
Research Models to Study Ferroptosis’s Impact in Neurodegenerative Diseases
 ,
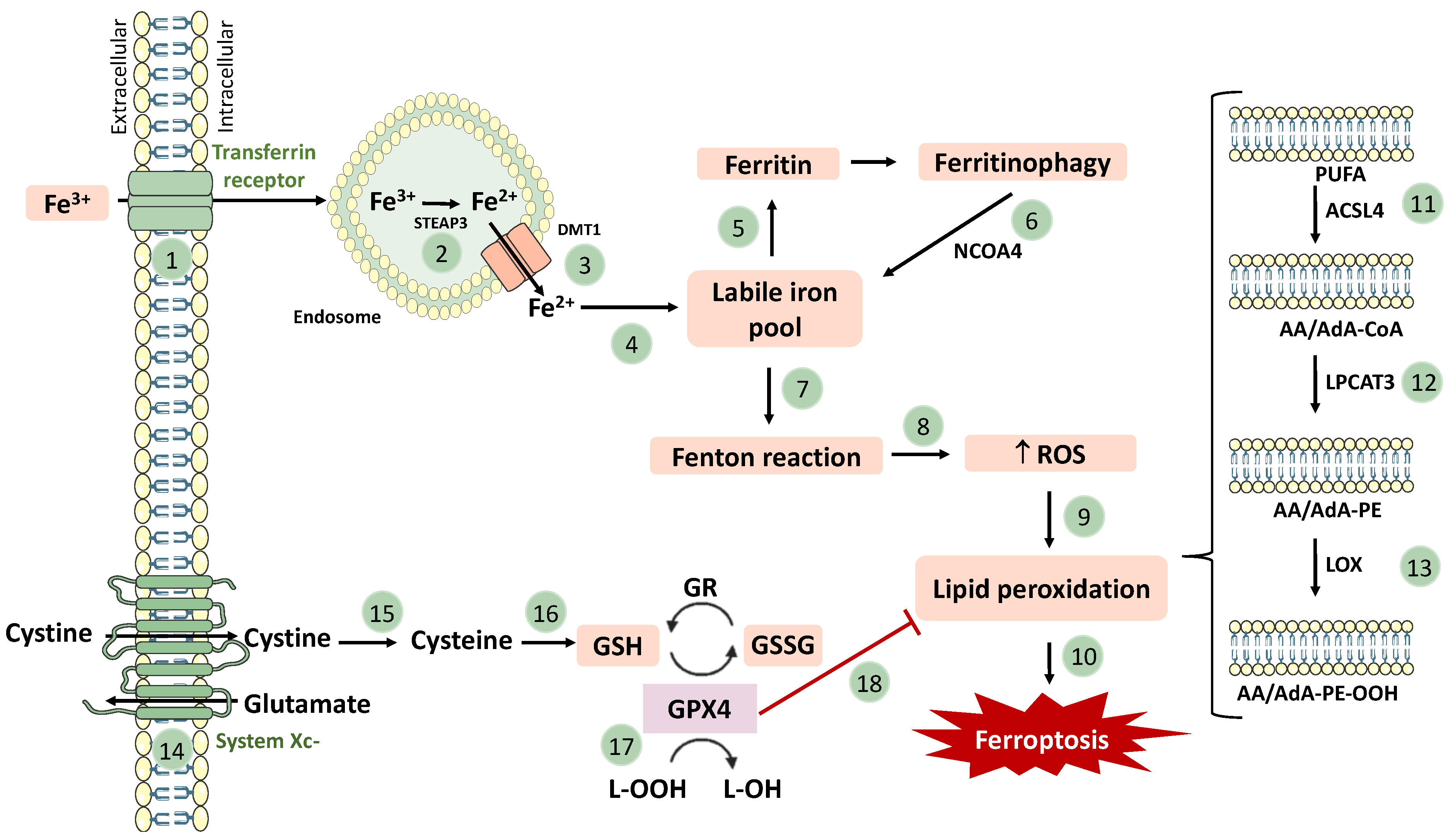
,  , , , , and
, , , , and
Abstract
: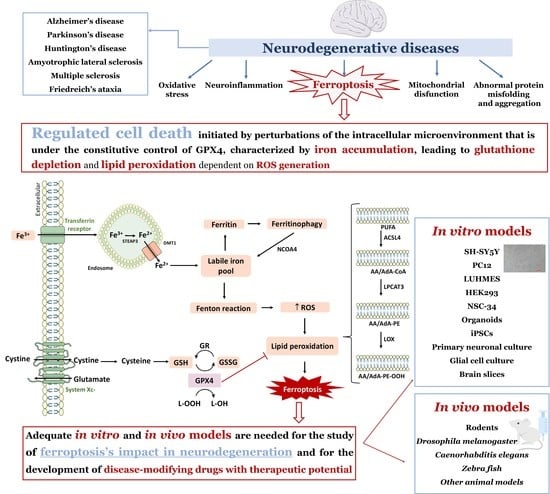
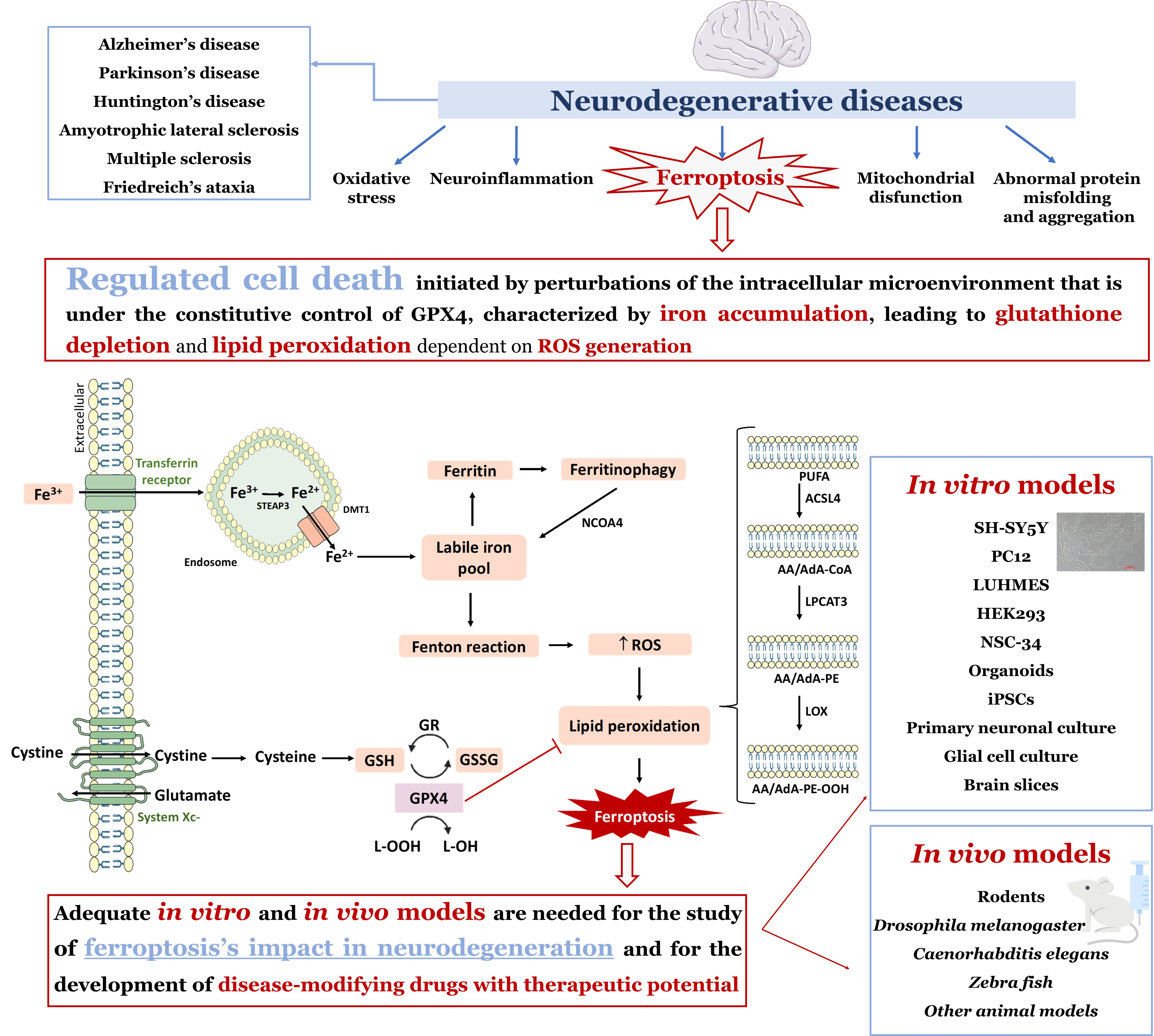
1. Neurodegenerative Diseases—An Overview
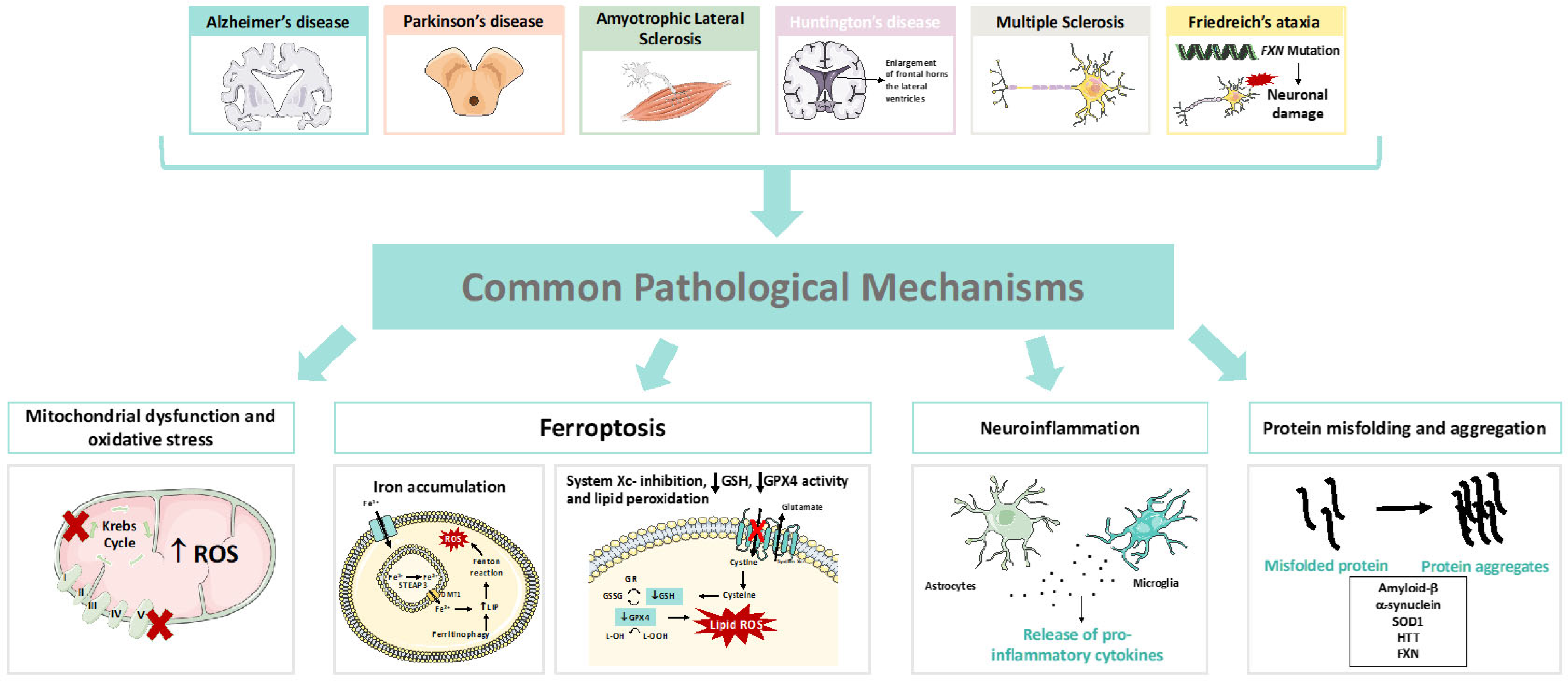
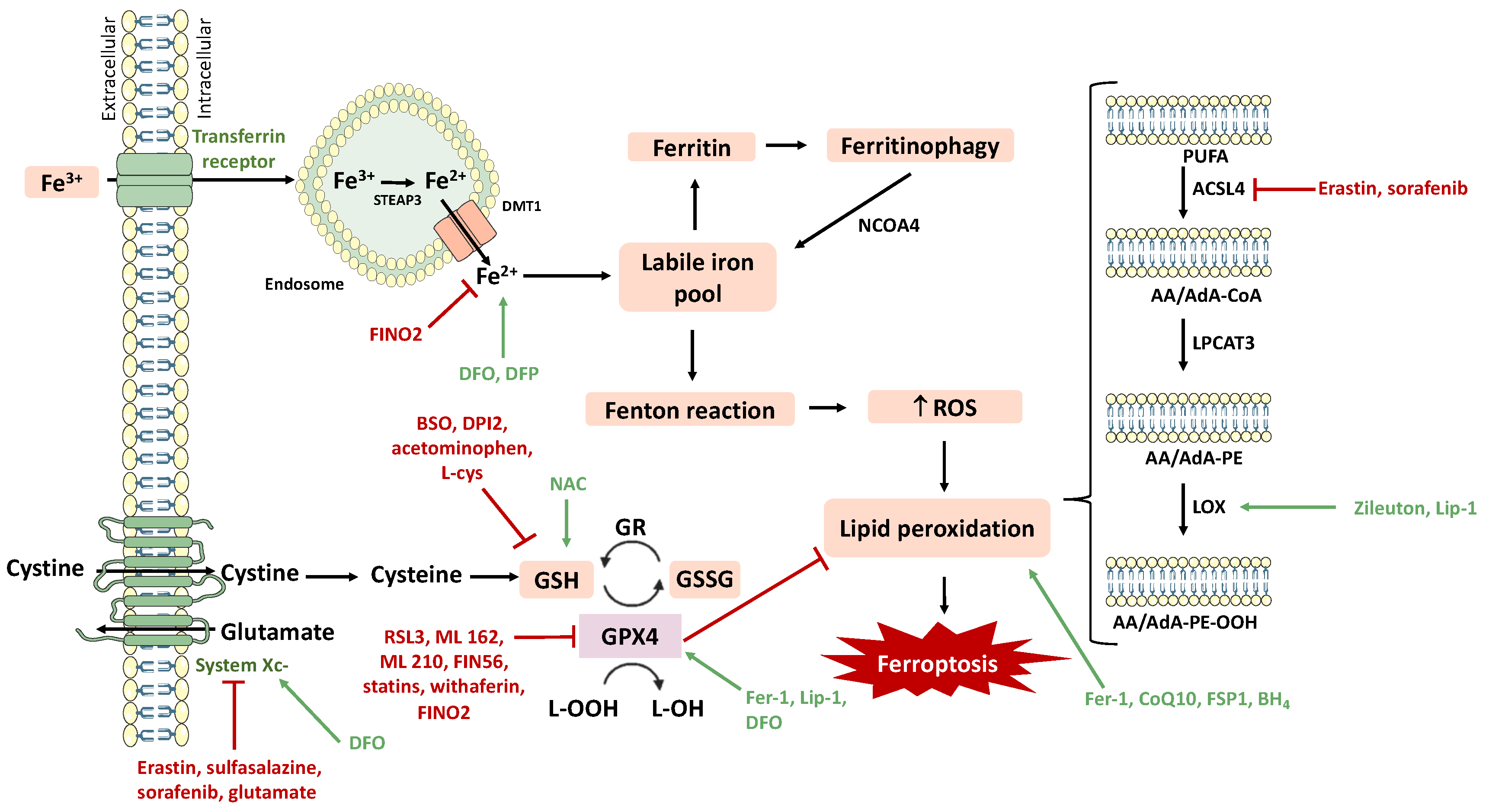
2. Neurodegeneration and Ferroptosis
3. Ferroptosis Inducers and Inhibitors
4. Models to Study Ferroptosis in Neurodegeneration
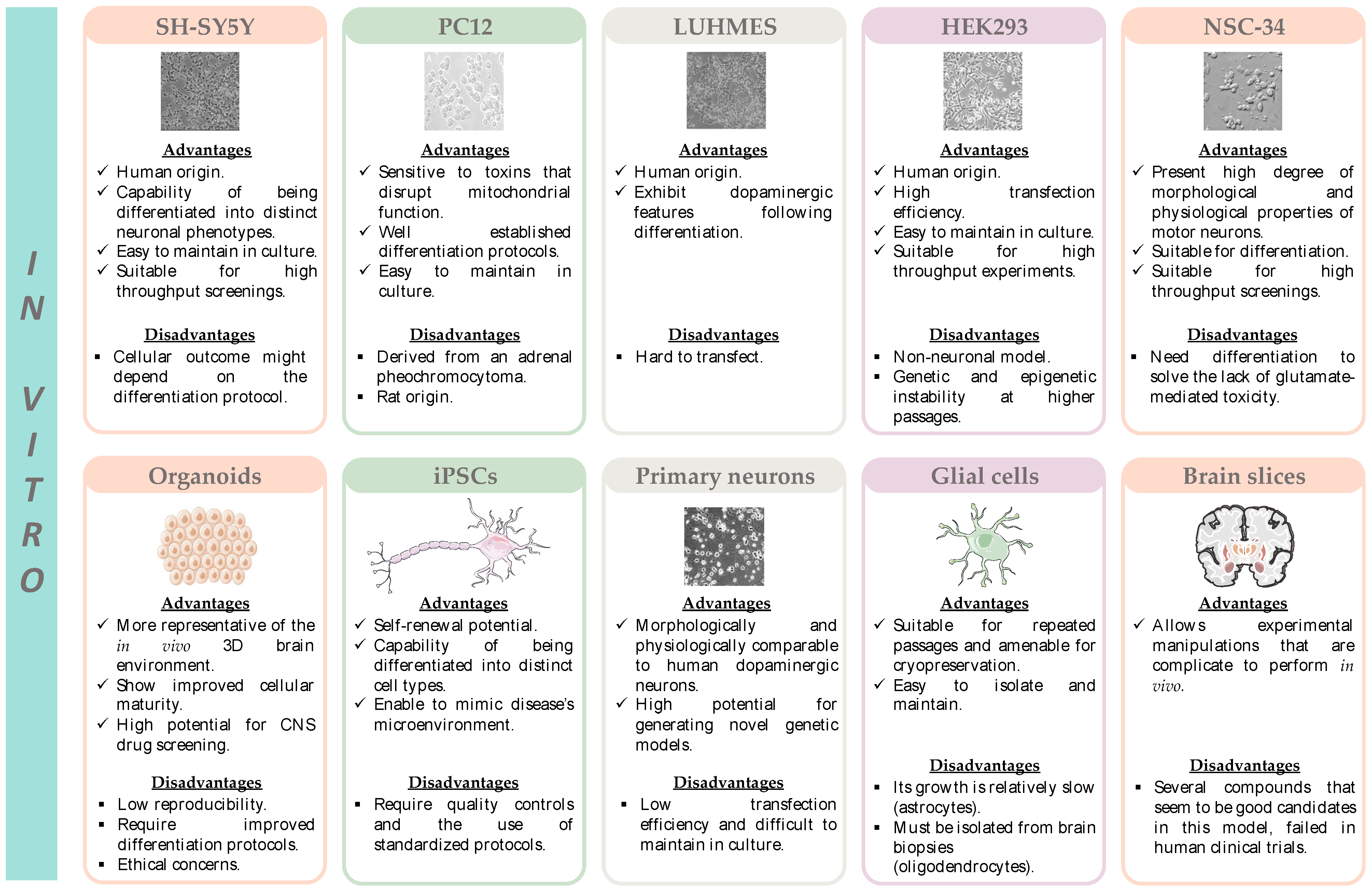
4.1. In Vitro Models
4.1.1. SH-SY5Y Cell Line
4.1.2. PC12 Cell Line
4.1.3. LUHMES Cell Line
4.1.4. HEK293 Cell Line
4.1.5. NSC-34 Cell Line
4.1.6. Organoids
4.1.7. Induced Pluripotent Stem Cell Line
4.1.8. Primary Neuronal Cultures
4.1.9. Glial Cell Culture

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Compounds | Main Conclusions | Reference |
|---|---|---|---|
| SH-SY5Y cells | Aggressor: MPP+ (5 mM, 48 h) Ferroptosis inhibitors: Fer-1 (1–5 μM, 48 h), DFO (100–500 μM, 48 h) and Trolox (100–500 μM, 48 h) | MPP+-induced cell death was blocked by ferroptosis inhibitors. | [100] |
| Aggressor: Erastin (20 μM, 24 h) Ferroptosis inhibitors: Fer-1 (2 μM, 24 h), DFO (100 μM, 24 h) and NAC (20 mM, 24 h) | Increased cell death promoted by erastin, which was reversed by ferroptosis inhibitors. | [101] | |
| Aggressor: ferric ammonium citrate (100–400 μM) Ferroptosis inhibitors: Fer-1, Lip-1 and vitamin E | The aggressor induced an increase in the generation of ROS and in the MDA levels, with a concomitant decrease in the GSH content. The ferroptosis inhibitors counteracted all these effects. | [103] | |
| Aggressor: Erastin (10 μM, 24 h) Ferroptosis inhibitor: Mitochondrial Ferritin | FtMt protected cells against erastin-induced cytotoxicity. | [106] | |
| PC12 cells | Aggressor: t-BHP (100 μM, 1 h) Ferroptosis inhibitors: Fer-1 (1 μM, 24 h) and DFO (100 μM, 24 h) | t-BHP promoted ROS generation and GPX4 decreased expression. These parameters were reversed by ferroptosis inhibitors. | [111] |
| Aggressor: MPP+ (0.4 mM, 24 h) Ferroptosis inhibitor: Fer-1 (2 μM, 30 min before the aggressor) | The aggressor decreased GPX4 expression and reduced GSH levels. The ferroptosis inhibitor attenuated MPP+-induced cytotoxic effects. | [112] | |
