

Kinetics of Water-Induced Amorphous Phase Separation in Amorphous Solid Dispersions via Raman Mapping
 , , ,
, , ,
Abstract
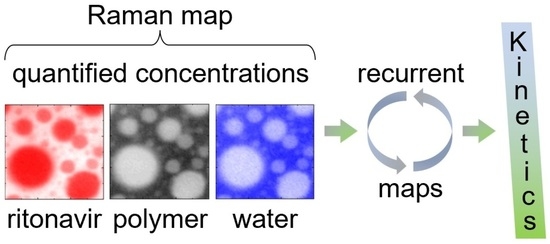
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Ball Milling
2.2.2. Vacuum Compression Molding (VCM)
2.2.3. Gravimetric Water-Sorption Measurements
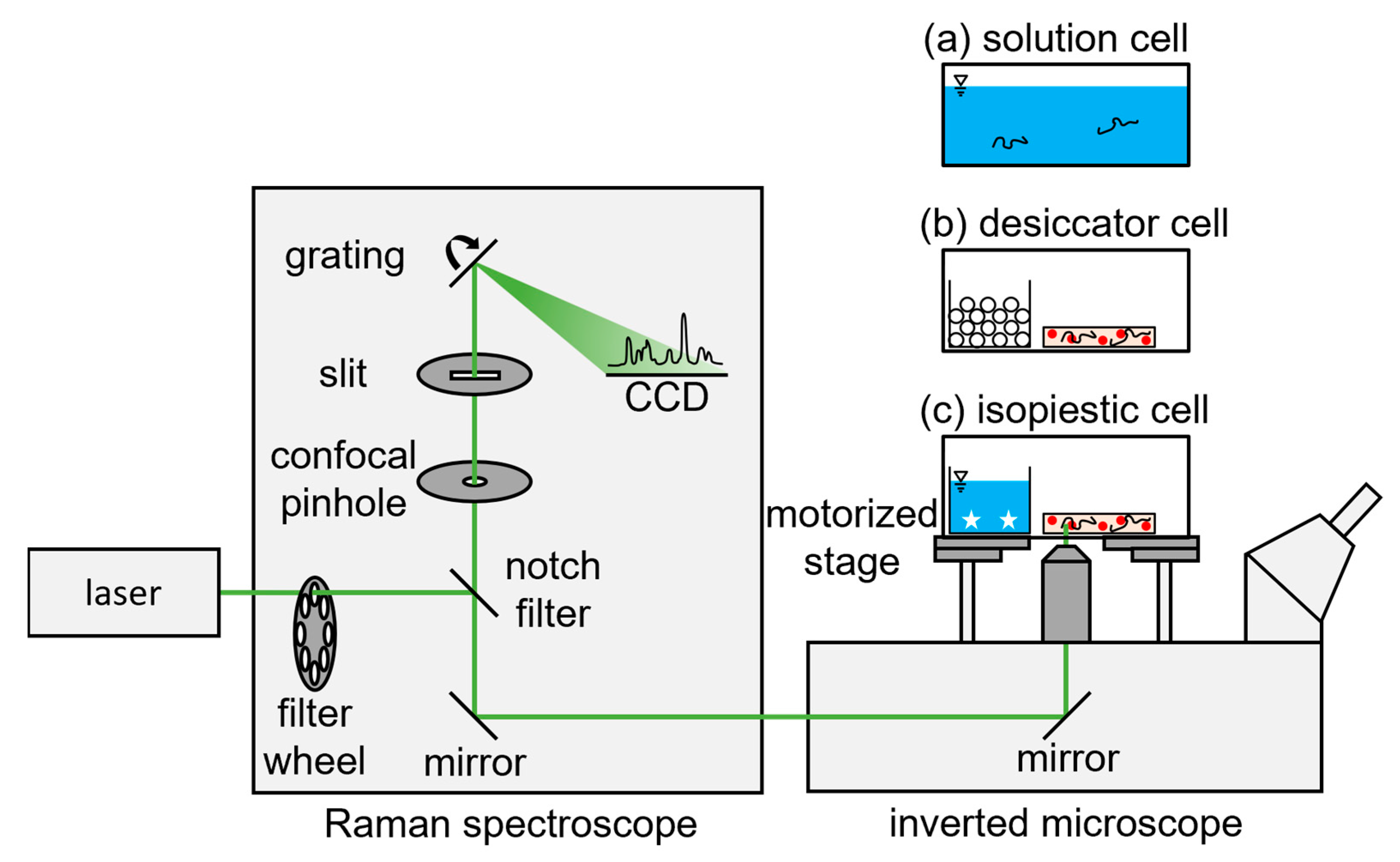
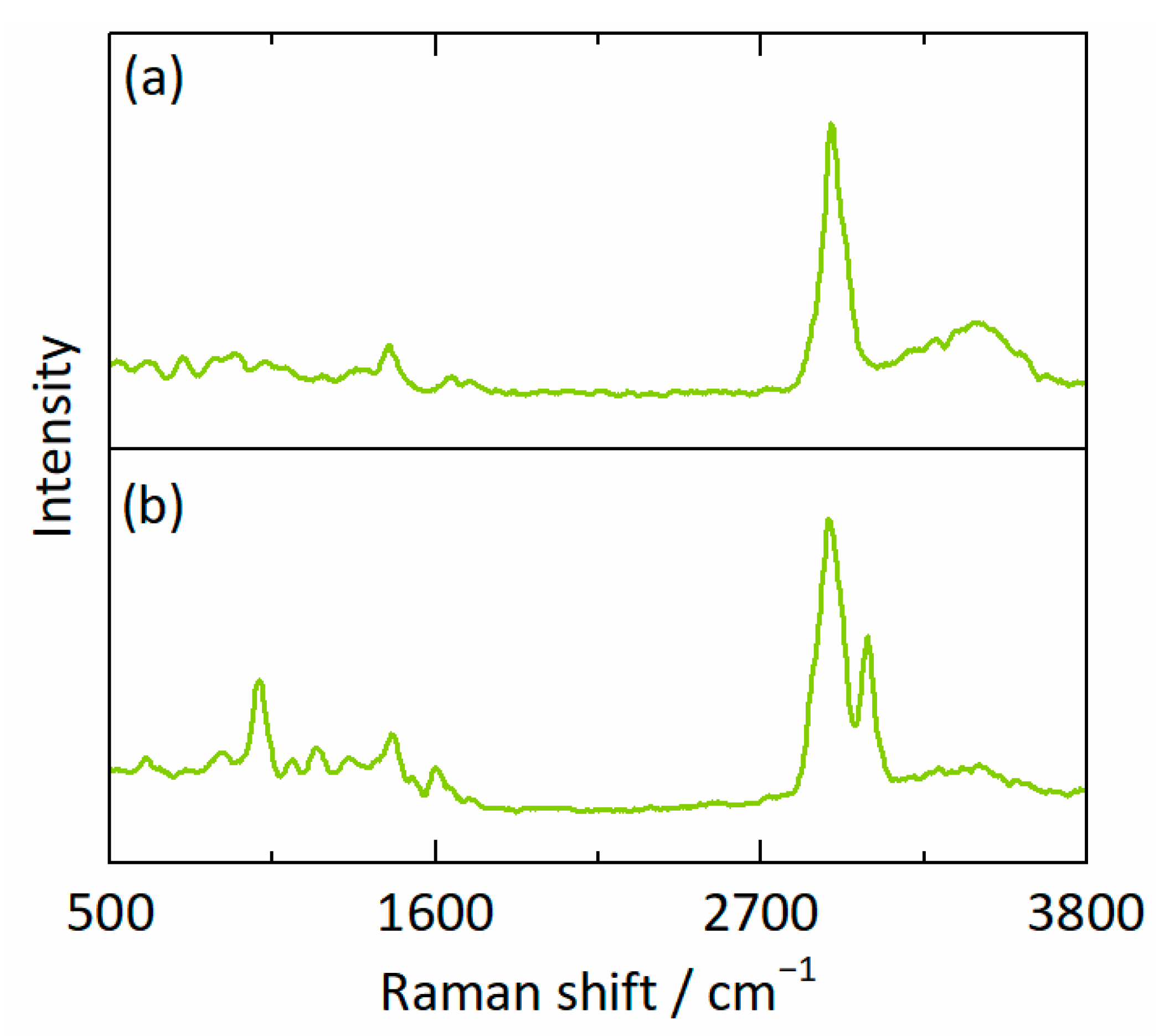
2.2.4. Confocal Raman Spectroscopy
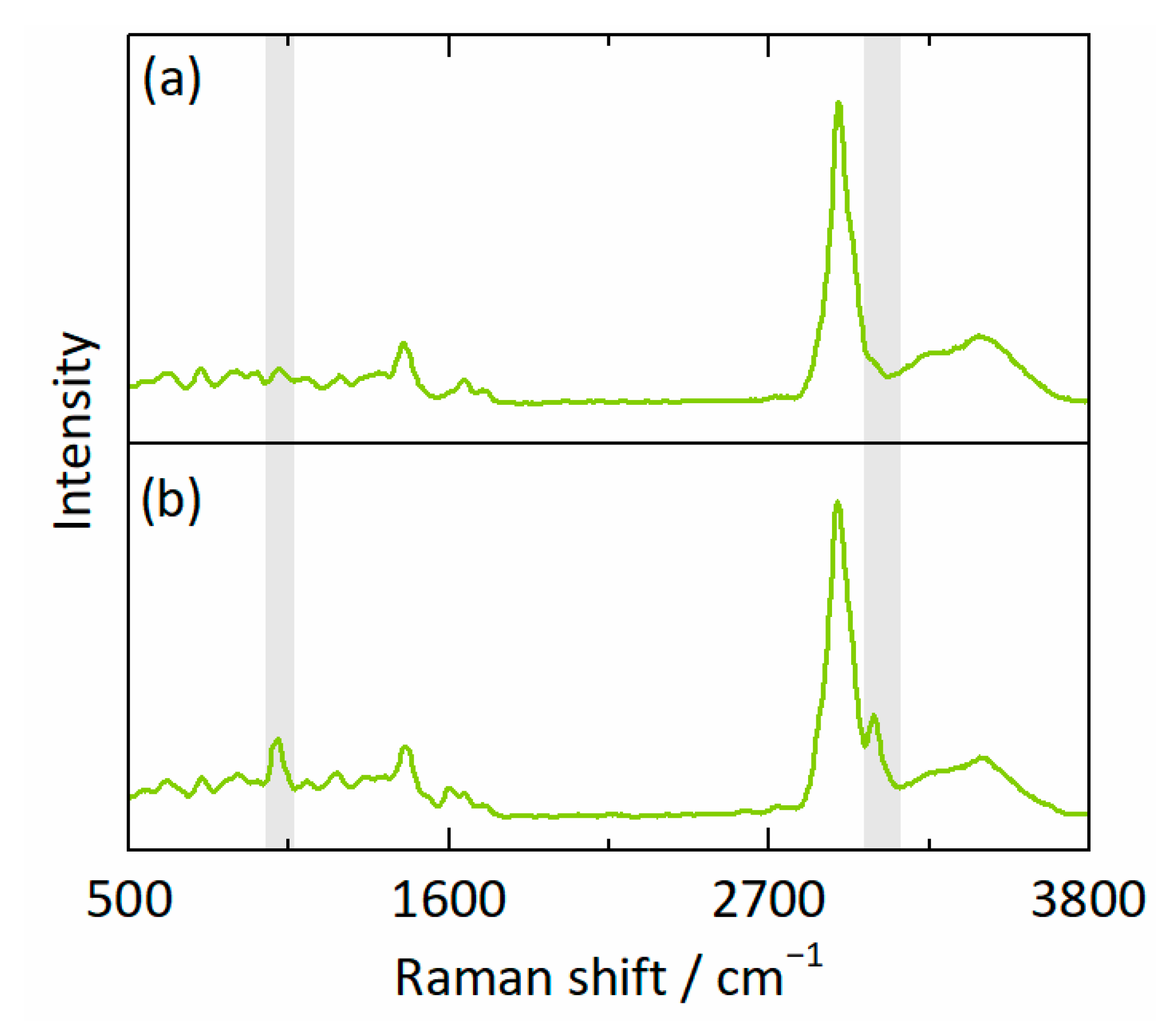
2.2.5. Calibration Model
3. Results and Discussion
3.1. Calibration Model
3.2. Homogeneity of Freshly Prepared ASDs
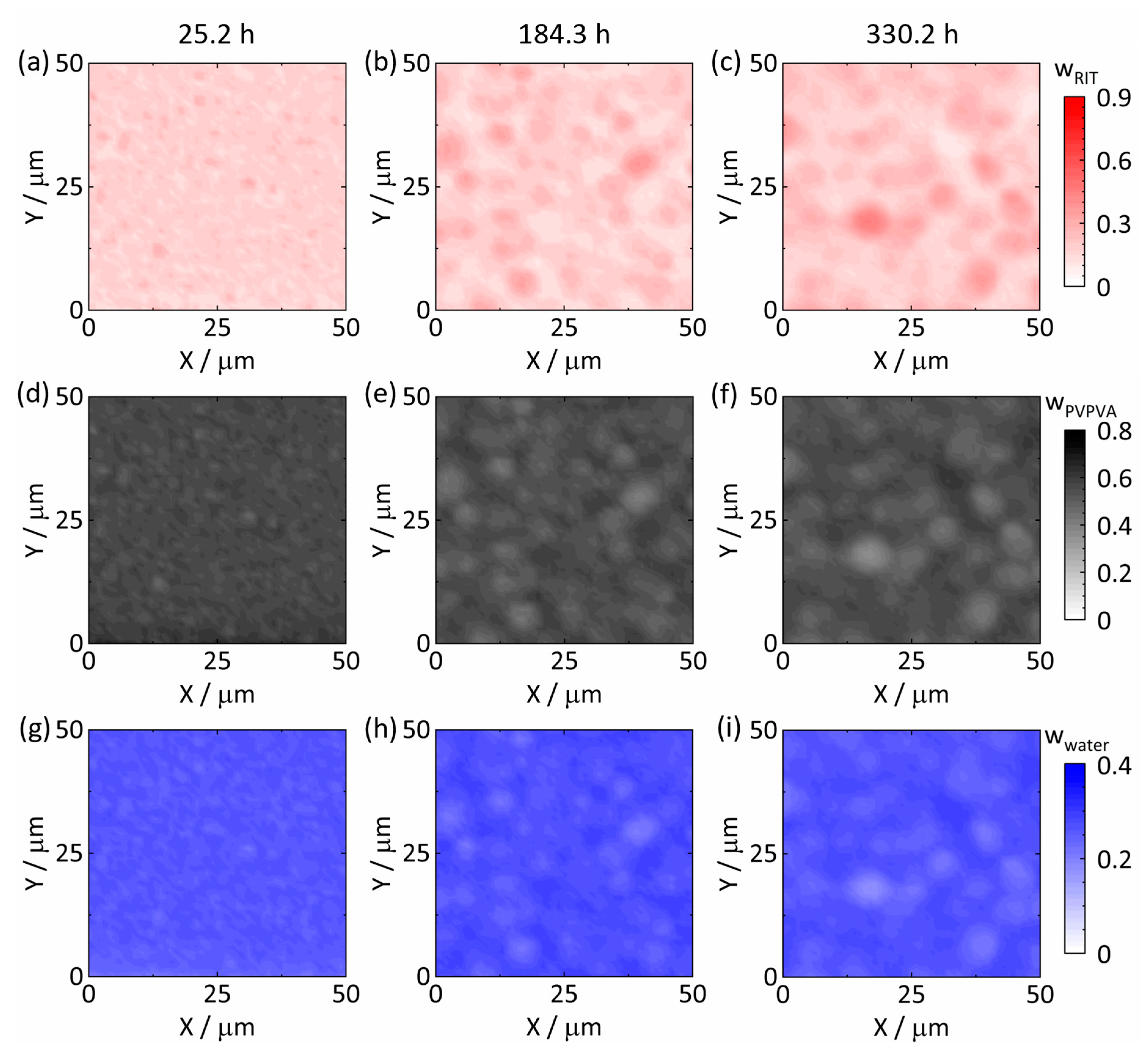
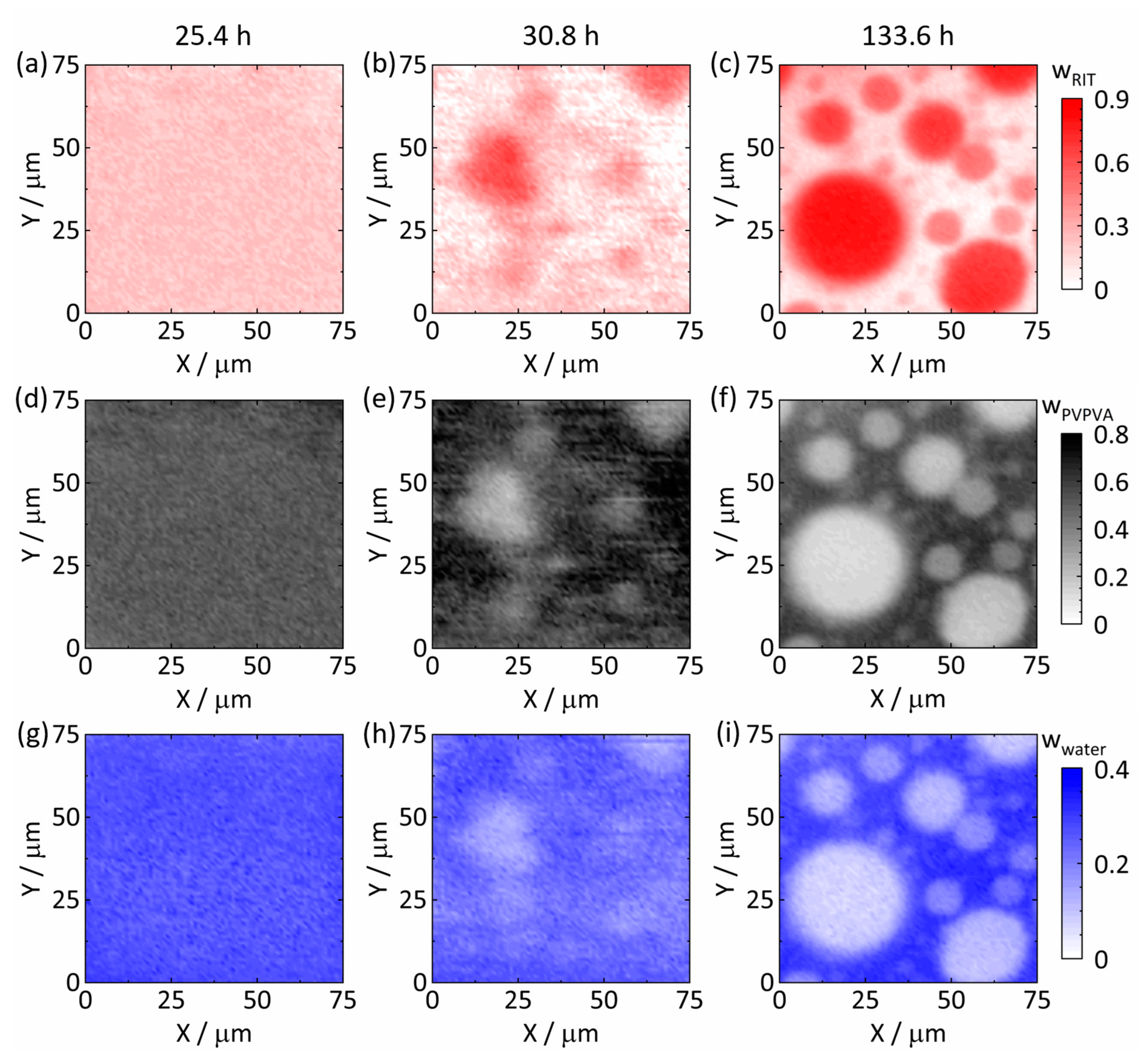
3.3. Raman Mapping of Amorphous Phase Separation
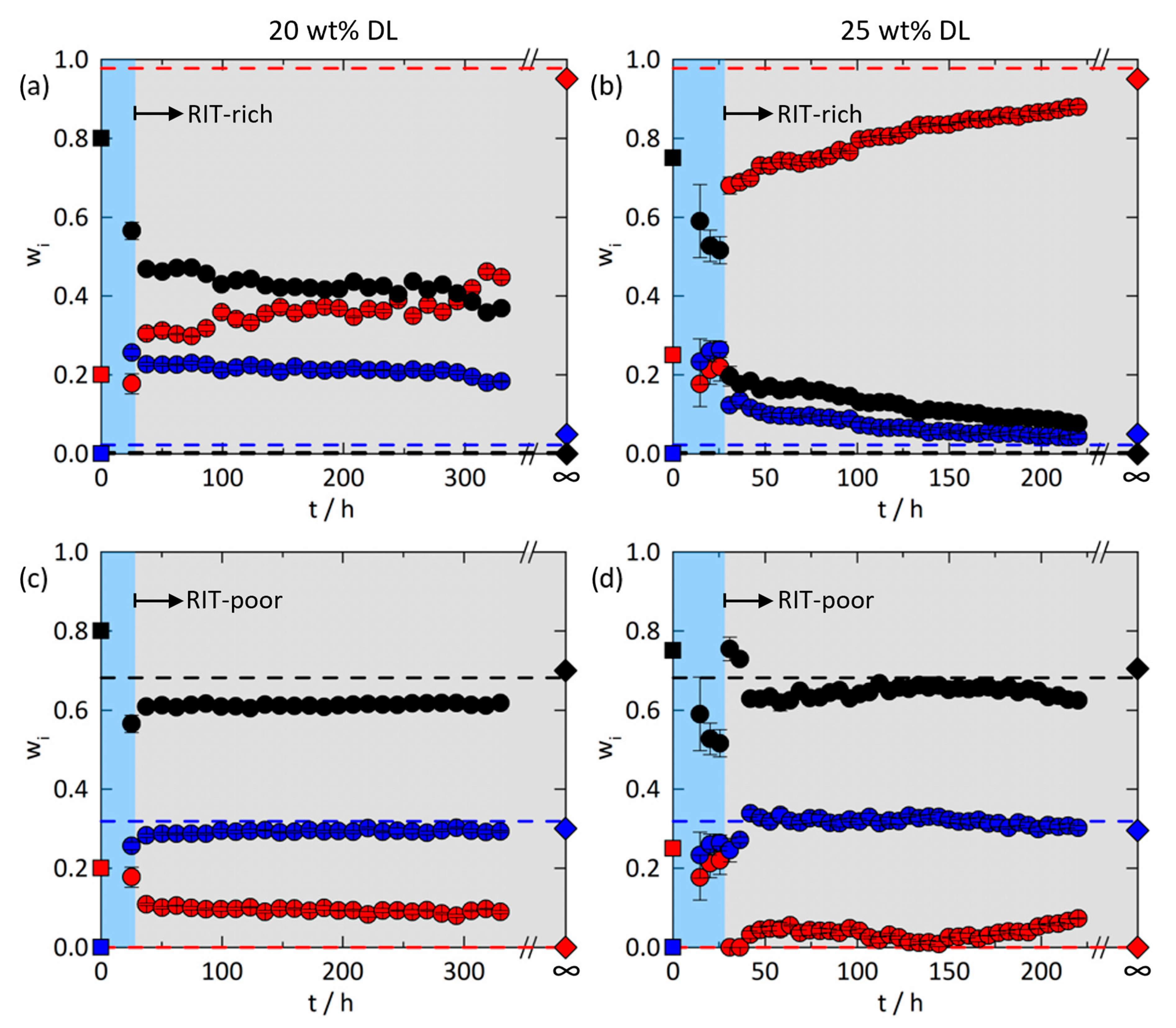
3.3.1. Amorphous Phase Separation in the ASD with 20 wt% DL
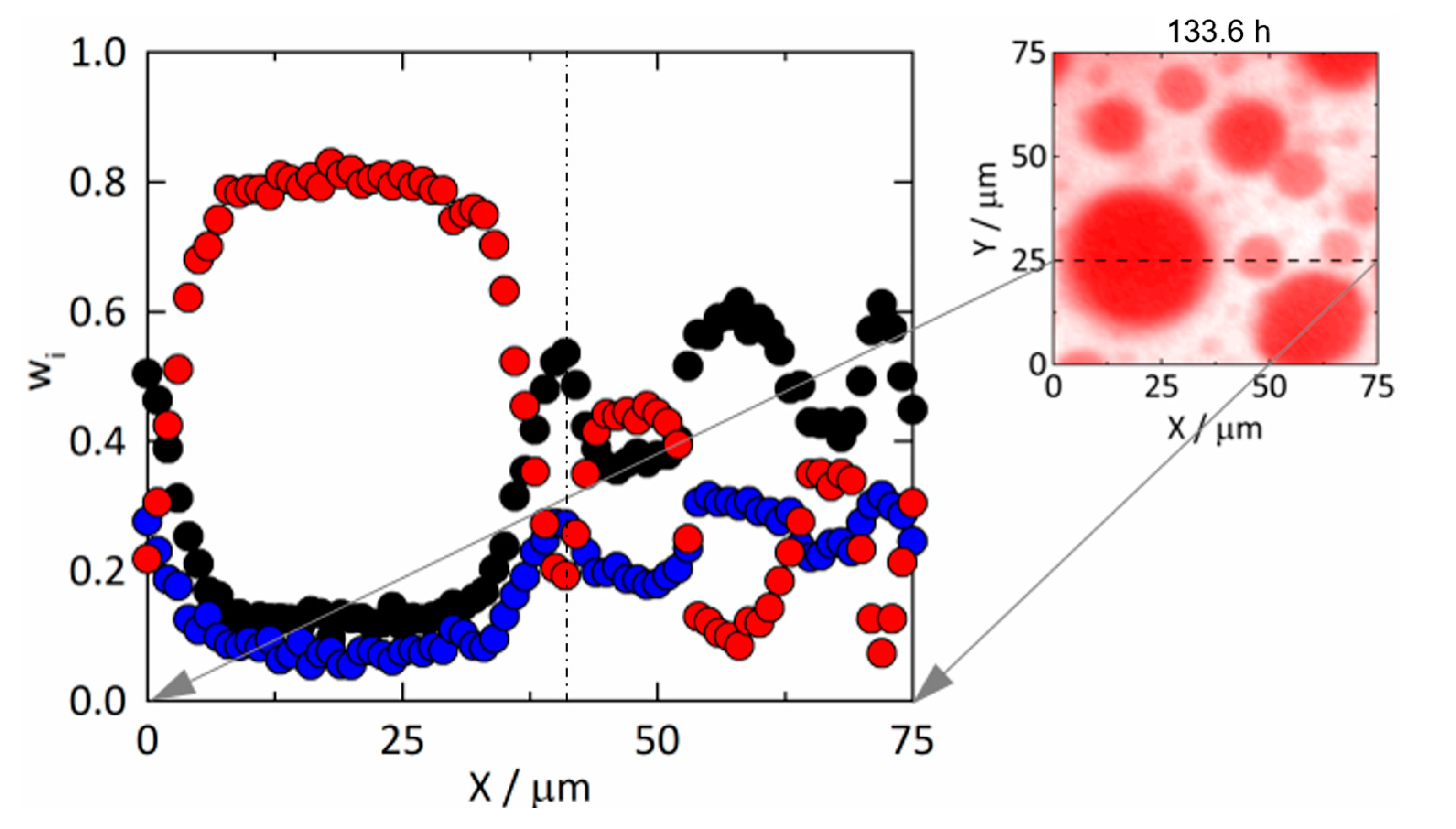
3.3.2. Amorphous Phase Separation in the ASD with 25 wt% DL
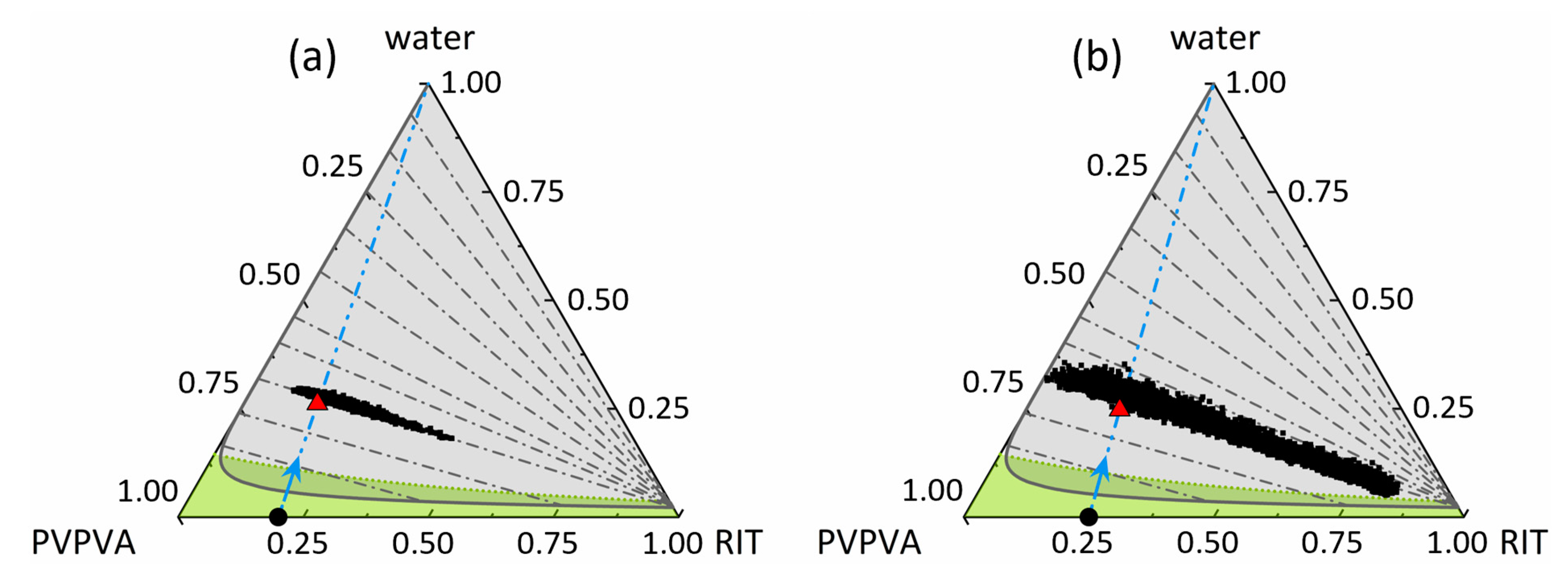
3.4. Amorphous Phase Separation in the Ternary Phase Diagram
3.5. Kinetics of Amorphous Phase Separation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Malick, A.W. (Eds.) Amorphous Solid Dispersions: Theory and Practice; Springer: New York, NY, USA, 2014; ISBN 978-1-4939-1598-9. [Google Scholar]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Amorphous Drugs: Benefits and Challenges, 1st ed.; Springer: Cham, Switzerland, 2018; ISBN 9783319720029. [Google Scholar]
