
Application of Physiologically Based Pharmacokinetic Modeling to Predict Drug–Drug Interactions between Elexacaftor/Tezacaftor/Ivacaftor and Tacrolimus in Lung Transplant Recipients

Abstract
:1. Introduction
2. Methods
2.1. Development of PBPK Model
2.1.1. Population Model
2.1.2. Ivacaftor Model
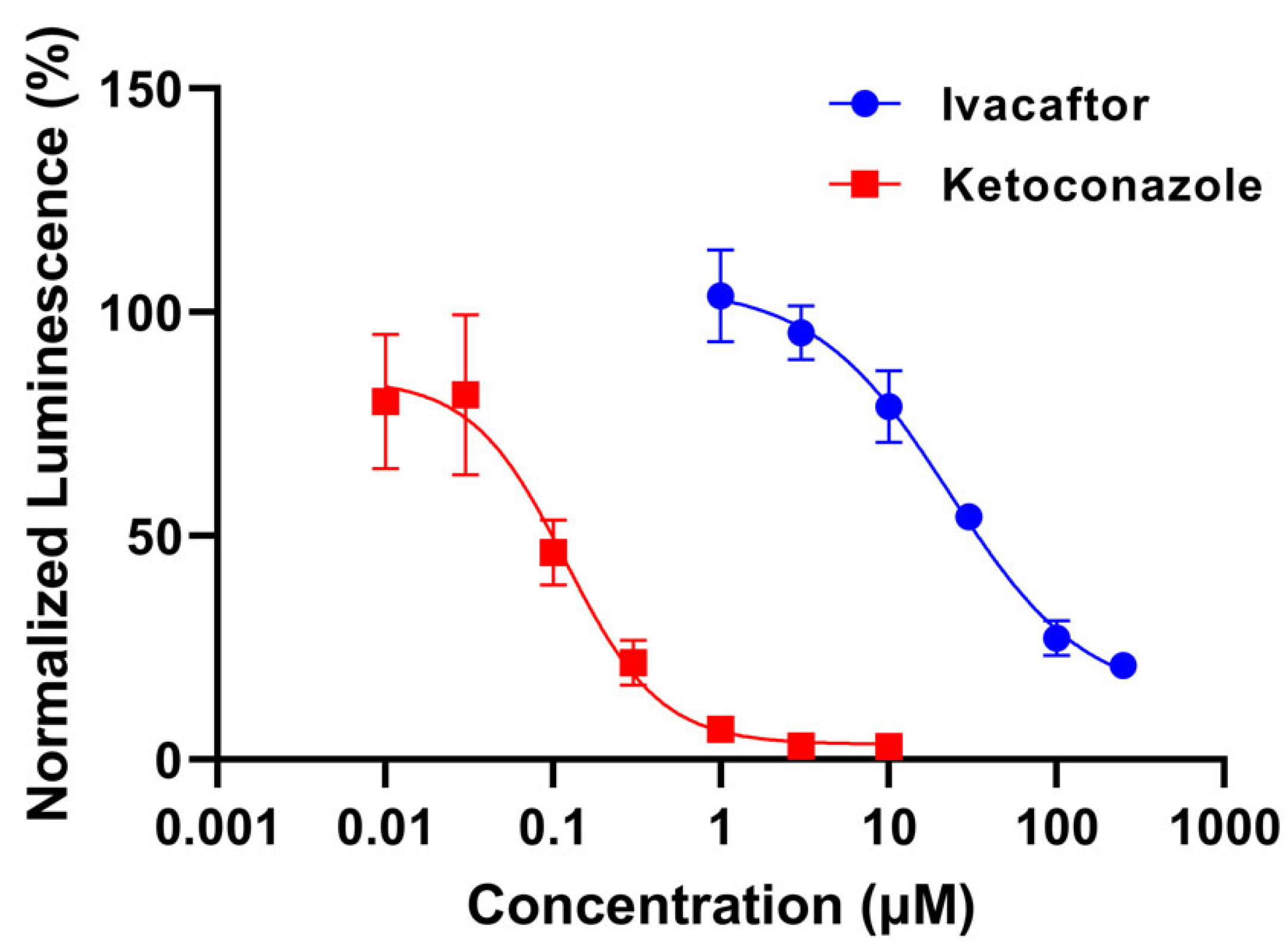
- Determination of CYP3A4 inhibition potential with P450 Glo luminescence assay
- Ivacaftor model development
2.1.3. Tacrolimus Model Development
2.2. PBPK Model Verification
2.2.1. Plasma or Blood Pharmacokinetic Simulations
2.2.2. DDI Simulations
2.3. Model Application
2.3.1. DDI Predictions of Tacrolimus with Ivacaftor
2.3.2. Sensitivity Analysis of Tacrolimus–Ivacaftor DDI
2.4. Clinical Case Series
3. Results
3.1. Model Development and Verification
3.1.1. Ivacaftor Inhibition Potential
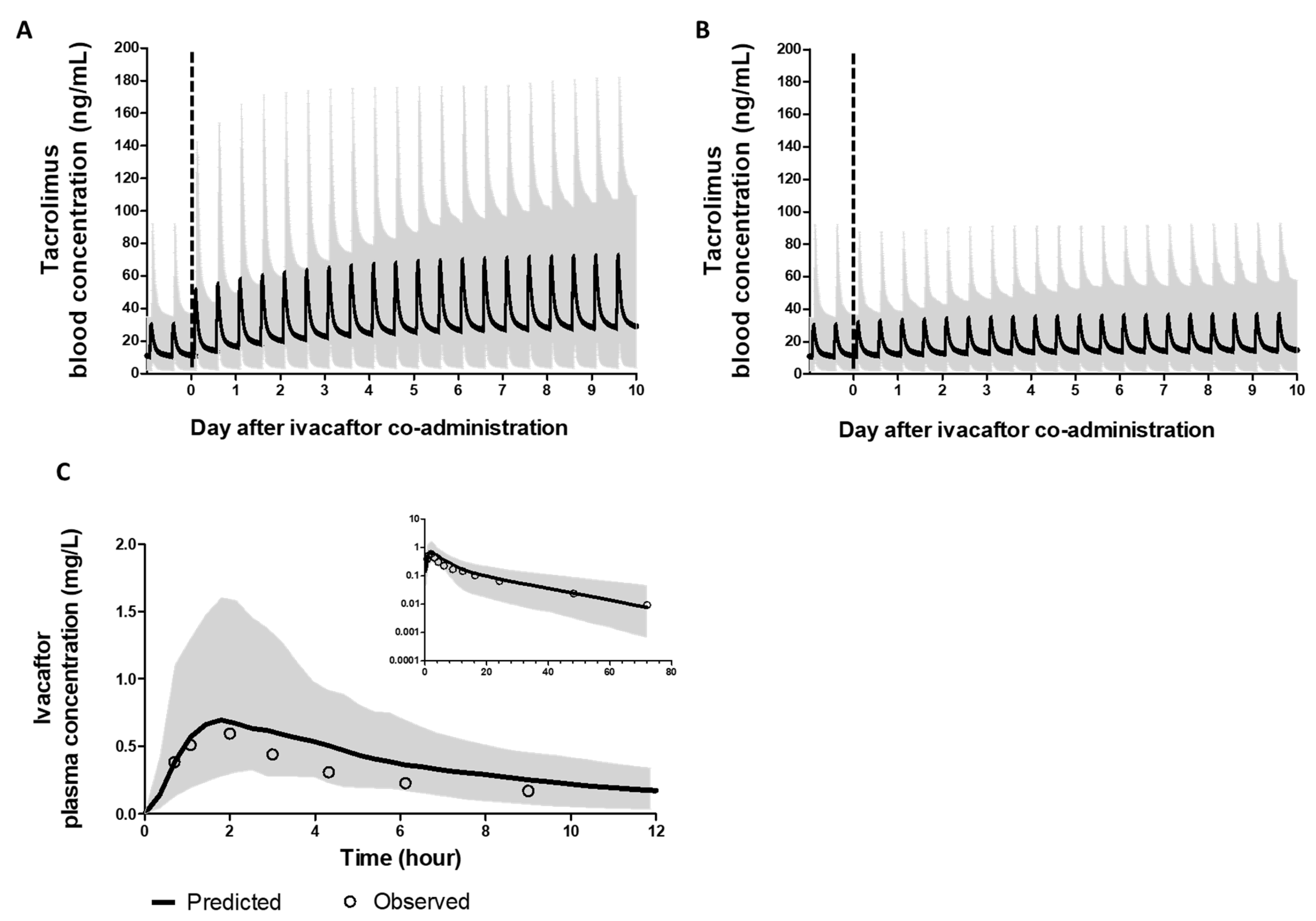
3.1.2. PBPK Models of Ivacaftor and Tacrolimus Recapitulated Clinically Observed PK Profiles
3.1.3. PBPK Models of Ivacaftor and Tacrolimus Recapitulated Observed DDI
3.2. Model Application
3.2.1. DDI Predictions of Tacrolimus with Ivacaftor
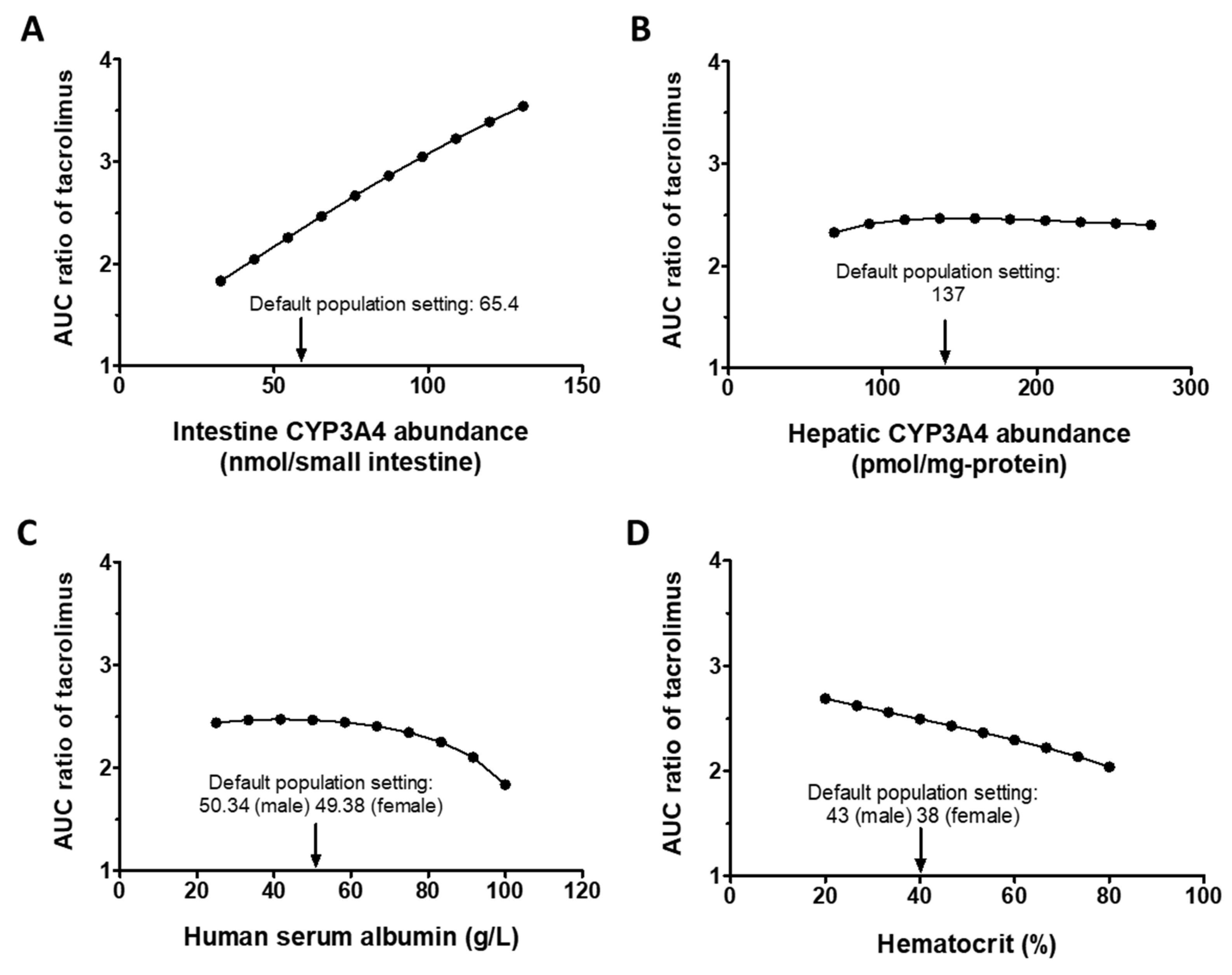
3.2.2. Sensitivity Analysis
