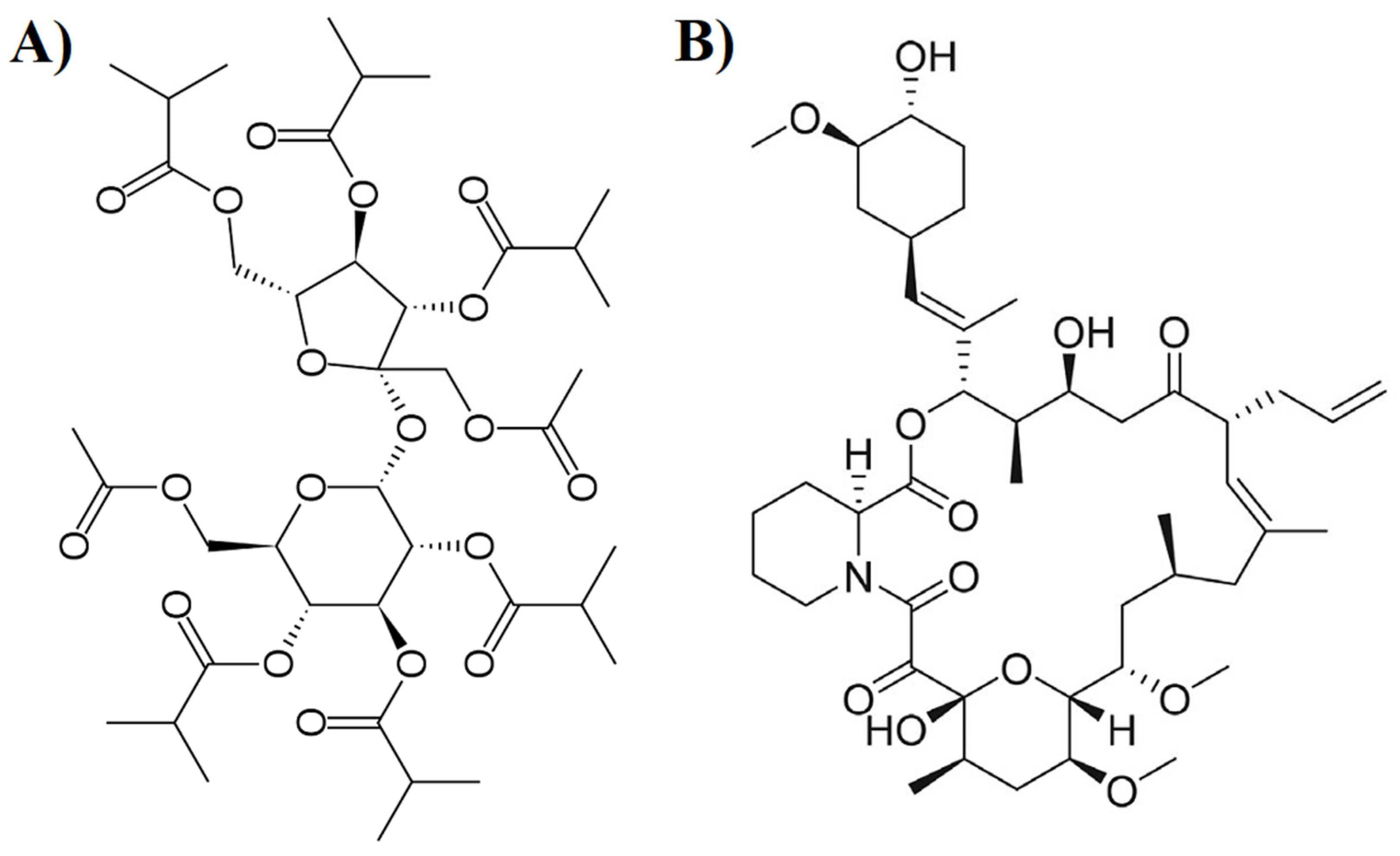
2.1. Materials
2.2. Preparation of CAD and ASD
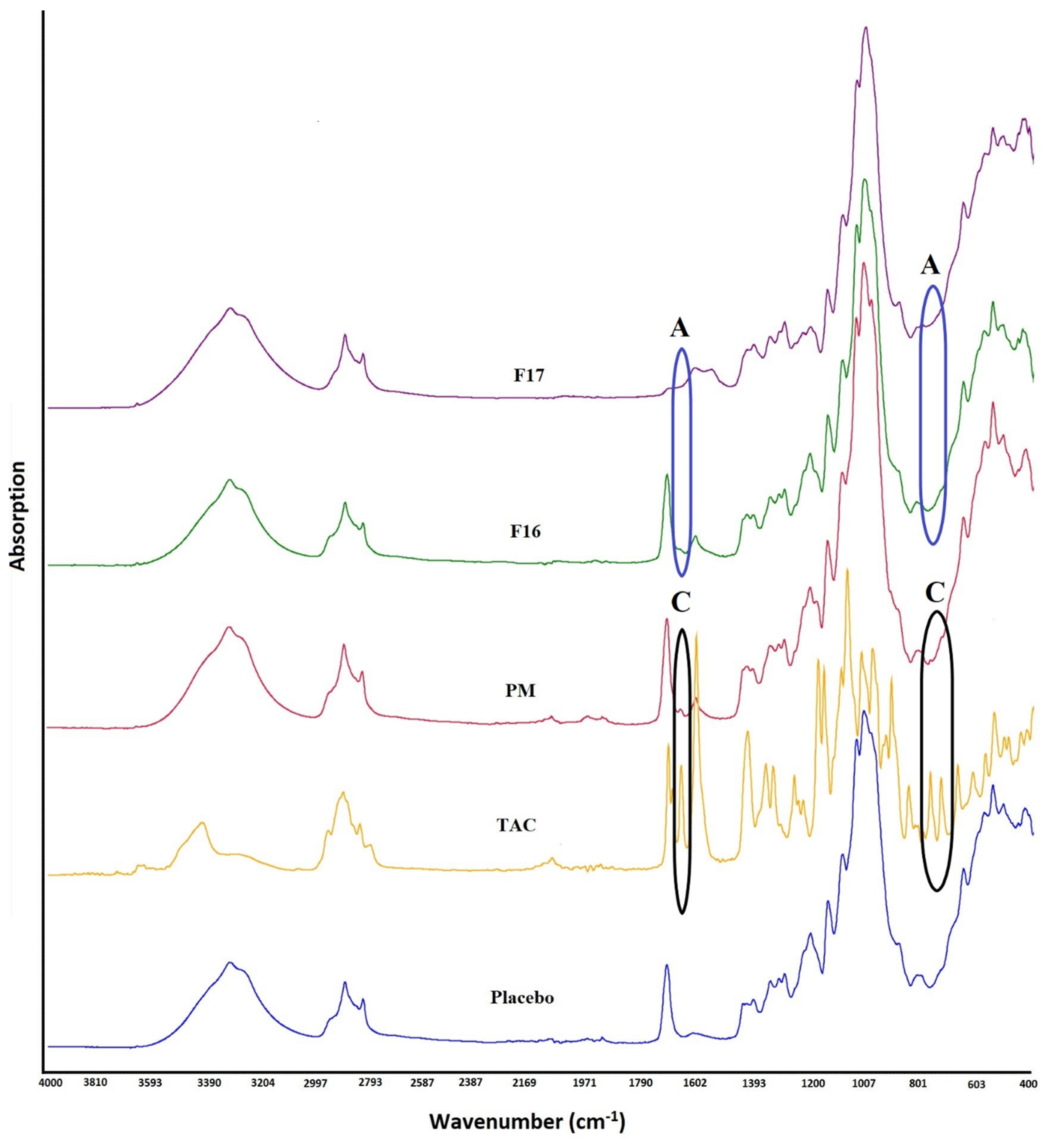
2.3. Fourier Transform Infrared Spectroscopy
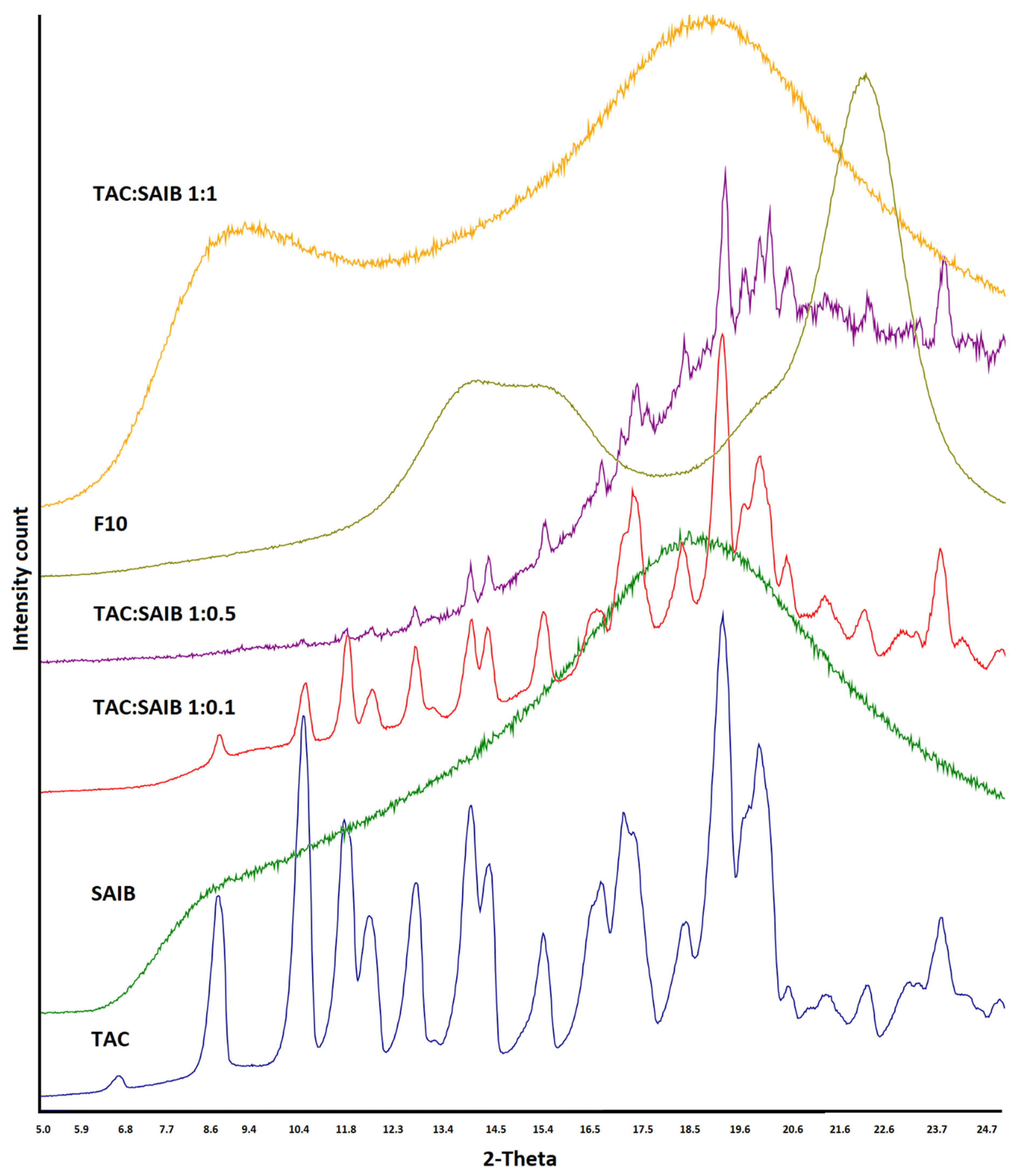
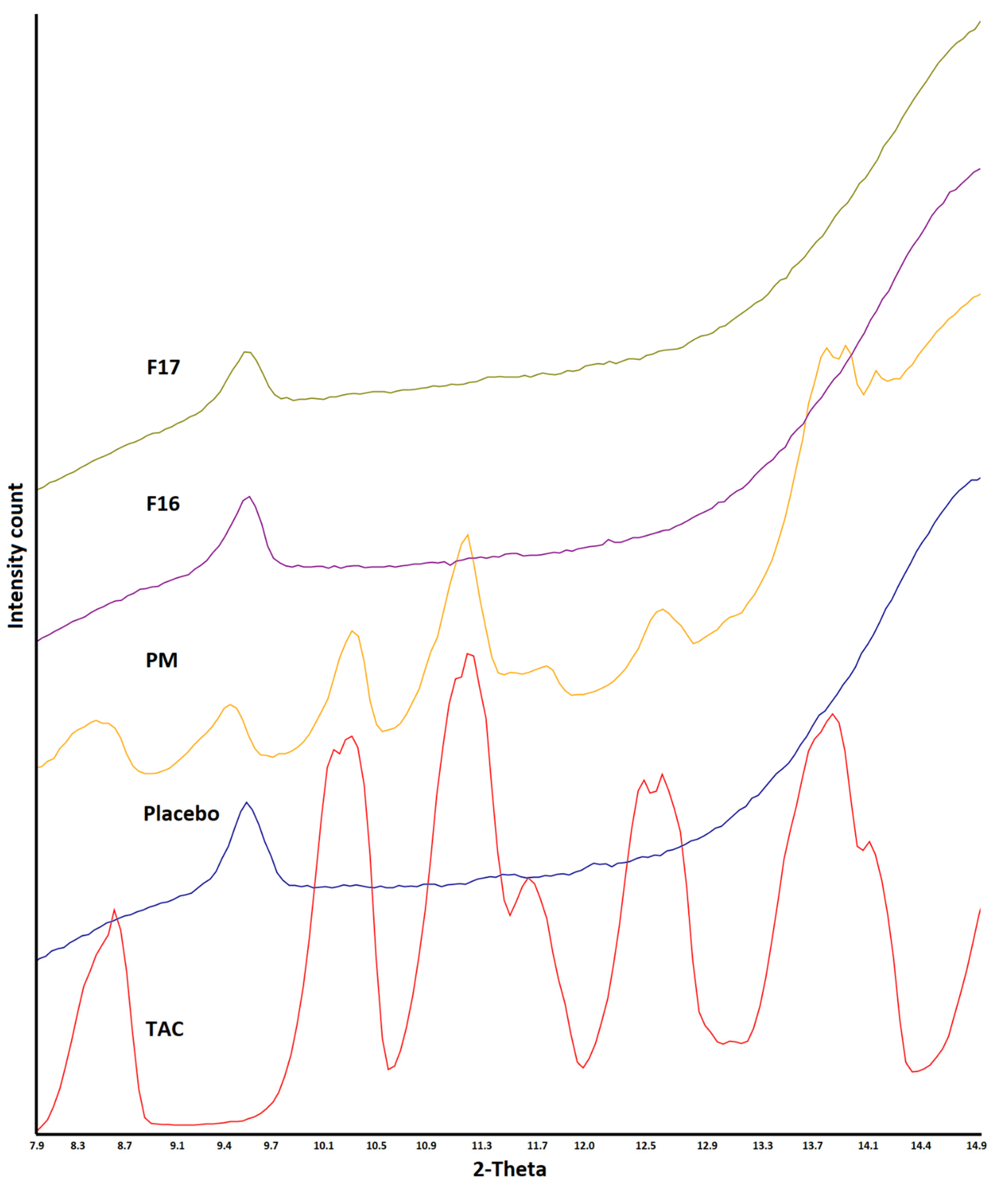
2.4. X-ray Powder Diffractometry
2.5. Differential Scanning Calorimetry
2.6. Dissolution
2.7. Stability
2.8. Pharmacokinetics
2.9. High-Performance Liquid Chromatography
2.10. Ultra-Performance Liquid Chromatography-Mass Spectroscopy
3. Results and Discussion
3.1. Fourier Transformed Infrared Spectroscopy
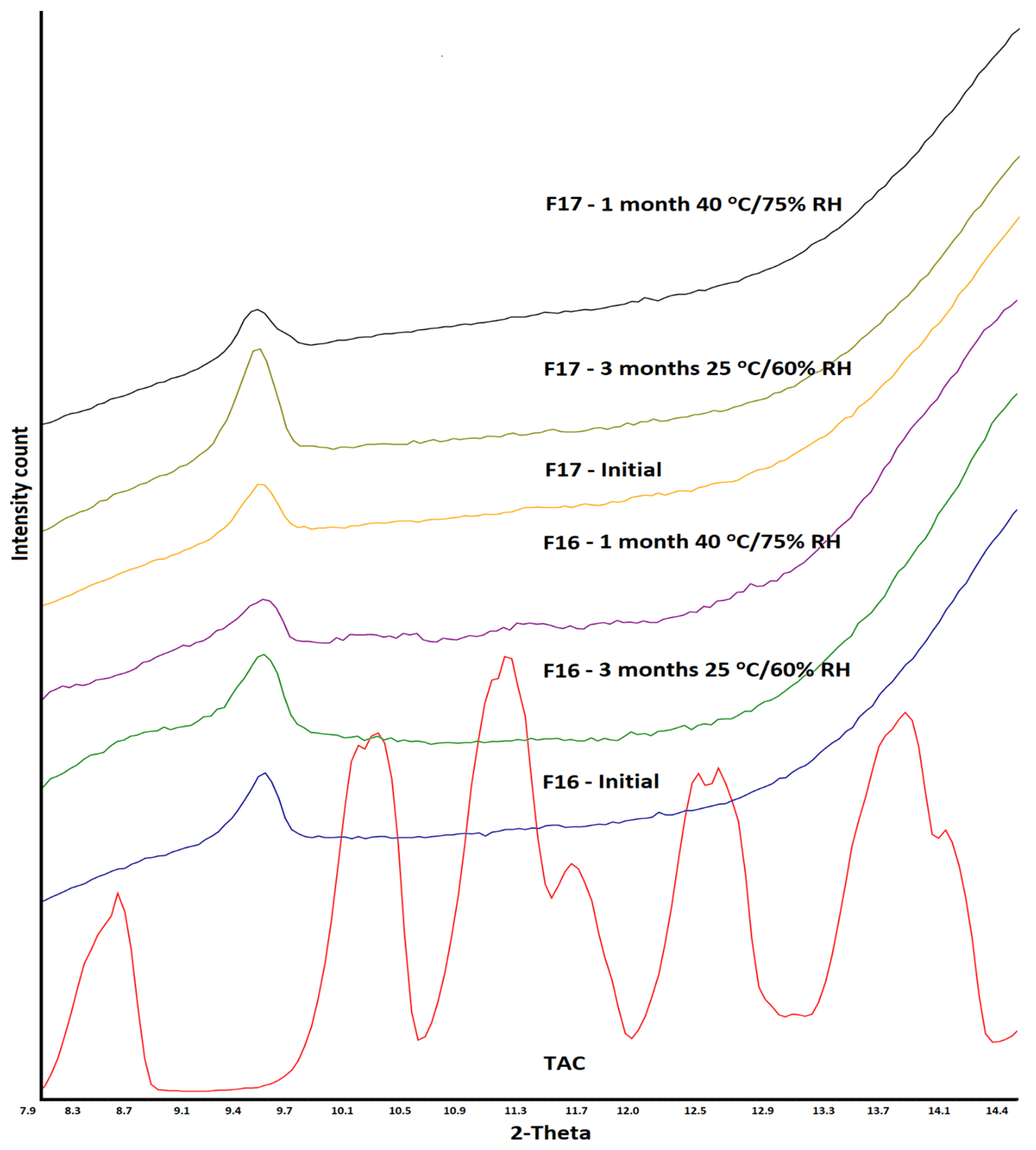
3.2. X-ray Powder Diffraction
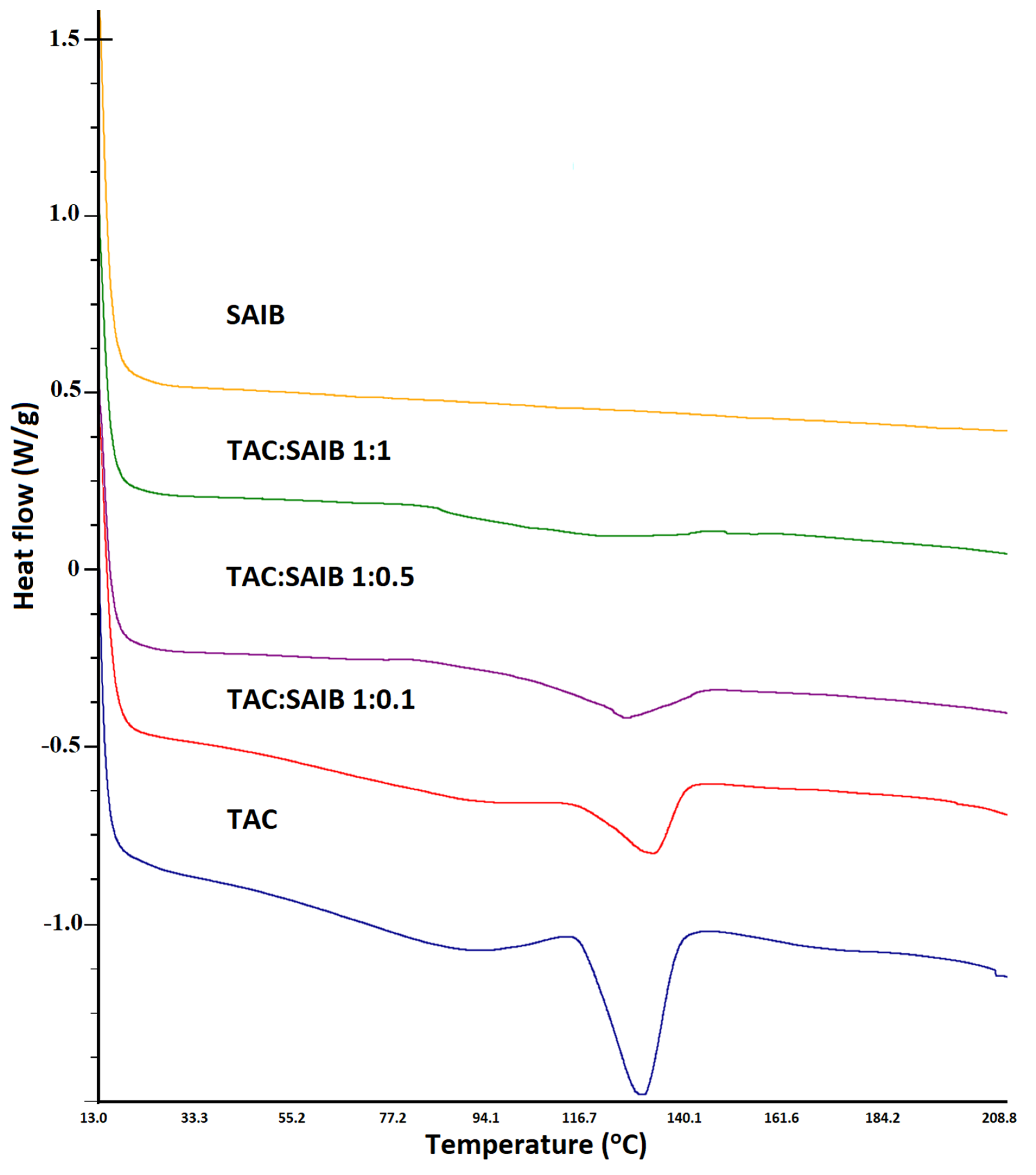
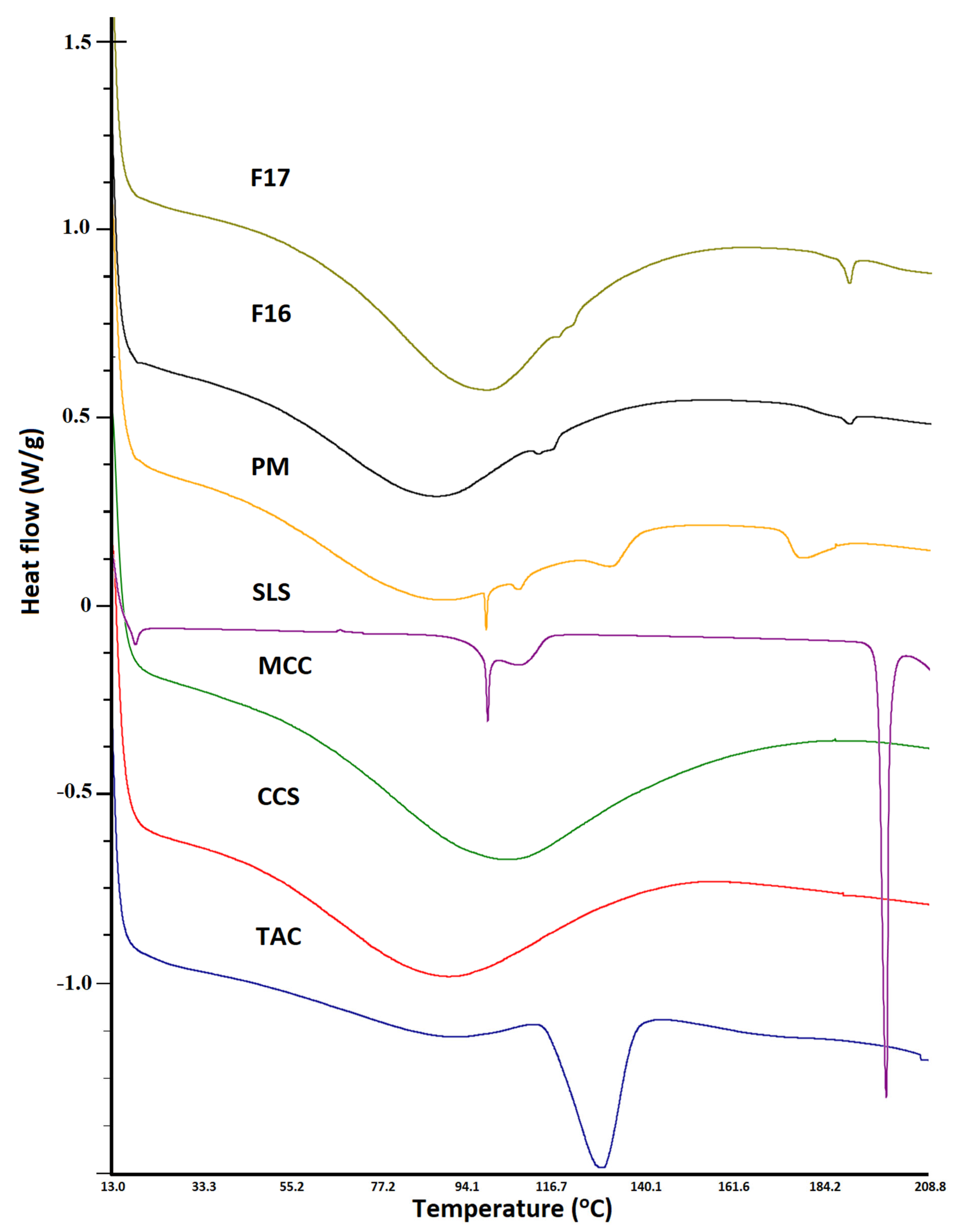
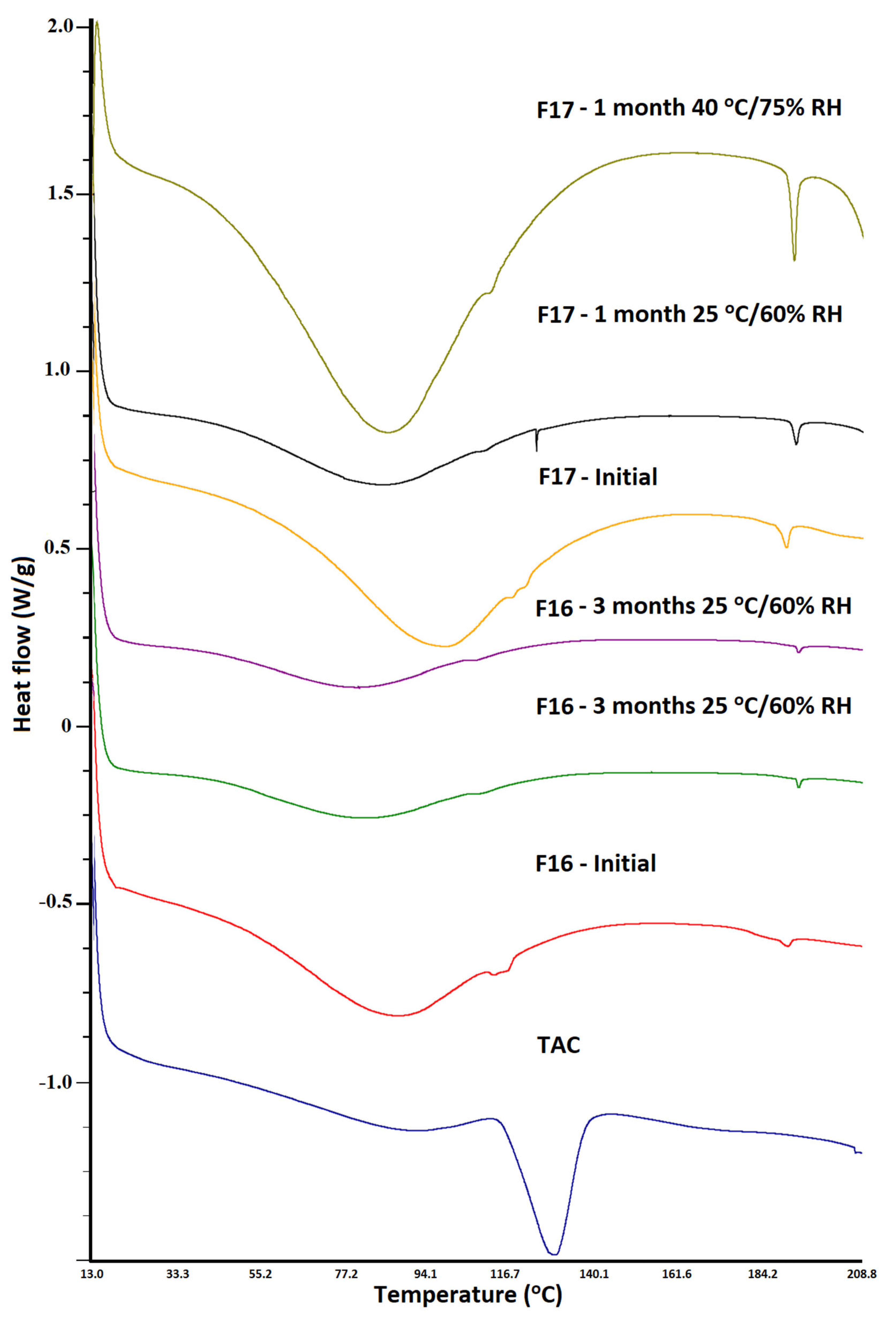
3.3. Differential Scanning Calorimetry
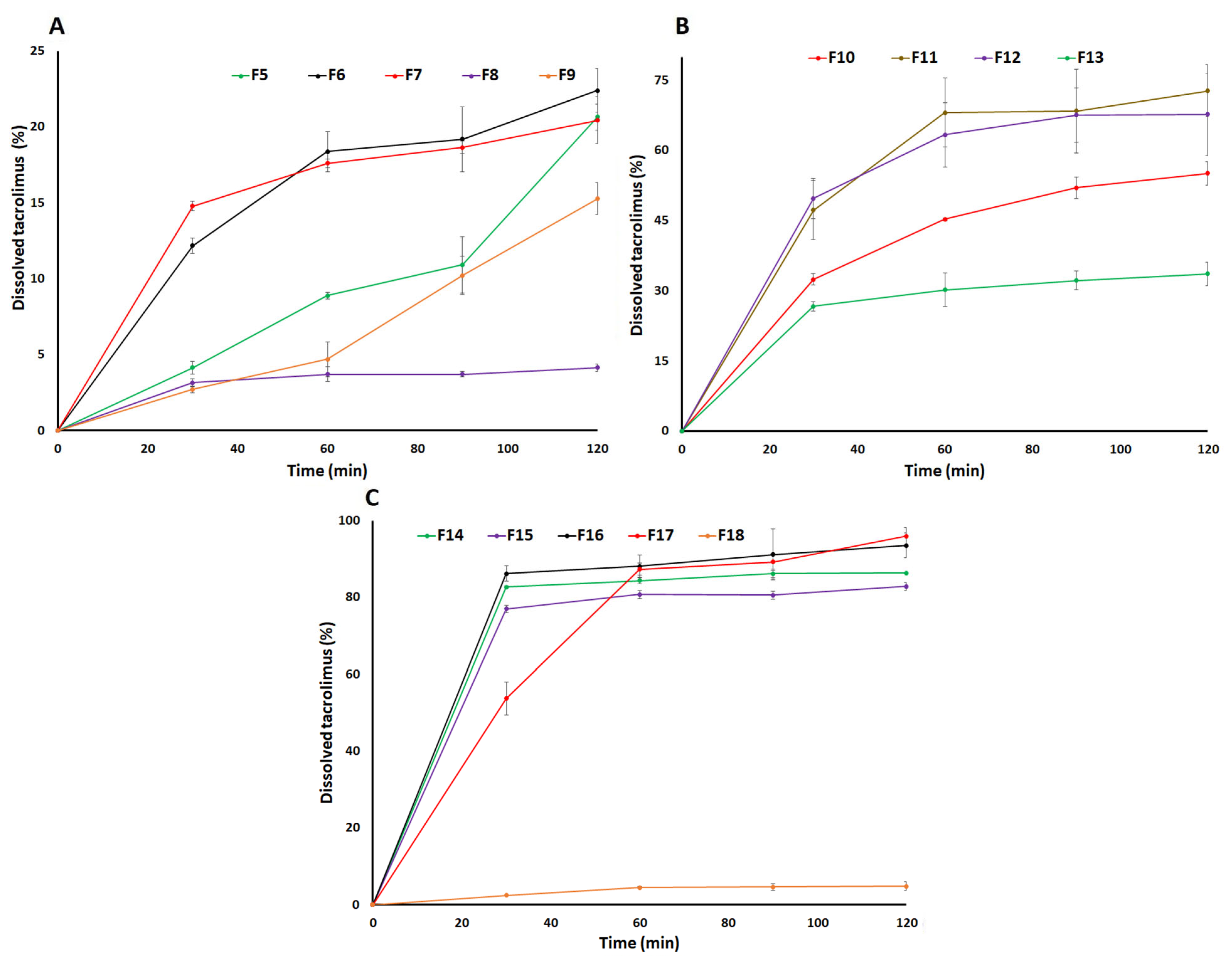
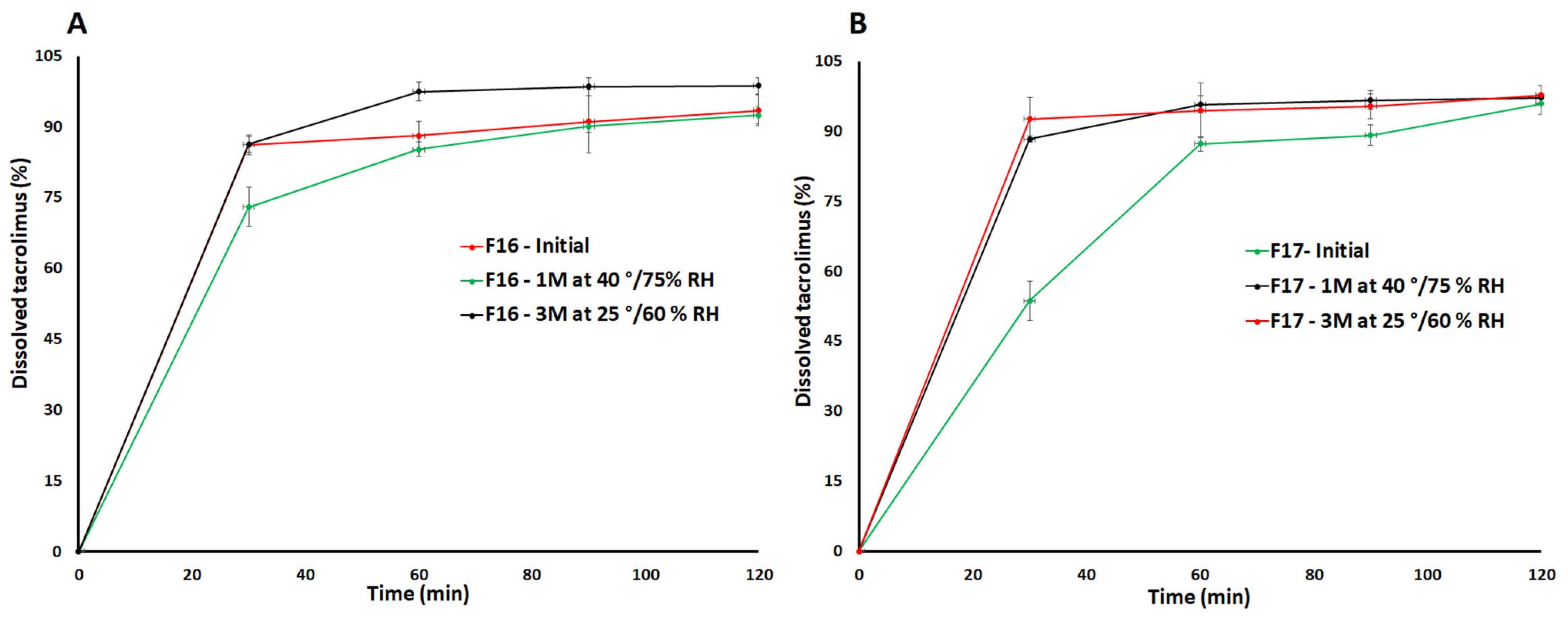
3.4. Dissolution
3.5. Stability
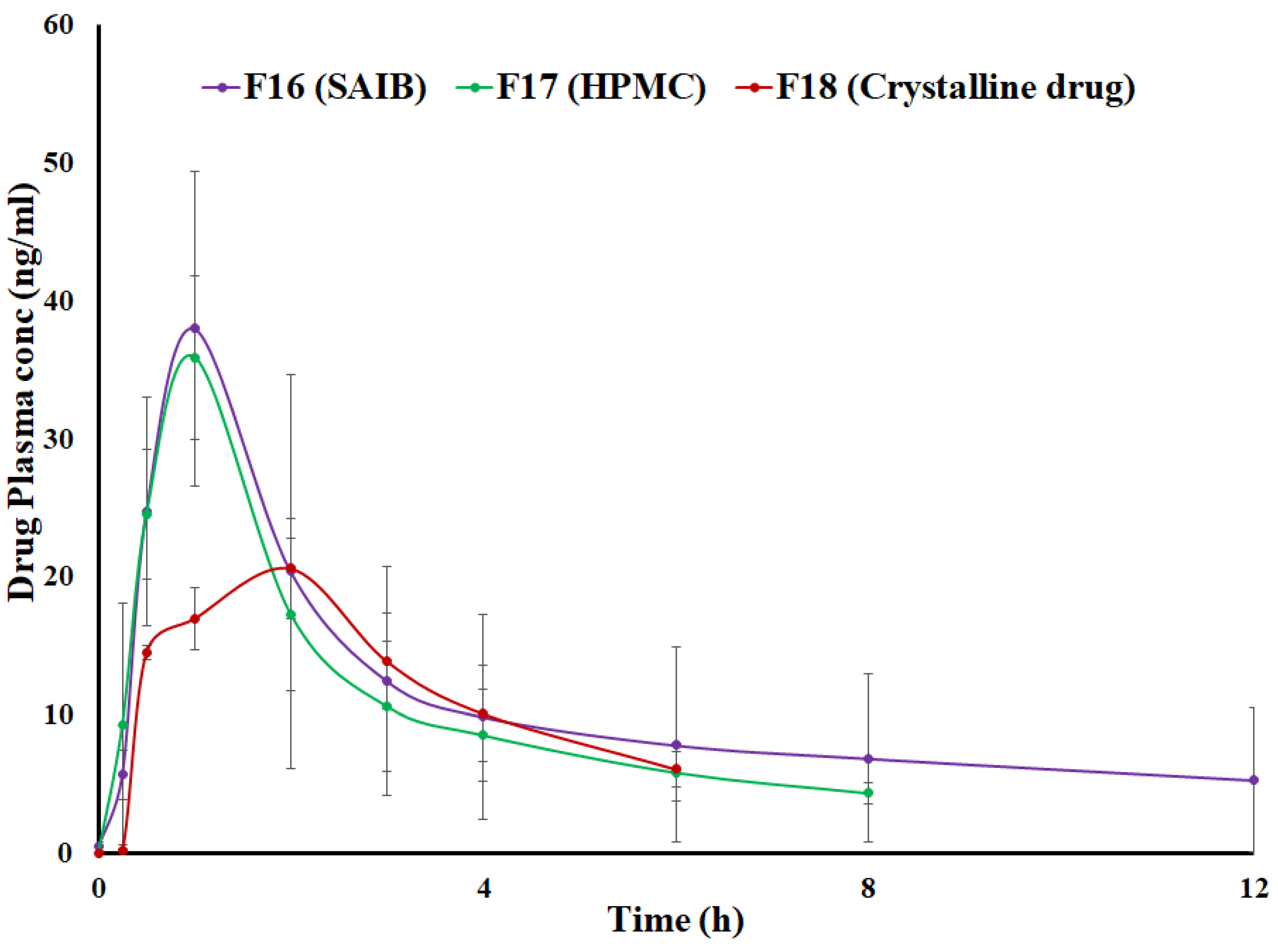