- Luebbert, C.; Huxoll, F.; Sadowski, G. Amorphous-Amorphous Phase Separation in API/Polymer Formulations. Molecules 2017, 22, 296. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP- and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Impact of Polymer Type and Relative Humidity on the Long-Term Physical Stability of Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 4374–4386. [Google Scholar] [CrossRef]
- Wolbert, F.; Nikoleit, K.; Steinbrink, M.; Luebbert, C.; Sadowski, G. The Shelf Life of ASDs: 1. Measuring the Crystallization Kinetics at Humid Conditions. Mol. Pharm. 2022, 19, 2483–2494. [Google Scholar] [CrossRef]
- Luebbert, C.; Wessner, M.; Sadowski, G. Mutual Impact of Phase Separation/Crystallization and Water Sorption in Amorphous Solid Dispersions. Mol. Pharm. 2018, 15, 669–678. [Google Scholar] [CrossRef]
- Newman, A.; Zografi, G. An Examination of Water Vapor Sorption by Multicomponent Crystalline and Amorphous Solids and Its Effects on Their Solid-State Properties. J. Pharm. Sci. 2019, 108, 1061–1080. [Google Scholar] [CrossRef]
- Vasanthavada, M.; Tong, W.-Q.; Joshi, Y.; Kislalioglu, M.S. Phase Behavior of Amorphous Molecular Dispersions II: Role of Hydrogen Bonding in Solid Solubility and Phase Separation Kinetics. Pharm. Res. 2005, 22, 440–448. [Google Scholar] [CrossRef]