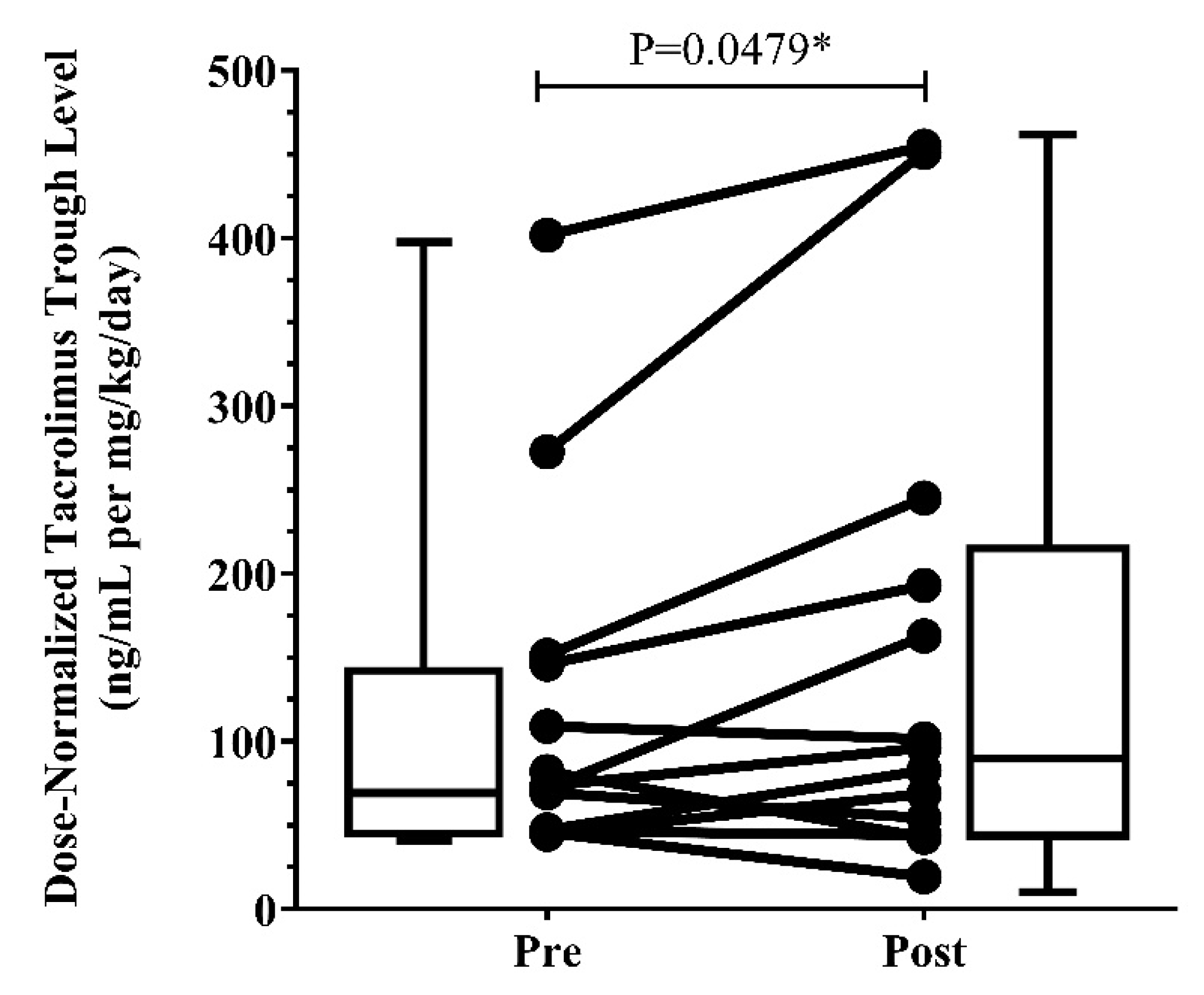
3.3. Clinical Presentation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Gavioli, E.; Guardado, N.; Haniff, F.; Deiab, N.; Vider, E. A current review of the safety of cystic fibrosis transmembrane conductance regulator modulators. J. Clin. Pharm. Ther. 2021, 46, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Ramos, K.; Pilewski, J.; Taylor-Cousar, J. Challenges in the use of highly effective modulator treatment for cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2021, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Lim, D.; Mackey, C.; Skinner, D.; Zhang, S.; McCormick, J.; Woodworth, B. Ivacaftor, a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator, Enhances Ciprofloxacin Activity Against Pseudomonas aeruginosa. Am. J. Rhinol. Allergy 2019, 33, 129–136. [Google Scholar] [CrossRef]
- Nichols, D.; Morgan, S.; Skalland, M.; Vo, A.; Van Dalfsen, J.; Singh, S.; Ni, W.; Hoffman, L.; McGeer, K.; Heltshe, S.; et al. Pharmacologic improvement of CFTR function rapidly decreases sputum pathogen density but lung infections generally persist. J. Clin. Investig. 2023. [Google Scholar] [CrossRef]
