
Pharmacokinetic Feasibility of Stability-Enhanced Solid-State (SESS) Tenofovir Disoproxil Free Base Crystal


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. LC-MS/MS Analysis of TEV in TEV, SESS-TD Crystal, and TDF Samples
2.4. Rat Plasma Protein Binding of TEV in Rat Plasma Spiked with TEV, the SESS-TD Crystal, or TDF
2.5. TEV Pharmacokinetics after Administration of TEV, SESS-TD Crystal, or TDF in Rats
2.6. TEV Pharmacokinetics after Administration of the SESS-TD Crystal and TDF Stored for 12 Months in Rats
2.7. Pharmacokinetic Parameters
2.8. Statistical Analysis
3. Results
3.1. Rat Plasma Protein Binding of TEV
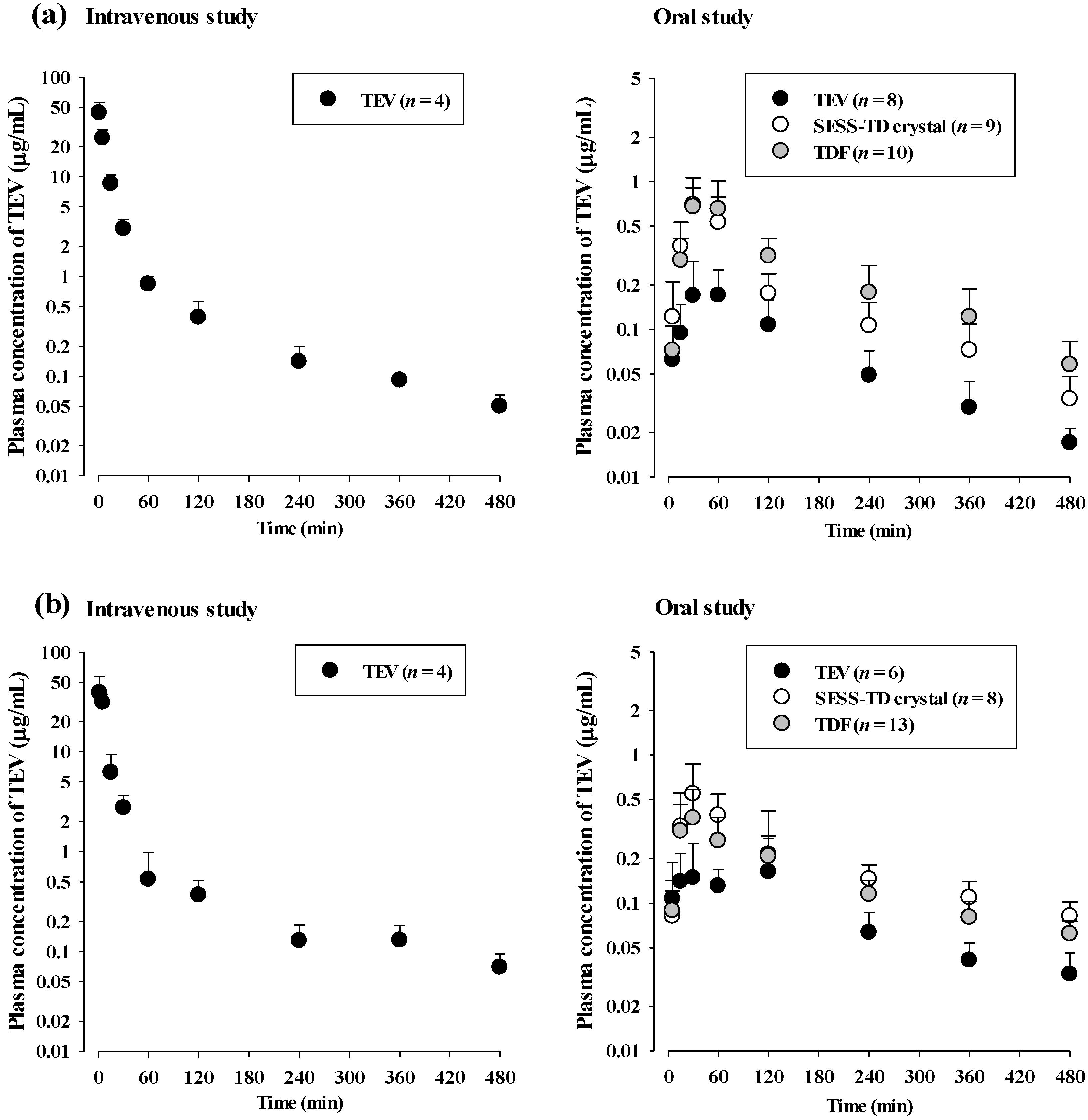
3.2. TEV Pharmacokinetics after Administration of TEV, the SESS-TD Crystal, and TDF in Rats
3.3. TEV Pharmacokinetics after Administration of the SESS-TD Crystal and TDF Stored for 12 Months in Rats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chapman, T.M.; McGavin, J.K.; Noble, S. Tenofovir disoproxil fumarate. Drugs 2003, 63, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Gelder, J.V.; Deferme, S.; Annaert, P.; Naesens, L.; Clercq, E.D.; Mooter, G.V.D.; Kinget, R.; Augustijns, P. Increased absorption of the antiviral ester prodrug tenofovir disoproxil in rat ileum by inhibiting its intestinal metabolism. Drug Metab. Dispos. 2000, 28, 1394–1396. [Google Scholar] [PubMed]
- Munger, J.D.; Rohloff, J.C.; Schultze, L.M.; inventors; Gilead Sciences Inc, assignee. Nucleotide Analog Composition and Synthesis Method. US Patent 5,935,946, 10 August 1999. pp. 935–946. [Google Scholar]
- Gomes, E.C.L.; Mussel, W.N.; Resende, J.M.; Fialho, S.L.; Barbosa, J.; Carignani, E.; Geppi, M.; Yoshida, M.I. Characterization of tenofovir disoproxil fumarate and its behavior under heating. Cryst Growth Des. 2015, 15, 1915–1922. [Google Scholar] [CrossRef]
- Crawford, K.W.; Brown Ripin, D.H.; Levin, A.D.; Campbell, J.R.; Flexner, C. Optimising the manufacture, formulation, and dose of antiretroviral drugs for more cost-efficient delivery in resource-limited settings: A consensus statement. Lancet Infect. Dis. 2012, 12, 550–560. [Google Scholar] [CrossRef]