- Krummnow, A.; Danzer, A.; Voges, K.; Dohrn, S.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Explaining the Release Mechanism of Ritonavir/PVPVA Amorphous Solid Dispersions. Pharmaceutics 2022, 14, 1904. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Nanoscale Infrared, Thermal, and Mechanical Characterization of Telaprevir-Polymer Miscibility in Amorphous Solid Dispersions Prepared by Solvent Evaporation. Mol. Pharm. 2016, 13, 1123–1136. [Google Scholar] [CrossRef]
- Saboo, S.; Bapat, P.; Moseson, D.E.; Kestur, U.S.; Taylor, L.S. Exploring the Role of Surfactants in Enhancing Drug Release from Amorphous Solid Dispersions at Higher Drug Loadings. Pharmaceutics 2021, 13, 735. [Google Scholar] [CrossRef]
- Purohit, H.S.; Taylor, L.S. Phase Behavior of Ritonavir Amorphous Solid Dispersions during Hydration and Dissolution. Pharm. Res. 2017, 34, 2842–2861. [Google Scholar] [CrossRef]
- Purohit, H.S.; Taylor, L.S. Miscibility of Itraconazole-Hydroxypropyl Methylcellulose Blends: Insights with High Resolution Analytical Methodologies. Mol. Pharm. 2015, 12, 4542–4553. [Google Scholar] [CrossRef] [PubMed]
- Purohit, H.S.; Ormes, J.D.; Saboo, S.; Su, Y.; Lamm, M.S.; Mann, A.K.P.; Taylor, L.S. Insights into Nano- and Micron-Scale Phase Separation in Amorphous Solid Dispersions Using Fluorescence-Based Techniques in Combination with Solid State Nuclear Magnetic Resonance Spectroscopy. Pharm. Res. 2017, 34, 1364–1377. [Google Scholar] [CrossRef]
- Han, Y.R.; Ma, Y.; Lee, P.I. Impact of phase separation morphology on release mechanism of amorphous solid dispersions. Eur. J. Pharm. Sci. 2019, 136, 104955. [Google Scholar] [CrossRef]