| Aggressor: RSL3 (3 μM, 12 h) and 6-OHDA (400 μM, 3 h) Ferroptosis inhibitor: Hino (20, 40 and 60 μM, 4 h) | Hino protected cells against RSL3-induced cell death. Hino protected cells against 6-OHDA-induced cell death and rescued cells from mitochondrial damage. Also, it reversed the increased ROS production and lipid peroxidation promoted by 6-OHDA, and increased GSH, GPX4 and SLC7A11 expression levels. Hino augmented the expression of TfR1 and FPN, and decreased FTH. | [113] | |
| Aggressor: Aβ25-35 (20 μM, 24 h) Ferroptosis inhibitor: senegenin (60 μM, 24 h) and Fer-1 (5 μM, 24 h) | Senegenin attenuated the oxidative damage caused by Aβ25-35 and also affected the expression of ferroptosis-related proteins. | [114] | |
| Aggressor: 6-OHDA (40 µM, 24 h) Ferroptosis inhibitor: Fer-1 (25 µM, 24 h) | 6-OHDA induced ferroptosis, and erastin aggravated the effects. Fer-1 was capable of counteracting 6-OHDA-mediated ferroptosis. | [115] | |
| Aggressor: H2O2 (0.6 mM, 24 h), 6-OHDA (1.0 mM, 24 h), Glutamate (100 mM, 24 h) and RSL3 (10 µM, 24 h) Ferroptosis inhibitor: OABL (1, 5 and 10 µM, 24 h) | Co-treatment of PC12 cells with OABL significantly rescued the cell injury caused by the chemical aggressors. | [116] | |
| LUHMES cells | Aggressor: Erastin (1.25 μM, 24 h) Ferroptosis inhibitors: DFO (200 μM, 24 h) | Erastin induced a significant cell death, which was reverted by DFO. | [120] |
| Aggressor: Erastin (2–6 μM) Ferroptosis inhibitors: DFP (100 μM, 1 h) and NAC (10 mM, 1 h) | Erastin was an extremely potent induced of cell death. Ferroptosis inhibitors attenuated the erastin-induced cell death. | [121] | |
| Aggressor: Erastin (12.5 mM, 4 h), RSL3 (0.625 mM, 4 h) and ML210 (0.625 mM, 4 h) Ferroptosis inhibitors: Fer-1 (5 μM, 4 h), deferoxiprone (10 mM, 24 h), DMPS (50 mM, 24 h) and DFP (50 mM, 24 h) | The ferroptosis inducers reduced cell viability and promoted lipid peroxidation, but Fer-1 reverted these effects. DFX, DMPS and DFP protected LUHMES neurons against RSL3-induced cell death. | [122] | |
| HEK293 cells | Aggressor: RSL3 | It was found a positive correlation between LOX overexpression and RSL3-induced ferroptosis, decreasing the LD50 from 6.8 µM in the wild-type cells to 0.6 µM in the 5-LOX overexpressing cells. | [124] |
| NSC-34 cells | Aggressor: RSL3 + ferric citrate (50 μg/mL) Ferroptosis inhibitor: Fer-1 | This combination induced a significant cell death, which was suppressed by Fer-1. | [128] |
| Organoids | Aggressor: Erastin (10 μM, 24 h) Ferroptosis inhibitors: Fer-1 (100 nM, 24 h), DFO (5 μM, 24 h) and SK4 (100 μM, 24 h) | Erastin induced a significant cell death, which was reversed by ferroptosis inhibitors. | [120] |