- Yuan, L.C.; Dahl, T.C.; Oliyai, R. Degradation kinetics of oxycarbonyloxymethyl prodrugs of phosphonates in solution. Pharm. Res. 2001, 18, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.H.; Woo, D.K.; Kang, S.K.; Chon, M.H.; Park, D.J.; Lee, S.K.; Choi, S.J.; inventors; Chong Kun Dang pharmaceutical Corp., assignee. The New Tenofovir Disoproxil Salt and the Preparation Method Thereof. Kr Patent 100,028,790, 11 September 2014. [Google Scholar]
- Watkins, M.E.; Wring, S.; Randolph, R.; Park, S.H.; Powell, K.; Lutz, L.; Nowakowski, M.; Ramabhadran, R.; Domanico, P.L. Development of a novel formulation that improves preclinical bioavailability of tenofovir disoproxil fumarate. J. Pharm. Sci. 2017, 106, 906–919. [Google Scholar] [CrossRef]
- Cundy, K.C.; Sueoka, C.; Lynch, G.R.; Griffin, L.; Lee, W.A.; Shaw, J.P. Pharmacokinetics and bioavailability of the anti-human immunodeficiency virus nucleotide analog 9-[(R)2-(phosphonomethoxy)propyl]adenine (PMPA) in dogs. Antimicrob Agents Chemother. 1998, 42, 687–690. [Google Scholar] [CrossRef]
- Phull, M.S.; Kankan, R.N.; Rao, D.R.; inventors; Cipla Ltd., assignee. Crystalline Form of Tenofovir Disoproxil and a Process for Its Preparation. US Patent 8,519,126 B2, 27 August 2013. [Google Scholar]
- An, J.H.; Kiyonga, A.N.; Yoon, W.; Ryu, H.C.; Kim, J.S.; Kang, C.; Park, M.; Yun, H.; Jung, K. Crystal structure analysis of the first discovered stability-enhanced solid state of tenofovir disoproxil free base using single crystal x-ray diffraction. Molecules 2017, 22, 1182. [Google Scholar] [CrossRef]
- Lee, E.H.; Smith, D.T.; Fanwick, P.E.; Byrn, S.R. Characterization and anisotropic lattice expansion/contraction of polymorphs of tenofovir disoproxil fumarate. Cryst Growth Des. 2010, 10, 2314–2322. [Google Scholar] [CrossRef]
- Cho, J.H.; Choi, Y.K.; Kim, H.S.; Kim, K.S.; Park, J.H.; Woo, J.S. Tenofovir Disoproxil Phosphate, and Pharmaceutical Composition Comprising Non-Metalic Salt Disintegrant and Non-Metalic Salt Lubricant. Kr Patent 100,661,153, 11 January 2016. [Google Scholar]
- Ji, J.H.; Ryu, H.C.; Yang, B.H. Antiviral Compounds in the Form of Solid and Method for Preparing Thereof. Kr Patent 101,548,724, 24 April 2014. [Google Scholar]
- Spinks, C.B.; Zidan, A.S.; Khan, M.A.; Habib, M.J.; Faustino, P.J. Pharmaceutical characterization of novel tenofovir liposomal formulations for enhanced oral drug delivery: In vitro pharmaceutics and Caco-2 permeability investigations. Clin. Pharmacol. 2017, 23, 29–38. [Google Scholar] [CrossRef]