- Purohit, H.S.; Taylor, L.S. Phase separation kinetics in amorphous solid dispersions upon exposure to water. Mol. Pharm. 2015, 12, 1623–1635. [Google Scholar] [CrossRef]
- Saboo, S.; Taylor, L.S. Water-induced phase separation of miconazole-poly (vinylpyrrolidone-co-vinyl acetate) amorphous solid dispersions: Insights with confocal fluorescence microscopy. Int. J. Pharm. 2017, 529, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating miscibility and molecular mobility of nifedipine-PVP amorphous solid dispersions using solid-state NMR spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef]
- Rumondor, A.C.F.; Wikström, H.; van Eerdenbrugh, B.; Taylor, L.S. Understanding the tendency of amorphous solid dispersions to undergo amorphous-amorphous phase separation in the presence of absorbed moisture. AAPS PharmSciTech 2011, 12, 1209–1219. [Google Scholar] [CrossRef]
- Li, N.; Gilpin, C.J.; Taylor, L.S. Understanding the Impact of Water on the Miscibility and Microstructure of Amorphous Solid Dispersions: An AFM-LCR and TEM-EDX Study. Mol. Pharm. 2017, 14, 1691–1705. [Google Scholar] [CrossRef]
- Wolbert, F.; Stecker, J.; Luebbert, C.; Sadowski, G. Viscosity of ASDs at humid conditions. Eur. J. Pharm. Biopharm. 2020, 154, 387–396. [Google Scholar] [CrossRef]