| iPSCs | Aggressor: RSL3 (0.01 to 10 μM, 24 h) Ferroptosis inhibitors: Vitamin E, Fer-1 (1 μM, 24 h) and DFO (100 μM, 24 h) | A concentration-dependent cell death was detected after exposure to the aggressor. Ferroptosis inhibitors reversed the aggressor-mediated cell death. | [136] |
| Primary neuronal cultures | Aggressor: Erastin (50 μM, 48 h) Ferroptosis inhibitor: DFO (50 μM) | Erastin promoted a decreased cell viability, increased the generation of ROS and downregulated GPX4 and system Xc- expression. All these effects were reversed by DFO. | [57] |
| Glial cell culture | Aggressor: Angiotensin II (10 μM) Ferroptosis inhibitors: Fer-1 (1 or 2 μM) | Angiotensin II caused an overproduction of ROS, a reduction in GSH levels, and the downregulation of GPX4, Nrf2 and heme oxygenase, all reverted by Fer-1. | [142] |
| Aggressor: GODs (100 μg/mL, 24 h) Ferroptosis inhibitors: Fer-1 and DFO | GODs promoted ferroptosis, which was reversed by Fer-1 and DFO. | [145] | |
| Aggressor: hemin (10–100 μM, 12 and 24 h) Ferroptosis inhibitors: Fer-1 (2 μM, 12 and 24 h) and DFO (100 μM, 12 and 24 h) | Hemin promoted an increase in ROS and MDA levels and induced the upregulation of genes involved in ferroptosis (such as the SLC7A11 gene). Ferroptosis inhibitors rescued cells from cell death. | [146] |
4.1.10. Brain Slices
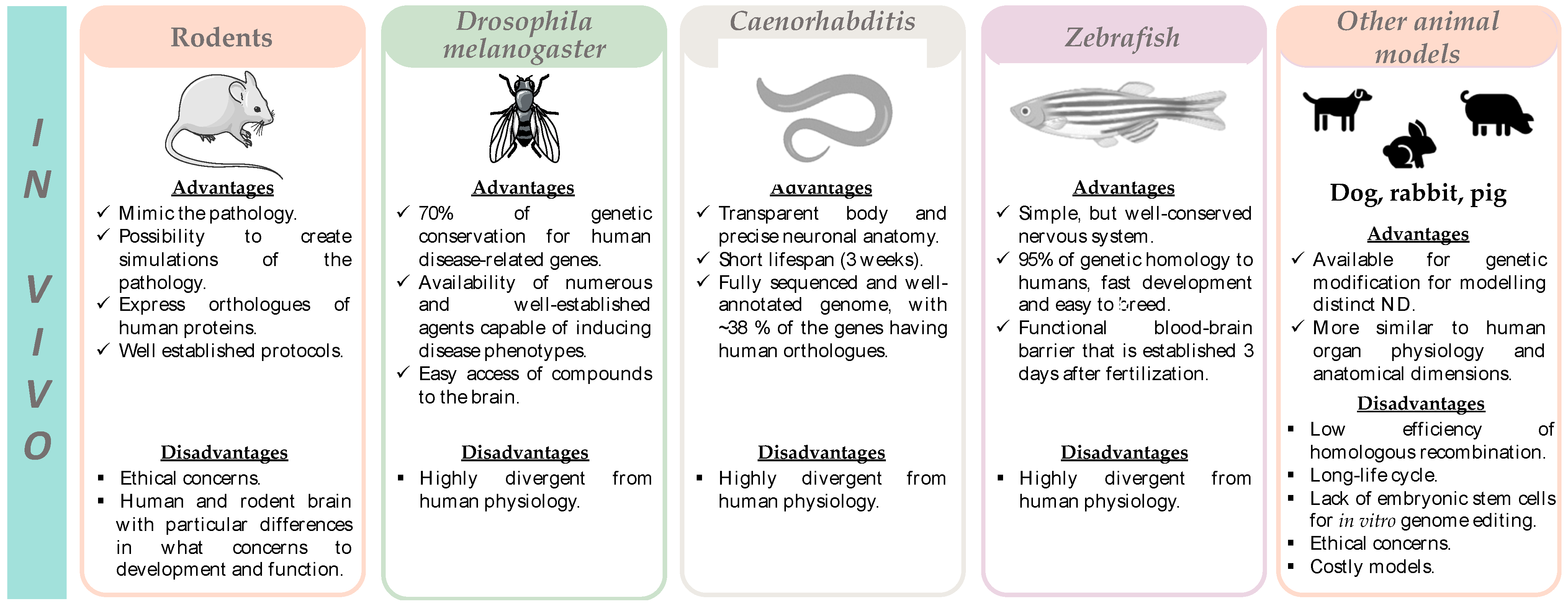
4.2. In Vivo Models
4.2.1. Rodents
4.2.2. Drosophila Melanogaster, Caenorhabditis Elegans, and Zebrafish
4.2.3. Other Animal Models
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef]
- Kovacs, G.G. Molecular Pathology of Neurodegenerative Diseases: Principles and Practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Concepts and Classification of Neurodegenerative Diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimers’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric Symptoms in Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer’s Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The Epidemiology of Parkinson’s Disease: Risk Factors and Prevention. Lancet. Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.-C. Ferroptosis and Its Potential Role in the Physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Moreau, C.; Danel, V.; Devedjian, J.C.; Grolez, G.; Timmerman, K.; Laloux, C.; Petrault, M.; Gouel, F.; Jonneaux, A.; Dutheil, M.; et al. Could Conservative Iron Chelation Lead to Neuroprotection in Amyotrophic Lateral Sclerosis? Antioxid. Redox Signal. 2018, 29, 742–748. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The Epidemiology of Amyotrophic Lateral Sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The Incidence and Prevalence of Huntington’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet. Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Ayala-Peña, S. Role of Oxidative DNA Damage in Mitochondrial Dysfunction and Huntington’s Disease Pathogenesis. Free Radic. Biol. Med. 2013, 62, 102–110. [Google Scholar] [CrossRef]
- Leray, E.; Moreau, T.; Fromont, A.; Edan, G. Epidemiology of Multiple Sclerosis. Rev. Neurol. 2016, 172, 3–13. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of Oligodendrocyte Generation in Multiple Sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; et al. Cyclophilin D Inactivation Protects Axons in Experimental Autoimmune Encephalomyelitis, an Animal Model of Multiple Sclerosis. Proc. Natl. Acad. Sci. USA. 2007, 104, 7558–7563. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Giunti, P. Friedreich’s Ataxia: Clinical Features, Pathogenesis and Management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.; Magrané, J. Mitochondrial Dysfunction in Neurons in Friedreich’s Ataxia. Mol. Cell. Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef]
- Tamarit, J.; Obis, È.; Ros, J. Oxidative Stress and Altered Lipid Metabolism in Friedreich Ataxia. Free Radic. Biol. Med. 2016, 100, 138–146. [Google Scholar] [CrossRef]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv. Ther. 2020, 37, 113–139. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.; de Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium Signaling and Molecular Mechanisms Underlying Neurodegenerative Diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of Gene Expression by Reactive Oxygen. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 67–101. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Scandalios, J.G. Genomic Responses to Oxidative Stress. In Reviews in Cell Biology and Molecular Medicine; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; ISBN 9783527600908. [Google Scholar]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Chakravarti, B.; Chakravarti, D.N. Oxidative Modification of Proteins: Age-Related Changes. Gerontology 2007, 53, 128–139. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Superoxide-Dependent Formation of Hydroxyl Radicals from Ferric-Complexes and Hydrogen Peroxide: An Evaluation of Fourteen Iron Chelators. Free Radic. Res. Commun. 1990, 9, 119–125. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Sheikh, S.; Haque, E.; Mir, S.S. Neurodegenerative Diseases: Multifactorial Conformational Diseases and Their Therapeutic Interventions. J. Neurodegener. Dis. 2013, 2013, 563481. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of Neuroinflammation in Neurodegenerative Diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remião, F.; Silva, R. Molecular Mechanisms of Ferroptosis and Their Involvement in Brain Diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, Y.; Lei, Y.; Liu, T.; Wan, Z.; Meng, H.; Wang, H. Ferroptosis Is Regulated by Mitochondria in Neurodegenerative Diseases. Neurodegener. Dis. 2020, 20, 20–34. [Google Scholar] [CrossRef]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Edwards, J.E.J.; Fu, Y.; Spellberg, B. Deferiprone Iron Chelation as a Novel Therapy for Experimental Mucormycosis. J. Antimicrob. Chemother. 2006, 58, 1070–1073. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco. Targets. Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a Potent Suppressor of Lymphoma Growth by Inhibition of the x(c)-Cystine Transporter: A New Action for an Old Drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System x(c)(-) in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Tan, S.; Sagara, Y.; Liu, Y.; Maher, P.; Schubert, D. The Regulation of Reactive Oxygen Species Production during Programmed Cell Death. J. Cell Biol. 1998, 141, 1423–1432. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, B.-Y.; Pang, Y.-L.; Shen, W.-Y.; Wang, X.; Zhao, C.-X.; Li, W.-X.; Liu, C.; Kong, X.-H.; Ning, G.-Z.; et al. Neuroprotective Effect of Deferoxamine on Erastininduced Ferroptosis in Primary Cortical Neurons. Neural Regen. Res. 2020, 15, 1539–1545. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO(2) Initiates Ferroptosis through GPX4 Inactivation and Iron Oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Liu, C.-X.; Song, R.; Li, Q.-L. Ferrostatin-1 Protects HT-22 Cells from Oxidative Toxicity. Neural Regen. Res. 2020, 15, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 Protects the Mouse Myocardium against Ischemia/Reperfusion Injury by Decreasing VDAC1 Levels and Restoring GPX4 Levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic Networks in Ferroptosis. Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic Studies of the Nrf2-Keap1 Signaling Pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Chen, K.; Jiang, X.; Wu, M.; Cao, X.; Bao, W.; Zhu, L.