- Prograf [Package Insert]; Astellas Pharma US, Inc.: Northbrook, IL, USA, 2022.
- Heizmann, P.; Eckert, M.; Ziegler, W. Pharmacokinetics and bioavailability of midazolam in man. Br. J. Clin. Pharmacol. 1983, 16, 43S–49S. [Google Scholar] [CrossRef]
- Staatz, C.; Tett, S. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin. Pharmacokinet. 2004, 43, 623–653. [Google Scholar] [CrossRef]
- Kalydeco [Package Insert]; Vertex Pharmaceuticals Inc.: Cambridge, MA, USA, 2020.
- U.S. Food and Drug Administration. Center for Drug Evaluation and Research. Multi-Discipline Review, Elexacaftor/Tezacaftor/Ivacaftor. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212273Orig1s000MultidisciplineR.pdf (accessed on 10 March 2023).
- Yeo, K.; Jamei, M.; Rostami-Hodjegan, A. Predicting drug-drug interactions: Application of physiologically based pharmacokinetic models under a systems biology approach. Expert Rev. Clin. Pharmacol. 2013, 6, 143–157. [Google Scholar] [CrossRef]
- Luzon, E.; Blake, K.; Cole, S.; Nordmark, A.; Versantvoort, C.; Berglund, E. Physiologically based pharmacokinetic modeling in regulatory decision-making at the European Medicines Agency. Clin. Pharmacol. Ther. 2017, 102, 98–105. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Owens, K.; Ritchie, T.; Ragueneau-Majlessi, I. What Can Be Learned from Recent New Drug Applications? A Systematic Review of Drug Interaction Data for Drugs Approved by the US FDA in 2015. Drug Metab. Dispos. Biol. Fate Chem. 2017, 45, 86–108. [Google Scholar] [CrossRef]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef]
- Hong, E.; Almond, L.; Chung, P.; Rao, A.; Beringer, P. Physiologically-Based Pharmacokinetic-Led Guidance for Patients with Cystic Fibrosis Taking Elexacaftor-Tezacaftor-Ivacaftor With Nirmatrelvir-Ritonavir for the Treatment of COVID-19. Clin. Pharmacol. Ther. 2022, 111, 1324–1333. [Google Scholar] [CrossRef]
- Hong, E.; Almond, L.; Chung, P.; Rao, A.; Beringer, P. Physiologically Based Pharmacokinetic Modeling To Guide Management of Drug Interactions between Elexacaftor-Tezacaftor-Ivacaftor and Antibiotics for the Treatment of Nontuberculous Mycobacteria. Antimicrob. Agents Chemother. 2022, 66, e01104-22. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation Patient Registry 2020 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2021.
- P450-GloTM Assays; ProMega Corporation: Madison, WI, USA, 2016.