- Feng, X.; Vo, A.; Patil, H.; Tiwari, R.V.; Alshetaili, A.S.; Pimparade, M.B.; Repka, M.A. The effects of polymer carrier, hot melt extrusion process and downstream processing parameters on the moisture sorption properties of amorphous solid dispersions. J. Pharm. Pharmacol. 2016, 68, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Raijada, D.; Rantanen, J. Raman spectroscopy in pharmaceutical product design. Adv. Drug Deliv. Rev. 2015, 89, 3–20. [Google Scholar] [CrossRef]
- Tres, F.; Treacher, K.; Booth, J.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Real time Raman imaging to understand dissolution performance of amorphous solid dispersions. J. Control. Release 2014, 188, 53–60. [Google Scholar] [CrossRef]
- Punčochová, K.; Ewing, A.V.; Gajdošová, M.; Pekárek, T.; Beránek, J.; Kazarian, S.G.; Štěpánek, F. The Combined Use of Imaging Approaches to Assess Drug Release from Multicomponent Solid Dispersions. Pharm. Res. 2017, 34, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Kazarian, S.G.; Ewing, A.V. Applications of Fourier transform infrared spectroscopic imaging to tablet dissolution and drug release. Expert Opin. Drug Deliv. 2013, 10, 1207–1221. [Google Scholar] [CrossRef]
- Luebbert, C.; Klanke, C.; Sadowski, G. Investigating phase separation in amorphous solid dispersions via Raman mapping. Int. J. Pharm. 2018, 535, 245–252. [Google Scholar] [CrossRef]
- Shi, C.; Li, L.; Zhang, G.G.Z.; Borchardt, T.B. Direct Visualization of Drug-Polymer Phase Separation in Ritonavir-Copovidone Amorphous Solid Dispersions Using in situ Synchrotron X-ray Fluorescence Imaging of Thin Films. Mol. Pharm. 2019, 16, 4751–4754. [Google Scholar] [CrossRef]