- Cho, J.H.; Choi, H.G. Development of novel tenofovir disoproxil phosphate salt with stability enhancement and bioequivalence to the commercial tenofovir disoproxil fumarate salt in rats and beagle dogs. Int. J. Pharm. 2020, 576, 118957. [Google Scholar] [CrossRef] [PubMed]
- Nye, L.C.; Gray, N.G.; McClure, M.; Boffit, M.; Holmes, E.; Plumb, R.S.; Wilson, I.D. Identification of a novel human circulating metabolite of tenofovir disoproxil fumarate with LC-MS/MS. Bioanalysis 2015, 7, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Mishra, T.; Singhal, P.; Goswami, S.; Shrivastav, P.S. Rapid and specific liquid chromatographic tandem mass spectrometric determination of tenofovir in human plasma and its fragmentation study. J. Chromatogr. Sci. 2009, 47, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Chen, Q.; Chen, Y.; Ji, S.; Wang, Y.; Sun, Y.; Qi, H.; Zhong, K.; Jiang, J.; Chen, X.; et al. A simple and fast LC-MS/MS method for the simultaneous determination of tenofovir alafenamide and tenofovir in human plasma. Biomed. Chromatogr. 2022, 36, e5273. [Google Scholar] [CrossRef]
- You, B.H.; Chin, Y.W.; Kim, H.; Choi, H.S.; Choi, Y.H. Houttuynia cordata extract increased systemic exposure and liver concentrations of metformin through OCTs and MATEs in rats. Phytother. Res. 2018, 32, 1004–1013. [Google Scholar] [CrossRef]
- Han, S.Y.; Chae, H.S.; You, B.H.; Chin, Y.W.; Kim, H.; Choi, H.S.; Choi, Y.H. Lonicera japonica extract increases metformin distribution in the liver without change of systemic exposed metformin in rats. J. Ethnopharmacol. 2019, 238, 111892. [Google Scholar] [CrossRef]
- Hawk, C.T.; Leary, S.L.; Morris, T.H. Formulary for Laboratory Animals, 3rd ed.; Blackwell: Ames, IA, USA, 2005. [Google Scholar]
- Lee, M.G.; Chiou, W.L. Evaluation of potential causes for the incomplete bioavailability of furosemide: Gastric first-pass metabolism. J. Pharmacokinet Biopharm. 1983, 11, 623–640. [Google Scholar] [CrossRef]
- Mallants, R.; Oosterwyck, K.V.; Vaeck, L.V.; Mols, R.; Clercq, E.D.; Augustijns, P. Multidrug resistance-associated protein 2 (MRP2) affects hepatobiliary elimination but not the intestinal disposition of tenofovir disoproxil fumarate and its metabolites. Xenobiotica 2005, 35, 1055–1066. [Google Scholar] [CrossRef]