- Page, K. Validation of Early Human Dose Prediction: A Key Metric for Compound Progression in Drug Discovery. Mol. Pharm. 2016, 13, 609–620. [Google Scholar] [CrossRef]
- Gertz, M.; Houston, J.; Galetin, A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab. Dispos. Biol. Fate Chem. 2011, 39, 1633–1642. [Google Scholar] [CrossRef]
- Gertz, M.; Harrison, A.; Houston, J.; Galetin, A. Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1147–1158. [Google Scholar] [CrossRef]
- Yang, D.; Mary, F.H.; Nina, I.; Connie, L.D.; Christopher, M.; Danny, D.S.; Kenneth, E.T. Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab. Dispos. 2006, 34, 836–847. [Google Scholar] [CrossRef]
- Möller, A.; Iwasaki, K.; Kawamura, A.; Teramura, Y.; Shiraga, T.; Hata, T.; Schäfer, A.; Undre, N. The disposition of 14c-labeled tacrolimus after intravenous and oral administration in healthy human subjects. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 633–636. [Google Scholar]
- Emoto, C.; Johnson, T.; Hahn, D.; Christians, U.; Alloway, R.; Vinks, A.; Fukuda, T. A Theoretical Physiologically-Based Pharmacokinetic Approach to Ascertain Covariates Explaining the Large Interpatient Variability in Tacrolimus Disposition. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 273–284. [Google Scholar] [CrossRef]
- Iwasaki, K. Metabolism of tacrolimus (FK506) and recent topics in clinical pharmacokinetics. Drug Metab. Pharmacokinet. 2007, 22, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Hashida, T.; Masuda, S.; Uemoto, S.; Saito, H.; Tanaka, K.; Inui, K. Pharmacokinetic and prognostic significance of intestinal MDR1 expression in recipients of living-donor liver transplantation. Clin. Pharmacol. Ther. 2001, 69, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, H.; Saikachi, Y.; Kobayashi, M.; Yamaguchi, M.; Oda, M.; Yuhki, Y.; Achiwa, K.; Tadano, K.; Takahashi, Y.; Aungst, B. Limited interaction between tacrolimus and P-glycoprotein in the rat small intestine. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2006, 28, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H. Confidence assessment of the Simcyp time-based approach and a static mathematical model in predicting clinical drug-drug interactions for mechanism-based CYP3A inhibitors. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1094–1104. [Google Scholar] [CrossRef]
- Galetin, A.; Ito, K.; Hallifax, D.; Houston, J.B. CYP3A4 substrate selection and substitution in the prediction of potential drug-drug interactions. J. Pharmacol. Exp. Ther. 2005, 314, 180–190. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutics Review(s), Ivacaftor. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203188Orig1s000ClinPharmR.pdf (accessed on 12 March 2023).
- Hong, Y.; Ishizuka, T.; Watanabe, A.; Tachibana, M.; Lee, M.; Ishizuka, H.; LaCreta, F.; Abutarif, M. Model-based assessments of CYP3A-mediated drug-drug interaction risk of milademetan. Clin. Transl. Sci. 2021, 14, 2220–2230. [Google Scholar] [CrossRef]
- Li, X.; Frechen, S.; Moj, D.; Lehr, T.; Taubert, M.; Hsin, C.; Mikus, G.; Neuvonen, P.; Olkkola, K.; Saari, T.; et al. A Physiologically Based Pharmacokinetic Model of Voriconazole Integrating Time-Dependent Inhibition of CYP3A4, Genetic Polymorphisms of CYP2C19 and Predictions of Drug-Drug Interactions. Clin. Pharmacokinet. 2020, 59, 781–808. [Google Scholar] [CrossRef]
- Davies, J.C.; Alton, E.W. Monitoring respiratory disease severity in cystic fibrosis. Respir. Care 2009, 54, 606–617. [Google Scholar] [CrossRef]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.; Zheng, J.; et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: The global lung function 2012 equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef]
- Marfo, K.; Altshuler, J.; Lu, A. Tacrolimus Pharmacokinetic and Pharmacogenomic Differences between Adults and Pediatric Solid Organ Transplant Recipients. Pharmaceutics 2010, 2, 291–299. [Google Scholar] [CrossRef]
- Trikafta [Package Insert]; Vertex Pharmaceuticals Inc.: Cambridge, MA, USA, 2019.