- Baird, J.A.; van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, G.G.Z.; Kjoller, K.; Dillon, E.; Purohit, H.S.; Taylor, L.S. Phase separation in surfactant-containing amorphous solid dispersions: Orthogonal analytical methods to probe the effects of surfactants on morphology and phase composition. Int. J. Pharm. 2022, 619, 121708. [Google Scholar] [CrossRef]
- LaFountaine, J.S.; Jermain, S.V.; Prasad, L.K.; Brough, C.; Miller, D.A.; Lubda, D.; McGinity, J.W.; Williams, R.O. Enabling thermal processing of ritonavir-polyvinyl alcohol amorphous solid dispersions by KinetiSol® Dispersing. Eur. J. Pharm. Biopharm. 2016, 101, 72–81. [Google Scholar] [CrossRef]
- Dohrn, S.; Luebbert, C.; Lehmkemper, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Solvent influence on the phase behavior and glass transition of Amorphous Solid Dispersions. Eur. J. Pharm. Biopharm. 2021, 158, 132–142. [Google Scholar] [CrossRef]
- Greenspan, L. Humidity Fixed Points of Binary Saturated Aqueous Solutions. J. Res. Natl. Bur. Stand. Sect. A 1977, 81A, 89–96. [Google Scholar] [CrossRef]
- Tuschel, D. Why Are the Raman Spectra of Crystalline and Amorphous Solids Different? Spectroscopy 2017, 32, 26–33. [Google Scholar]