- Ray, A.S.; Fordyce, M.W.; Hitchcock, M.J.M. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antiviral Res. 2016, 125, 63–70. [Google Scholar] [CrossRef]
- Geboers, S.; Haenen, S.; Mols, R.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Intestinal behavior of the ester prodrug tenofovir DF in humans. Int. J. Pharm. 2015, 485, 131–137. [Google Scholar] [CrossRef]
- Van Gelder, J.; Deferme, S.; Naesens, L.; De Clercq, E.; van den Mooter, G.; Kinget, R.; Augustijns, P. Intestinal absorption enhancement of the ester prodrug tenofovir disoproxil fumarate through modulation of the biochemical barrier by defined ester mixtures. Drug Metab. Dispos. 2002, 30, 924–930. [Google Scholar] [CrossRef]
- Bijsterbosch, M.K.; Duursma, A.M.; Bouma, J.M.W.; Gruber, M. The plasma volume of the wistar rat in relation to the body weight. Experientia 1981, 37, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E.; Deresinski, S. Tenofovir disoproxil fumarate. Clin. Infect. Dis. 2003, 37, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Venter, W.D.F.; Fabian, J.; Feldman, C. An overview of tenofovir and renal disease for the HIV-treating clinician. S. Afr. J. HIV Med. 2018, 19, 817. [Google Scholar] [CrossRef]



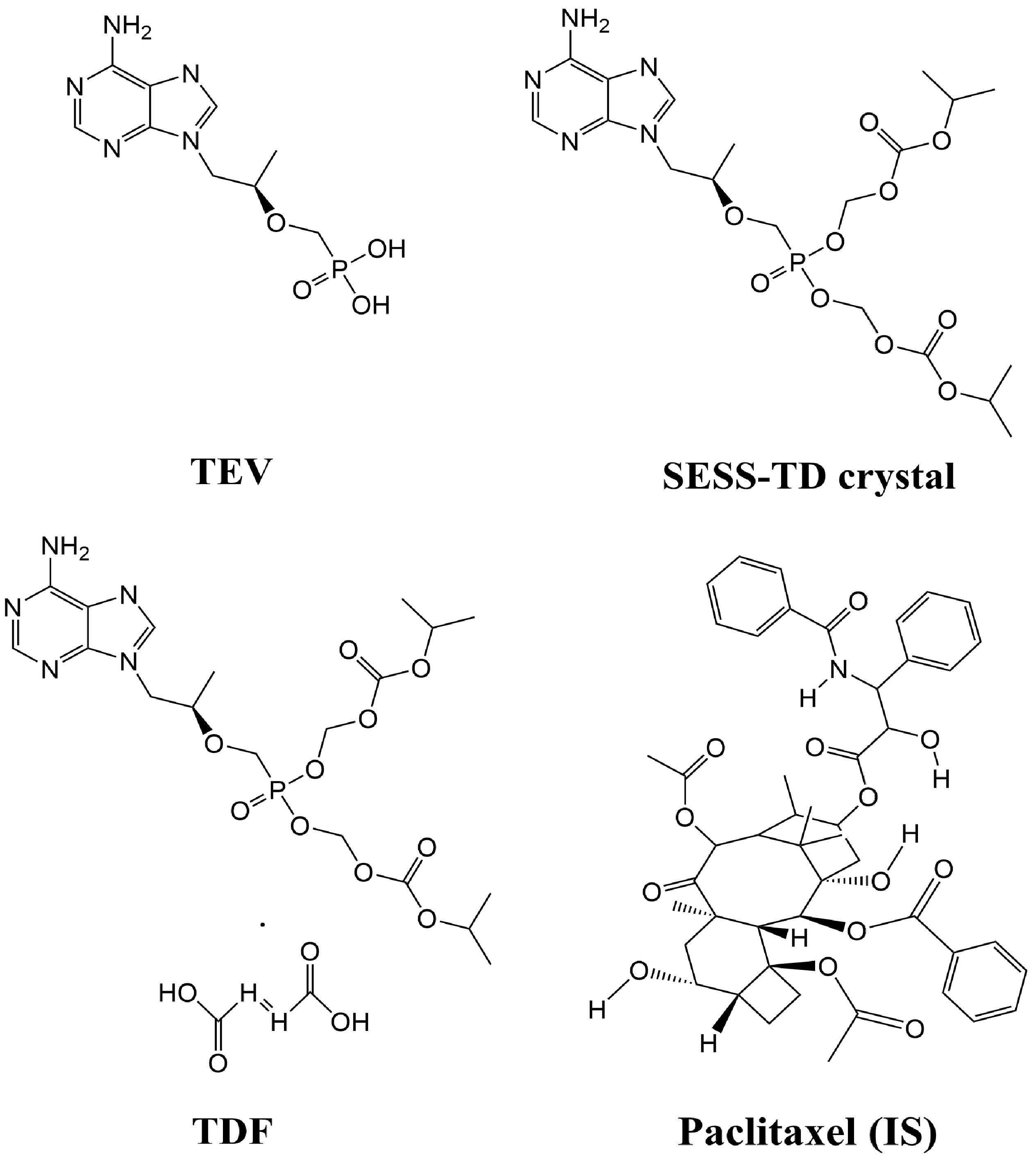{kind=link}
{kind=link}
| TEV | SESS-TD Crystal | TDF | |
|---|---|---|---|
| Chemical formula | C9H14N5O4P [13] | C19H30N5O10P [11,14] | C23H34N5O14P [4] |
| Formula weight (amu) | 287.21 [13] | 519.45 [11] | 635.52 [11] |
| Crystal system | - | Orthorhombic [11] | TD/FA 1:1 salt [11] |
| pKa | 3.75 [15] | - | 3.75 [11] |
| log P | 1.3 [15] | 0.68 [11] | 1.25 [11] |
| Solubility (mg/mL) | |||
| distilled water | 5.20 [15] | 5.88 [14] | 6.20 [14] |
| pH 1.2 | 43.4 [16] | 34.9 [14] | 35.0 [14] |
| pH 4.0 | - | 5.12 [14] | 4.88 [14] |
| pH 6.8 | - | 6.91 [14] | 6.45 [14] |
| Stability (%) | |||
| 15% relative humidity | - | 100 [11] | 99.90 [11] |
| 35% relative humidity | - | 100 [11] | 99.67 [11] |
| 55% relative humidity | - | 99.99 [11] | 99.44 [11] |
| 75% relative humidity | - | 99.98 [11] | 99.21 [11] |
| 95% relative humidity | - | 99.96 [11] | 98.60 [11] |