- Mignat, C. Clinically significant drug interactions with new immunosuppressive agents. Drug Saf. 1997, 16, 267–278. [Google Scholar] [CrossRef]
- Assan, R.; Fredj, G.; Larger, E.; Feutren, G.; Bismuth, H. FK 506/fluconazole interaction enhances FK 506 nephrotoxicity. Diabete Metab. 1994, 20, 49–52. [Google Scholar]
- Gaïes, E.; Salouage, I.; Sahnoun, R.; Trabelsi, S.; Jebabli, N.; Lakhal, M.; Klouz, A. Interaction between azole antifugals drugs and tacrolimus in four kidney transplant patients. J. Mycol. Med. 2011, 21, 46–50. [Google Scholar] [CrossRef]
- Wolter, K.; Wagner, K.; Philipp, T.; Fritschka, E. Interaction between FK 506 and clarithromycin in a renal transplant patient. Eur. J. Clin. Pharmacol. 1994, 47, 207–208. [Google Scholar] [CrossRef]
- Doligalski, C.; McKinzie, C.; Yang, A.; Lobo, L.; Coakley, R. Poor tolerability of cystic fibrosis transmembrane conductance regulator modulator therapy in lung transplant recipients. Pharmacotherapy 2022, 42, 580–584. [Google Scholar] [CrossRef]
- Bekersky, I.; Dressler, D.; Mekki, Q. Effect of time of meal consumption on bioavailability of a single oral 5 mg tacrolimus dose. J. Clin. Pharm. 2001, 41, 289–297. [Google Scholar] [CrossRef]
- Ragan, H.; Autry, E.; Bomersback, T.; Hewlett, J.; Kormelink, L.; Safirstein, J.; Shanley, L.; Lubsch, L. The use of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis postliver transplant: A case series. Pediatr. Pulmonol. 2022, 57, 411–417. [Google Scholar] [CrossRef]
- Orum, M.B.; Ronsholt, F.F.; Jeppesen, M.; Bendstrup, E.; Katzenstein, T.L.; Ott, P.; Perch, M.; Pressler, T.; Qvist, T.; Jensen-Fangel, S. Outcome of elexacaftor/tezacaftor/ivacaftor therapy in patients with cystic fibrosis and solid organ transplantation. Pediatr. Pulmonol. 2023, 58, 602–605. [Google Scholar] [CrossRef]
- Benninger, L.A.; Trillo, C.; Lascano, J. CFTR modulator use in post lung transplant recipients. J. Heart Lung Transpl. 2021, 40, 1498–1501. [Google Scholar] [CrossRef]
- Ramos, K.J.; Guimbellot, J.S.; Valapour, M.; Bartlett, L.E.; Wai, T.H.; Goss, C.H.; Pilewski, J.M.; Faro, A.; Diamond, J.M.; Group, C.S. Use of elexacaftor/tezacaftor/ivacaftor among cystic fibrosis lung transplant recipients. J. Cyst. Fibros. 2022, 21, 745–752. [Google Scholar] [CrossRef]
- Yvonne, S.L.; Amy, L.S.D.; Sean, D.Q.; Federico, M.F.; Jiong, Z.; Jatinder, L.; Erin, G.S.; Kenneth, E.T. Co-Regulation of CYP3A4 and CYP3A5 and Contribution to Hepatic and Intestinal Midazolam Metabolism. Mol. Pharmacol. 2002, 62, 162–172. [Google Scholar] [CrossRef]
- Berkowitz, N.; Schulman, L.; McGregor, C.; Markowitz, D. Gastroparesis after lung transplantation. Potential role in postoperative respiratory complications. Chest 1995, 108, 1602–1607. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Table of Substrates, Inhibitors and Inducers (Updated 2022). Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 29 December 2022).
- DDI-Predictor. Quantitative Prediction of Drug Drug Interactions-Ddi-Predictor Academic Version. Available online: https://www.Ddi-predictor.Org (accessed on 3 October 2022).
- Noxafil [Package Insert]; Merck & Co., Inc.: Whitehouse Station, NJ, USA, 2015.
- Yamada, M.; Inoue, S.; Sugiyama, D.; Nishiya, Y.; Ishizuka, T.; Watanabe, A.; Watanabe, K.; Yamashita, S.; Watanabe, N. Critical Impact of Drug-Drug Interactions via Intestinal CYP3A in the Risk Assessment of Weak Perpetrators Using Physiologically Based Pharmacokinetic Models. Drug Metab. Dispos. Biol. Fate Chem. 2020, 48, 288–296. [Google Scholar] [CrossRef] [PubMed]
- De Sutter, P.; Van Haeverbeke, M.; Van Braeckel, E.; Van Biervliet, S.; Van Bocxlaer, J.; Vermeulen, A.; Gasthuys, E. Altered intravenous drug disposition in people living with cystic fibrosis: A meta-analysis integrating top-down and bottom-up data. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Modrykamien, A. Anemia post-lung transplantation: Mechanisms and approach to diagnosis. Chronic Respir. Dis. 2010, 7, 29–34. [Google Scholar] [CrossRef]