3.6. Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Coty, J.B.; Martin, C.; Telò, I.; Spitzer, D. Use of Spray Flash Evaporation (SFE) technology to improve dissolution of poorly soluble drugs: Case study on furosemide nanocrystals. Int. J. Pharm. 2020, 589, 119827. [Google Scholar] [CrossRef]
- Al-Ani, A.J.; Sugden, P.; Wilson, C.C.; Castro-Dominguez, B. Elusive Seed Formation via Electrical Confinement: Control of a Novel Cocrystal in Cooling Crystallization. Cryst. Growth Des. 2021, 21, 3310–3315. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.; dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef]
- Sigfridsson, K.; Lundqvist, A.J.; Strimfors, M. Particle size reduction for improvement of oral absorption of the poorly soluble drug UG558 in rats during early development. Drug Dev. Ind. Pharm. 2009, 35, 1479–1486. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Grohganz, H.; Rades, T.; Löbmann, K. Development of a screening method for co-amorphous formulations of drugs and amino acids. Eur. J. Pharm. Sci. 2016, 95, 28–35. [Google Scholar] [CrossRef]
- Korhonen, O.; Pajula, K.; Laitinen, R. Rational excipient selection for co-amorphous formulations. Expert Opin. Drug Deliv. 2017, 14, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.; Mohammad, A.; Akhtar, S.; Siddiqui, A.; Korang-Yeboah, M.; Khan, M.A. Chemometric Model Development and Comparison of Raman and (13)C Solid-State Nuclear Magnetic Resonance-Chemometric Methods for Quantification of Crystalline/Amorphous Warfarin Sodium Fraction in the Formulations. J. Pharm. Sci. 2015, 104, 2550–2558. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.F.; Abdelhaleem Ali, A.M.; Maher, E.M. Formulation and in-vitro evaluation of fast dissolving tablets containing a poorly soluble antipsychotic drug. Int. J. Drug Deliv. 2015, 7, 113–125. [Google Scholar]
- Ueda, H.; Muranushi, N.; Sakuma, S.; Ida, Y.; Endoh, T.; Kadota, K.; Tozuka, Y. A Strategy for Co-former Selection to Design Stable Co-amorphous Formations Based on Physicochemical Properties of Non-steroidal Inflammatory Drugs. Pharm. Res. 2016, 33, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Suo, Z.; Peng, X.; Gan, N.; Zhao, L.; Tang, P.; Wei, X.; Li, H. Microcrystalline cellulose as an effective crystal growth inhibitor for the ternary Ibrutinib formulation. Carbohydr. Polym. 