| Parameters | Intravenous | Parameters | Oral | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TEV (n = 4) | TEV (n = 8) | SESS-TD Crystal (n = 9) | TDF (n = 10) | ||||||||||
| Body weight (g) | 301 | ± | 8.54 | Body weight (g) | 311 | ± | 9.91 | 317 | ± | 20.6 | 319 | ± | 22.6 |
| AUC0–t (μg min/mL) | 554 | ± | 35.4 | AUC0–t (μg min/mL) * | 30.8 | ± | 11.3 | 79.0 | ± | 17.0 | 111 | ± | 34.2 |
| AUC0–inf (μg min/mL) | 564 | ± | 37.6 | AUC0–inf (μg min/mL) * | 34.0 | ± | 11.6 | 87.5 | ± | 19.4 | 127 | ± | 41.7 |
| AUMC0–t (μg min2/mL) | 20,296 | ± | 4017 | AUMC0–t (μg min2/mL) * | 4411 | ± | 2122 | 10,556 | ± | 3265 | 17,554 | ± | 6575 |
| AUMC0-inf (μg min2/mL) | 27,577 | ± | 3980 | AUMC0–inf (μg min2/mL) * | 6264 | ± | 2543 | 16,496 | ± | 6378 | 29,848 | ± | 16,997 |
| t1/2 (min) | 166 | ± | 25.9 | t1/2 (min) | 115 | ± | 20.9 | 153 | ± | 46.3 | 167 | ± | 76.2 |
| Vss (mL/kg) | 1917 | ± | 47.2 | Cmax (μg/mL) * | 0.234 | ± | 0.100 | 0.823 | ± | 0.299 | 0.865 | ± | 0.210 |
| CL (mL/min/kg) | 35.6 | ± | 2.37 | Tmax (min) a | 45 (15–60) | 30 (15–60) | 45 (15–60) | ||||||
| CLR (mL/min/kg) | 23.4 | ± | 1.51 | CL/F (mL/min/kg) * | 667 | ± | 275 | 239 | ± | 55.3 | 177 | ± | 66.6 |
| CLNR (mL/min/kg) | 12.2 | ± | 1.00 | CLoral (mL/min/kg) | 40.3 | ± | 16.6 | 37.1 | ± | 8.58 | 39.6 | ± | 14.9 |
| MRT0-t (min) | 36.5 | ± | 5.85 | MRT0-t (min) | 138 | ± | 27.2 | 134 | ± | 31.2 | 156 | ± | 20.8 |
| MRT0-inf (min) | 48.7 | ± | 3.82 | MRT0-inf (min) | 183 | ± | 37.2 | 188 | ± | 32.1 | 225 | ± | 68.6 |
| Ae0–24h (%) | 65.9 | ± | 1.10 | Ae0–24h (%) | 66.9 | ± | 8.15 | 62.3 | ± | 54.9 | 61.3 | ± | 11.4 |
| GI24h (%) | 7.00 | ± | 3.79 | GI24h (%) * | 30.8 | ± | 8.48 | 9.91 | ± | 4.40 | 10.6 | ± | 5.05 |
| Absolute F (%) | 6.03 | 16.0 | 21.6 | ||||||||||
| Relative F (%) | 266 | 359 | |||||||||||
| Funabs | 30.4 | 8.79 | 9.09 | ||||||||||
| Parameters | Intravenous | Parameters | Oral | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TEV (n = 4) | TEV (n = 6) | SESS-TD Crystal (n = 8) | TDF (n = 13) | ||||||||||
| Body weight (g) | 323 | ± | 28.7 | Body weight (g) | 304 | ± | 25.3 | 312 | ± | 33.5 | 318 | ± | 28.6 |
| AUC0–t (μg min/mL) | 516 | ± | 86.8 | AUC0–t (μg min/mL) * | 38.8 | ± | 13.5 | 87.2 | ± | 27.0 | 68.2 | ± | 21.0 |