-Q. Ferroptosis, a Potential Therapeutic Target in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 704298. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Bellinger, M.T.; Seale, L.A.; Takemoto, A.S.; Raman, A.V.; Miki, T.; Manning-Boğ, A.B.; Berry, M.J.; White, L.R.; Ross, G.W. Glutathione Peroxidase 4 Is Associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson’s Brain. Mol. Neurodegener. 2011, 6, 8. [Google Scholar] [CrossRef]
- Dexter, D.; Carter, C.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Lipid Peroxidation as Cause of Nigral Cell Death in Parkinson’s Disease. Lancet 1986, 2, 639–640. [Google Scholar] [CrossRef]
- De Farias, C.C.; Maes, M.; Bonifácio, K.L.; Bortolasci, C.C.; de Souza Nogueira, A.; Brinholi, F.F.; Matsumoto, A.K.; do Nascimento, M.A.; de Melo, L.B.; Nixdorf, S.L.; et al. Highly Specific Changes in Antioxidant Levels and Lipid Peroxidation in Parkinson’s Disease and Its Progression: Disease and Staging Biomarkers and New Drug Targets. Neurosci. Lett. 2016, 617, 66–71. [Google Scholar] [CrossRef]
- Vallerga, C.L.; Zhang, F.; Fowdar, J.; McRae, A.F.; Qi, T.; Nabais, M.F.; Zhang, Q.; Kassam, I.; Henders, A.K.; Wallace, L.; et al. Analysis of DNA Methylation Associates the Cystine–Glutamate Antiporter SLC7A11 with Risk of Parkinson’s Disease. Nat. Commun. 2020, 11, 1238. [Google Scholar] [CrossRef]
- Wu, L.; Liu, M.; Liang, J.; Li, N.; Yang, D.; Cai, J.; Zhang, Y.; He, Y.; Chen, Z.; Ma, T. Ferroptosis as a New Mechanism in Parkinson’s Disease Therapy Using Traditional Chinese Medicine. Front. Pharmacol. 2021, 12, 659584. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Rathore, K.I.; Schulz, K.; Ponka, P.; Arosio, P.; David, S. Dysregulation of Iron Homeostasis in the CNS Contributes to Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2009, 29, 610–619. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased Lipid Peroxidation in Sera of ALS Patients: A Potential Biomarker of Disease Burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of Lipid Peroxidation and Mitochondrial Function Improves Neuropathology in Huntington’s Disease Mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Klepac, N.; Relja, M.; Klepac, R.; Hećimović, S.; Babić, T.; Trkulja, V. Oxidative Stress Parameters in Plasma of Huntington’s Disease Patients, Asymptomatic Huntington’s Disease Gene Carriers and Healthy Subjects: A Cross-Sectional Study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain Mitochondrial Iron Accumulates in Huntington’s Disease, Mediates Mitochondrial Dysfunction, and Can Be Removed Pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-L.