- End, A.; Ringl, H.; Grimm, M.; Schlick, W.; Schreiner, W.; Klepetko, W.; Wolner, E. Chronic anemia after lung transplantation: Treatment with human recombinant erythropoietin. Transplantation 1994, 57, 1142. [Google Scholar] [CrossRef]
- Bugge, T.; Perch, M.; Rezahosseini, O.; Crone, C.; Jensen, K.; Schultz, H.; Bredahl, P.; Hornum, M.; Nielsen, S.; Lund, T. Post-Transplantation Anemia and Risk of Death Following Lung Transplantation. Transplant. Proc. 2022, 54, 2329–2336. [Google Scholar] [CrossRef]
- Shah, N.; Bulitta, J.; Kinzig, M.; Landersdorfer, C.; Jiao, Y.; Sutaria, D.; Tao, X.; Höhl, R.; Holzgrabe, U.; Kees, F.; et al. Novel Population Pharmacokinetic Approach to Explain the Differences between Cystic Fibrosis Patients and Healthy Volunteers via Protein Binding. Pharmaceutics 2019, 11, 286. [Google Scholar] [CrossRef]
- Courville, A.; DiVito, M.; Moyer, L.; Rossinoff, A.; Royster, C.; Psota, T.; Ayres, E.; Zambell, K. Increase in body mass index from normal weight to overweight in a cross-sectional sample of healthy research volunteers. Nutr. Res. 2014, 34, 1052–1057. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Source |
|---|---|---|
| Physiochemical properties | ||
| Molecular weight (g/mol) | 804.02 | Drug label |
| Log Po:w | 3.3 | Gertz et al. [21] |
| Compound type | Neutral | Gertz et al. [21] |
| B/P | 35 | Gertz et al. [21] |
| fup | 0.013 | Gertz et al. [21] |
| Absorption | ||
| Absorption model | First-order model | |
| Caco-2 permeability (10−6 cm/s) | 13.1 | Gertz et al. [22] |
| Scalar | 2.157 | Gertz et al. [22] |
| fugut | 1 | Default |
| ka (h−1) | 3.68 | Emoto et al. [25] |
| fa | 1.0 | Emoto et al. [25] |
| Lag time (h) | 0.43 | Emoto et al. [25] |
| Qgut (L/h) | 13.3 | Predicted by Simcyp |
| Peff,man (×10−4 cm/s) | 3.52 | Predicted by Simcyp |
| Distribution | ||
| Distribution model | Minimal PBPK model | |
| kin (h−1) | 0.68 | Emoto et al. [25] |
| kout (h−1) | 0.10 | Emoto et al. [25] |
| Vsac (l/kg) | 10.8 | Emoto et al. [25] |
| Vss (l/kg) | 18.0 | Predicted by Simcyp Method 1 |
| Elimination | ||
| 13-O-desmethylation | ||
| CYP3A4 Vmax (pmol/min/pmol CYP) | 8 | Dai et al. [23] |
| CYP3A4 Km (μM) | 0.21 | Dai et al. [23] |
| CYP3A5 Vmax (pmol/min/pmol CYP) | 17 | Dai et al. [23] |
| CYP3A5 Km (μM) | 0.21 | Dai et al. [23] |
| 12-hydroxylation | ||
| CYP3A4 Vmax (pmol/min/pmol CYP) | 0.6 | Dai et al. [23] |
| CYP3A4 Km (μM) | 0.29 | Dai et al. [23] |
| CYP3A5 Vmax (pmol/min/pmol CYP) | 1.4 | Dai et al. [23] |
| CYP3A5 Km (μM) | 0.35 | Dai et al. [23] |
| CYP3A4, 3A5 ISEF | 0.24 | Simcyp default |
| Renal clearance (L/h) | 0 | Moller et al. [24] |
| PK Study | PK Parameters | |||||
|---|---|---|---|---|---|---|
| Drug | Regimen | Simulated | Observed | |||
| Cmax (ng/mL) | AUC * (ng∙h/mL) | Cmax (ng/mL) | AUC * (ng∙h/mL) | |||
| ivacaftor | 150 mg q12h oral | Mean | 1536 | 12,184 | 1270 | 12,100 |
| SD | 1085 | 6641 | 353 | 4170 | ||
| Simulated/ observed | 1.2 | 1.0 | ||||
| tacrolimus | 0.05 mg/kg single dose oral | Mean | 37.5 | 398 | 37.8 | 307 |
| SD | 28.1 | 320 | 16.0 | 251 | ||
| Simulated/ observed | 1.0 | 1.3 | ||||
| tacrolimus | 0.1 mg/kg single dose iv infusion (4 h) | Mean | 16.2 | 404 | 21.4 | 378 |
| SD | 7.2 | 204 | 8.0 | 109 | ||
| Simulated/ observed | 0.8 | 1.1 | ||||
| Drug | PK Parameters | Simulated GMR (90% CI) | Observed * GMR (90% CI) | Ratio (Simulated/Observed) |
|---|---|---|---|---|
| Midazolam +/− ivacaftor | Cmax Ratio | 1.51 (1.48, 1.55) | 1.38 (1.26, 1.52) | 1.09 |
| AUC Ratio | 1.68 (1.64, 1.71) | 1.54 (1.39, 1.69) | 1.09 | |
| Ethinylestradiol +/− ivacaftor | Cmax Ratio | 1.11 (1.10, 1.12) | 1.22 (1.10, 1.36) | 0.91 |
| AUC Ratio | 1.14 (1.13, 1.15) | 1.07 (1.00, 1.14) | 1.07 |