- Alsmeyer, F.; Koß, H.-J.; Marquardt, W. Indirect Spectral Hard Modeling for the Analysis of Reactive and Interacting Mixtures. Appl. Spectrosc. AS 2004, 58, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Alsmeyer, F.; Marquardt, W. Automatic Generation of Peak-Shaped Models. Appl. Spectrosc. AS 2004, 58, 986–994. [Google Scholar] [CrossRef]
- Tellinghuisen, J. Inverse vs. classical calibration for small data sets. Fresenius J. Anal. Chem. 2000, 368, 585–588. [Google Scholar] [CrossRef]
- Krutchkoff, R.G. Classical and Inverse Regression Methods of Calibration. Technometrics 1967, 9, 425. [Google Scholar] [CrossRef]
- Kriesten, E.; Alsmeyer, F.; Bardow, A.; Marquardt, W. Fully automated indirect hard modeling of mixture spectra. Chemom. Intell. Lab. Syst. 2008, 91, 181–193. [Google Scholar] [CrossRef]
- Kriesten, E.; Mayer, D.; Alsmeyer, F.; Minnich, C.B.; Greiner, L.; Marquardt, W. Identification of unknown pure component spectra by indirect hard modeling. Chemom. Intell. Lab. Syst. 2008, 93, 108–119. [Google Scholar] [CrossRef]
- Brooks, D.E.; Bamberger, S.B.; Harris, J.M.; Vanalstine, J.; Snyder, R.S. Demixing Kinetics of Phase Separated Polymer Solutions in Microgravity. In Microgravity Science and Applications Flight Programs, January–March 1987, Selected Papers; NASA: Washington, DC, USA, 1988; Volume 1, pp. 119–131. [Google Scholar]
- Sattler, K.; Feindt, H.J. Thermal Separation Processes: Principles and Design; VCH: Weinheim, NY, USA, 1995; ISBN 9783527286225. [Google Scholar]
- Ostwald, W. Lehrbuch der Allgemeinen Chemie, 2nd ed.; Engelmann: Leipzig, Germany, 1896. [Google Scholar]