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced Expression of the Ferroptosis Inhibitor Glutathione Peroxidase-4 in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; Lenoël, I.; Stys, P.K.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J. Neurosci. 2020, 40, 9327–9341. [Google Scholar] [CrossRef]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in Blood of Patients with Friedreich’s Ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 Induction Prevents Ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef]
- Bradley, J.L.; Homayoun, S.; Hart, P.E.; Schapira, A.H.V.; Cooper, J.M. Role of Oxidative Damage in Friedreich’s Ataxia. Neurochem. Res. 2004, 29, 561–567. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione Peroxidase 4: A New Player in Neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef]
- Reed, T.T. Lipid Peroxidation and Neurodegenerative Disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In Vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple Neurotransmitter Synthesis by Human Neuroblastoma Cell Lines and Clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Adem, A.; Mattsson, M.E.; Nordberg, A.; Påhlman, S. Muscarinic Receptors in Human SH-SY5Y Neuroblastoma Cell Line: Regulation by Phorbol Ester and Retinoic Acid-Induced Differentiation. Brain Res. 1987, 430, 235–242. [Google Scholar] [CrossRef]
- Lopes, F.M.; Schröder, R.; da Frota, M.L.C.J.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between Proliferative and Neuron-like SH-SY5Y Cells as an in Vitro Model for Parkinson Disease Studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V.; Gallego, C.; Comella, J.X. Sequential Treatment of SH-SY5Y Cells with Retinoic Acid and Brain-Derived Neurotrophic Factor Gives Rise to Fully Differentiated, Neurotrophic Factor-Dependent, Human Neuron-like Cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef]
- Xie, H.; Hu, L.; Li, G. SH-SY5Y Human Neuroblastoma Cell Line: In Vitro Cell Model of Dopaminergic Neurons in Parkinson’s Disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar]
- Ito, K.; Eguchi, Y.; Imagawa, Y.; Akai, S.; Mochizuki, H.; Tsujimoto, Y. MPP+ Induces Necrostatin-1- and Ferrostatin-1-Sensitive Necrotic Death of Neuronal SH-SY5Y Cells. Cell Death Discov. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Geng, N.; Shi, B.-J.; Li, S.-L.; Zhong, Z.-Y.; Li, Y.-C.; Xua, W.-L.; Zhou, H.; Cai, J.-H. Knockdown of Ferroportin Accelerates Erastin-Induced Ferroptosis in Neuroblastoma Cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef]