| AUC0–inf (μg min/mL) | 533 | ± | 84.1 | AUC0–inf (μg min/mL) * | 47.8 | ± | 15.9 | 122 | ± | 35.9 | 94.6 | ± | 23.1 |
| AUMC0–t (μg min2/mL) | 21,127 | ± | 6633 | AUMC0–t (μg min2/mL) * | 6544 | ± | 2332 | 15,305 | ± | 3943 | 11,901 | ± | 2561 |
| AUMC0–inf (μg min2/mL) | 33,997 | ± | 10,787 | AUMC0–inf (μg min2/mL) * | 13,002 | ± | 4764 | 48,469 | ± | 22,049 | 36,955 | ± | 17,401 |
| t1/2 (min) | 170 | ± | 28.8 | t1/2 (min) | 174 | ± | 40.4 | 291 | ± | 97.8 | 281 | ± | 100 |
| Vss (mL/kg) | 2549 | ± | 1275 | Cmax (μg/mL) * | 0.222 | ± | 0.116 | 0.554 | ± | 0.286 | 0.394 | ± | 0.204 |
| CL (mL/min/kg) | 38.2 | ± | 6.18 | Tmax (min) a,+ | 90 (15–120) | 30 (30–90) | 30 (15–120) | ||||||
| CLR (mL/min/kg) | 27.3 | ± | 5.24 | CL/F (mL/min/kg) * | 464 | ± | 167 | 179 | ± | 60.9 | 222 | ± | 48.3 |
| CLNR mL/min/kg) | 10.9 | ± | 2.16 | CLoral (mL/min/kg) | 41.5 | ± | 15.0 | 40.9 | ± | 13.9 | 39.4 | ± | 8.57 |
| MRT0–t (min) | 41.4 | ± | 12.1 | MRT0–t (min) | 169 | ± | 18.9 | 177 | ± | 11.9 | 279 | ± | 26.1 |
| MRT0–inf (min) | 65.1 | ± | 24.5 | MRT0–inf (min) | 272 | ± | 36.1 | 390 | ± | 110 | 389 | ± | 137 |
| Ae0–24 h (%) | 71.4 | ± | 4.29 | Ae0–24 h (%) | 67.7 | ± | 8.40 | 69.6 | ± | 10.3 | 64.7 | ± | 9.91 |
| GI24 h (%) | 3.56 | ± | 2.72 | GI24 h (%) * | 42.5 | ± | 10.1 | 20.1 | ± | 9.20 | 23.6 | ± | 13.7 |
| Absolute F (%) | 8.95 | 22.9 | 17.7 | ||||||||||
| Relative F (%) | 256 | 198 | |||||||||||
| Funabs | 42.2 | 19.3 | 23.0 | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, B.H.; Bae, M.; Han, S.Y.; Jung, J.; Jung, K.; Choi, Y.H. Pharmacokinetic Feasibility of Stability-Enhanced Solid-State (SESS) Tenofovir Disoproxil Free Base Crystal. Pharmaceutics 2023, 15, 1392. https://doi.org/10.3390/pharmaceutics15051392
You BH, Bae M, Han SY, Jung J, Jung K, Choi YH. Pharmacokinetic Feasibility of Stability-Enhanced Solid-State (SESS) Tenofovir Disoproxil Free Base Crystal. Pharmaceutics. 2023; 15(5):1392. https://doi.org/10.3390/pharmaceutics15051392
Chicago/Turabian StyleYou, Byoung Hoon, Mingoo Bae, Seung Yon Han, Jieun Jung, Kiwon Jung, and Young Hee Choi. 2023. "Pharmacokinetic Feasibility of Stability-Enhanced Solid-State (SESS) Tenofovir Disoproxil Free Base Crystal" Pharmaceutics 15, no. 5: 1392. https://doi.org/10.3390/pharmaceutics15051392