- Ostwald, W. Studien über die Bildung und Umwandlung fester Körper. Z. Für Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Kerč, J.; Srčić, S. Thermal analysis of glassy pharmaceuticals. Thermochim. Acta 1995, 248, 81–95. [Google Scholar] [CrossRef]
- Floudas, G.; Paluch, M.; Grzybowski, A.; Ngai, K.L. Molecular Dynamics of Glass-Forming Systems: Effects of Pressure; Springer: Berlin/Heidelberg, Germany, 2011; ISBN 9783642049026. [Google Scholar]
- Borrmann, D.; Danzer, A.; Sadowski, G. Predicting the Water Sorption in ASDs. Pharmaceutics 2022, 14, 1181. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krummnow, A.; Danzer, A.; Voges, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Kinetics of Water-Induced Amorphous Phase Separation in Amorphous Solid Dispersions via Raman Mapping. Pharmaceutics 2023, 15, 1395. https://doi.org/10.3390/pharmaceutics15051395
Krummnow A, Danzer A, Voges K, Kyeremateng SO, Degenhardt M, Sadowski G. Kinetics of Water-Induced Amorphous Phase Separation in Amorphous Solid Dispersions via Raman Mapping. Pharmaceutics. 2023; 15(5):1395. https://doi.org/10.3390/pharmaceutics15051395
Chicago/Turabian StyleKrummnow, Adrian, Andreas Danzer, Kristin Voges, Samuel O. Kyeremateng, Matthias Degenhardt, and Gabriele Sadowski. 2023. "Kinetics of Water-Induced Amorphous Phase Separation in Amorphous Solid Dispersions via Raman Mapping" Pharmaceutics 15, no. 5: 1395. https://doi.org/10.3390/pharmaceutics15051395