2020, 229, 115476. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e71–e174. [Google Scholar] [CrossRef]
- Hamed, R.; Mohamed, E.M.; Sediri, K.; Khan, M.A.; Rahman, Z. Development of stable amorphous solid dispersion and quantification of crystalline fraction of lopinavir by spectroscopic-chemometric methods. Int. J. Pharm. 2021, 602, 120657. [Google Scholar] [CrossRef] [PubMed]
- Zidan, A.S.; Rahman, Z.; Sayeed, V.; Raw, A.; Yu, L.; Khan, M.A. Crystallinity evaluation of tacrolimus solid dispersions by chemometric analysis. Int. J. Pharm. 2012, 423, 341–350. [Google Scholar] [CrossRef]
- Maher, E.M.; Ali, A.M.; Salem, H.F.; Abdelrahman, A.A. In vitro/in vivo evaluation of an optimized fast dissolving oral film containing olanzapine co-amorphous dispersion with selected carboxylic acids. Drug Deliv. 2016, 23, 3088–3100. [Google Scholar] [CrossRef]
- Wang, R.; Han, J.; Jiang, A.; Huang, R.; Fu, T.; Wang, L.; Zheng, Q.; Li, W.; Li, J. Involvement of metabolism-permeability in enhancing the oral bioavailability of curcumin in excipient-free solid dispersions co-formed with piperine. Int. J. Pharm. 2019, 561, 9–18. [Google Scholar] [CrossRef]
- Anane-Adjei, A.B.; Jacobs, E.; Nash, S.C.; Askin, S.; Soundararajan, R.; Kyobula, M.; Booth, J.; Campbell, A. Amorphous solid dispersions: Utilization and challenges in preclinical drug development within AstraZeneca. Int. J. Pharm. 2022, 614, 121387. [Google Scholar] [CrossRef]
- Kang, J.H.; Chon, J.; Kim, Y.I.; Lee, H.J.; Oh, D.W.; Lee, H.G.; Han, C.S.; Kim, D.W.; Park, C.W. Preparation and evaluation of tacrolimus-loaded thermosensitive solid lipid nanoparticles for improved dermal distribution. Int. J. Nanomed. 2019, 14, 5381–5396. [Google Scholar] [CrossRef]
- Rahman, Z.; Siddiqui, A.; Bykadi, S.; Khan, M.A. Determination of tacrolimus crystalline fraction in the commercial immediate release amorphous solid dispersion products by a standardized X-ray powder diffraction method with chemometrics. Int. J. Pharm. 2014, 475, 462–470. [Google Scholar] [CrossRef]