| Drug | PK Parameters | Simulated GMR (90% CI) | Observed * GMR (90% CI) | Ratio (Simulated/Observed) |
|---|---|---|---|---|
| tacrolimus +/− posaconazole | Cmax Ratio | 2.4 (2.2, 2.5) | 2.0 (2.0, 2.4) | 1.20 |
| AUC Ratio | 4.4 (4.0, 4.8) | 4.5 (4.0, 5.2) | 0.98 | |
| tacrolimus +/− voriconazole | Cmax Ratio | 2.0 (1.9, 2.1) | 2.0 (1.9, 2.5) | 1.00 |
| AUC Ratio | 3.2 (3,0, 3.4) | 3.0 (2.7, 3.8) | 1.07 |
| Characteristic | Included Patients (N = 13) | |
|---|---|---|
| Age | ||
| Median (25%, 75%) | 37 (36.5, 52.5) | |
| Female sex—no. (%) | 6 (46) | |
| Percentage of predicted FEV1 | ||
| Median (25%, 75%) | 92 (70.5, 104.5) | |
| Body mass index | ||
| Median—kg/m2 (25%, 75%) | 22.5 (20.6, 23.75) | |
| CFTR Mutation—no. (%) | ||
| F508del/F508del | 6 (46) | |
| F508del/minimal function | 5 (38) | |
| F508del/residual function | 1 (8) | |
| Other genotype | 1 (8) | |
| Comorbidities—no. (%) | ||
| Chronic rhinosinusitis | 13 (100) | |
| Cystic fibrosis-related diabetes | 8 (61) | |
| Gastrointestinal manifestations | 7 (54) | |
| Two or more comorbidities | 9 (69) | |
| Duration since transplant and ETI start | ||
| Median—years (25%, 75%) | 10.27 (4.19, 15.26) | |
| ETI dose—no. (%) | ||
| Full | 9 (69) | |
| Reduced | 4 (31) | |
| On other CYP3A4 modulators—no. (%) | 4 (31) | |
| Pre-ETI | Post-ETI | ||||||
|---|---|---|---|---|---|---|---|
| TAC Dose | Laboratory Examinations | TAC Dose | Laboratory Examinations | ||||
| Patient Number | WN Daily Dose (mg/kg/day) | TAC Trough Concentration (ng/mL) | TAC DN Trough Level (ng/mL per mg/kg/day) | WN Daily Dose (mg/kg/day) | TAC Trough Concentration (ng/mL) | TAC DN Trough Level (ng/mL per mg/kg/day) | Percent Difference DN Trough (%) |
| 1 | 0.178 | 8.40 | 47.25 | 0.171 | 14.10 | 82.64 | 74.90 |
| 2 | 0.028 | 7.70 | 272.58 | 0.022 | 9.75 | 451.10 | 65.49 |
| 3 | 0.051 | 7.50 | 146.00 | 0.050 | 9.73 | 193.04 | 32.22 |
| 4 | 0.165 | 11.40 | 69.29 | 0.156 | 8.45 | 54.29 | −21.64 |
| 5 | 0.026 | 10.40 | 401.70 | 0.025 | 11.30 | 454.83 | 13.23 |
| 6 | 0.037 | 5.60 | 151.20 | 0.039 | 9.50 | 245.10 | 62.10 |
| 7 | 0.160 | 7.30 | 45.68 | 0.176 | 3.40 | 19.30 | −57.76 |
| 8 | 0.097 | 7.10 | 73.41 | 0.093 | 8.90 | 96.12 | 30.93 |
| 9 | 0.061 | 6.60 | 108.97 | 0.060 | 6.10 | 101.40 | −6.95 |
| 10 | 0.139 | 11.40 | 81.98 | 0.130 | 5.58 | 42.88 | −47.69 |
| 11 * | 0.118 | NA | 46.37 | 0.106 | 6.70 | 62.88 | 35.60 |
| 12 * | 0.132 | NA | 44.69 | 0.132 | 9.05 | 68.78 | 53.90 |
| 13 | 0.096 | 6.90 | 72.22 | 0.096 | 15.70 | 162.86 | 125.51 |
| Median | 0.096 | 7.50 | 73.41 | 0.096 | 9.05 | 96.12 | 32.33 |
| IQR (25%, 75%) | 0.044, 0.15 | 6.90, 10.40 | 46.81, 148.6 | 0.044, 0.14 | 6.40, 10.53 | 58.29, 219.10 | −14.30, 63.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, E.; Carmanov, E.; Shi, A.; Chung, P.S.; Rao, A.P.; Forrester, K.; Beringer, P.M. Application of Physiologically Based Pharmacokinetic Modeling to Predict Drug–Drug Interactions between Elexacaftor/Tezacaftor/Ivacaftor and Tacrolimus in Lung Transplant Recipients. Pharmaceutics 2023, 15, 1438. https://doi.org/10.3390/pharmaceutics15051438
Hong E, Carmanov E, Shi A, Chung PS, Rao AP, Forrester K, Beringer PM. Application of Physiologically Based Pharmacokinetic Modeling to Predict Drug–Drug Interactions between Elexacaftor/Tezacaftor/Ivacaftor and Tacrolimus in Lung Transplant Recipients. Pharmaceutics. 2023; 15(5):1438. https://doi.org/10.3390/pharmaceutics15051438
Chicago/Turabian StyleHong, Eunjin, Eugeniu Carmanov, Alan Shi, Peter S. Chung, Adupa P. Rao, Kevin Forrester, and Paul M. Beringer. 2023. "Application of Physiologically Based Pharmacokinetic Modeling to Predict Drug–Drug Interactions between Elexacaftor/Tezacaftor/Ivacaftor and Tacrolimus in Lung Transplant Recipients" Pharmaceutics 15, no. 5: 1438. https://doi.org/10.3390/pharmaceutics15051438