- Ponnammal, P.; Kanaujia, P.; Yani, Y.; Ng, W.K.; Tan, R.B.H. Orally Disintegrating Tablets Containing Melt Extruded Amorphous Solid Dispersion of Tacrolimus for Dissolution Enhancement. Pharmaceutics 2018, 10, 35. [Google Scholar] [CrossRef]
- Yamashita, K.; Nakate, T.; Okimoto, K.; Ohike, A.; Tokunaga, Y.; Ibuki, R.; Higaki, K.; Kimura, T. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int. J. Pharm. 2003, 267, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kurimoto, I.; Yoshihara, K.; Umejima, H.; Ito, N.; Watanabe, S.; Sako, K.; Kikuchi, A. Effect of aminoalkyl methacrylate copolymer E/HCl on in vivo absorption of poorly water-soluble drug. Drug Dev. Ind. Pharm. 2013, 39, 1698–1705. [Google Scholar] [CrossRef]
- Jung, H.J.; Ahn, H.I.; Park, J.Y.; Ho, M.J.; Lee, D.R.; Cho, H.R.; Park, J.S.; Choi, Y.S.; Kang, M.J. Improved oral absorption of tacrolimus by a solid dispersion with hypromellose and sodium lauryl sulfate. Int. J. Biol. Macromol. 2016, 83, 282–287. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, Y.I.; Kim, D.W.; Yousaf, A.M.; Kim, J.O.; Woo, J.S.; Yong, C.S.; Choi, H.G. Development of novel fast-dissolving tacrolimus solid dispersion-loaded prolonged release tablet. Eur. J. Pharm. Sci. 2014, 54, 1–7. [Google Scholar] [CrossRef]
- Tsunashima, D.; Yamashita, K.; Ogawara, K.; Sako, K.; Higaki, K. Preparation of extended release solid dispersion formulations of tacrolimus using ethylcellulose and hydroxypropylmethylcellulose by solvent evaporation method. J. Pharm. Pharmacol. 2016, 68, 316–323. [Google Scholar] [CrossRef]
- HMP Global Learning Network—Immunosuppressant Recalled, 3 April 2020. Available online: https://www.hmpgloballearningnetwork.com/site/pln/content/immunosuppressant-recalled-0 (accessed on 1 February 2023).
- Fierce Pharma. European Medicines Agency Aggress to Precautionary Recall of Advagraf 0.5 mg Capsule Batches. 2011. Available online: https://www.fiercepharma.com/pharma/european-medicines-agency-agrees-to-precautionary-recall-of-advagraf-0-5-mg-capsule-batches (accessed on 1 February 2023).
- Dharani, S.; Sediri, K.; Cook, P.; Arunagiri, R.; Khan, M.A.; Rahman, Z. Preparation and Characterization of Stable Amorphous Glassy Solution of BCS II and IV Drugs. AAPS PharmSciTech 2022, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Badran, M.M.; Alomrani, A.; Aljuffali, I.A.; Alghonaim, M.; Al-Muhsen, S.; Halwani, R.; Alshamsan, A. Reutilization of Tacrolimus Extracted from Expired Prograf® Capsules: Physical, Chemical, and Pharmacological Assessment. AAPS PharmSciTech 2016, 17, 978–987. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance. 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 2 February 2023).
- Wu, X.; Hayes, D., Jr.; Zwischenberger, J.B.; Kuhn, R.J.; Mansour, H.M. Design and physicochemical characterization of advanced spray-dried tacrolimus multifunctional particles for inhalation. Drug Des. Dev. Ther. 2013, 7, 59–72. [Google Scholar] [CrossRef]
- PubChem. Sodium Dodecyl Sulfate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Sodium-dodecyl-sulfate (accessed on 2 February 2023).
- Freire, F.; Aragão, C.; de Lima e Moura, T.; Raffin, F. Compatibility study between chlorpropamide and excipients in their physical mixtures. J. Therm. Anal. Calorim. 2009, 97, 355–357. [Google Scholar] [CrossRef]
- USP43-NF38. Tacrolimus Capsules. 2022; p. 4196. Available online: https://online.uspnf.com/uspnf/document/1_GUID-3E9022BA-83DC-47EC-80DC-45D54BE1C4B1_3_en-US?source=Search%20Results&highlight=tacrolimus (accessed on 4 February 2023).
- Westermarck, S.; Juppo, A.M.; Kervinen, L.; Yliruusi, J. Microcrystalline cellulose and its microstructure in pharmaceutical processing. Eur. J. Pharm. Biopharm. 1999, 48, 199–206. [Google Scholar] [CrossRef]
- Gao, Y.; Glennon, B.; He, Y.; Donnellan, P. Dissolution Kinetics of a BCS Class II Active Pharmaceutical Ingredient: Diffusion-Based Model Validation and Prediction. ACS Omega 2021, 6, 8056–8067. [Google Scholar] [CrossRef]
- Rahman, Z.; Khan, M.A. Hunter screening design to understand the product variability of solid dispersion formulation of a peptide antibiotic. Int. J. Pharm. 2013, 456, 572–582. [Google Scholar] [CrossRef]
- Prograf® FDA Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/050708s027,050709s021lbl.pdf (accessed on 2 February 2023).
- Hirai, D.; Tsunematsu, H.; Kimura, S.I.; Itai, S.; Fukami, T.; Iwao, Y. Theoretical evaluation of supersaturation of amorphous solid dispersion formulations with different drug/polymer combinations using mathematical modeling. Int. J. Pharm. 2022, 625, 122110. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Khames, A. Investigation of the effect of solubility increase at the main absorption site on bioavailability of BCS class II drug (risperidone) using liquisolid technique. Drug Deliv. 2017, 24, 328–338. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance for Industry—Statistical Approaches to Establishing Bioequivalence. 2001. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/statistical-approaches-establishing-bioequivalenc (accessed on 2 February 2023).
- Jiang, W.; Makhlouf, F.; Schuirmann, D.J.; Zhang, X.; Zheng, N.; Conner, D.; Yu, L.X.; Lionberger, R. A Bioequivalence Approach for Generic Narrow Therapeutic Index Drugs: Evaluation of the Reference-Scaled Approach and Variability Comparison Criterion. AAPS J. 2015, 17, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Jiang, W.; Zhang, X.; Lionberger, R.; Makhlouf, F.; Schuirmann, D.J.; Muldowney, L.; Chen, M.L.; Davit, B.; Conner, D.; et al. Novel bioequivalence approach for narrow therapeutic index drugs. Clin. Pharmacol. Ther. 2015, 97, 286–291. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Tacrolimus (mg) | SAIB (mg) | SLS (mg) | MCC (mg) | LMH (mg) | HPMC (mg) | CCS (mg) | Dosage Form |
|---|---|---|---|---|---|---|---|---|
| F1 | 5 | 5 | 0 | 0 | 0 | 0 | 0 | Capsule |
| F2 | 5 | 5 | 0 | 0 | 25 | 0 | 0 | Capsule |
| F3 | 5 | 5 | 0 | 0 | 50 | 0 | 0 | Capsule |
| F4 | 5 | 5 | 0 | 0 | 100 | 0 | 0 | Capsule |
| F5 | 5 | 5 | 0 | 25 | 0 | 0 | 0 | Capsule |
| F6 | 5 | 5 | 0 | 50 | 0 | 0 | 0 | Capsule |
| F7 | 5 | 5 | 0 | 100 | 0 | 0 | 0 | Capsule |
| F8 | 5 | 0.5 | 0 | 50 | 0 | 0 | 0 | Capsule |
| F9 | 5 | 3.75 | 0 | 50 | 0 | 0 | 0 | Capsule |
| F10 | 5 | 5 | 0 | 50 | 0 | 0 | 10 | Capsule |
| F11 | 5 | 7.5 | 0 | 50 | 0 | 0 | 10 | Capsule |
| F12 | 5 | 10 | 0 | 50 | 0 | 0 | 10 | Capsule |
| F13 | 5 | 7.5 | 5 | 50 | 0 | 0 | 10 | Capsule |
| F14 | 5 | 7.5 | 5 | 50 | 0 | 0 | 15 | Capsule |
| F15 | 5 | 7.5 | 5 | 50 | 0 | 0 | 20 | Capsule |
| F16 | 5 | 7.5 | 5 | 50 | 0 | 0 | 15 | Tablet |
| F17 | 5 | 0 | 5 | 50 | 0 | 7.5 | 15 | Tablet |
| F18 | 5 | 0 | 5 | 50 | 0 | 0 | 15 | Tablet |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, E.M.; Dharani, S.; Nutan, M.T.H.; Cook, P.; Arunagiri, R.; Khan, M.A.; Rahman, Z. Application of Sucrose Acetate Isobutyrate in Development of Co-Amorphous Formulations of Tacrolimus for Bioavailability Enhancement. Pharmaceutics 2023, 15, 1442. https://doi.org/10.3390/pharmaceutics15051442
Mohamed EM, Dharani S, Nutan MTH, Cook P, Arunagiri R, Khan MA, Rahman Z. Application of Sucrose Acetate Isobutyrate in Development of Co-Amorphous Formulations of Tacrolimus for Bioavailability Enhancement. Pharmaceutics. 2023; 15(5):1442. https://doi.org/10.3390/pharmaceutics15051442
Chicago/Turabian StyleMohamed, Eman M., Sathish Dharani, Mohammad T. H. Nutan, Phillip Cook, Rajendran Arunagiri, Mansoor A. Khan, and Ziyaur Rahman. 2023. "Application of Sucrose Acetate Isobutyrate in Development of Co-Amorphous Formulations of Tacrolimus for Bioavailability Enhancement" Pharmaceutics 15, no. 5: 1442. https://doi.org/10.3390/pharmaceutics15051442