- Sun, Y.; He, L.; Wang, T.; Hua, W.; Qin, H.; Wang, J.; Wang, L.; Gu, W.; Li, T.; Li, N.; et al. Activation of P62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol. Neurobiol. 2020, 57, 4628–4641. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, U.; Banerjee, A.; Chakrabarti, S. Alpha-Synuclein, Ferroptosis and Neurodegeneration: Investigating a Complex Relationship. Alzheimer’s Dement. 2020, 16, e041982. [Google Scholar] [CrossRef]
- Cong, L.; Dong, X.; Wang, Y.; Deng, Y.; Li, B.; Dai, R. On the Role of Synthesized Hydroxylated Chalcones as Dual Functional Amyloid-β Aggregation and Ferroptosis Inhibitors for Potential Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2019, 166, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.R.; Dimayuga, E.R.; Keller, J.N.; Maragos, W.F. Enhanced Toxicity to the Catecholamine Tyramine in Polyglutamine Transfected SH-SY5Y Cells. Neurochem. Res. 2005, 30, 527–531. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Chang, S.-Y.; Wu, Q.; Gou, Y.-J.; Jia, L.; Cui, Y.-M.; Yu, P.; Shi, Z.-H.; Wu, W.-S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Wang, W.-L.; Dai, R.; Yan, H.-W.; Han, C.-N.; Liu, L.-S.; Duan, X.-H. Current Situation of PC12 Cell Use in Neuronal Injury Study. Int. J. Biotechnol. Wellness Ind. 2015, 4, 61–66. [Google Scholar]
- De Los Rios, C.; Cano-Abad, M.F.; Villarroya, M.; López, M.G. Chromaffin Cells as a Model to Evaluate Mechanisms of Cell Death and Neuroprotective Compounds. Pflugers Arch. 2018, 470, 187–198. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Maruta, R.; Takaki, K.; Kotani, E.; Kato, Y.; Yoshimura, R.; Endo, Y.; Whitty, C.; Pernstich, C.; Gandhi, R.; et al. Sustained Neurotrophin Release from Protein Nanoparticles Mediated by Matrix Metalloproteinases Induces the Alignment and Differentiation of Nerve Cells. Biomolecules 2019, 9, 510. [Google Scholar] [CrossRef]
- Wiatrak, B.; Kubis-Kubiak, A.; Piwowar, A.; Barg, E. PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions. Cells 2020, 9, 958. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of Ferroptosis and Mitochondrial Dysfunction by Oxidative Stress in PC12 Cells. Sci. Rep. 2018, 8, 574. [Google Scholar] [CrossRef]
- Bai, L.; Yan, F.; Deng, R.; Gu, R.; Zhang, X.; Bai, J. Thioredoxin-1 Rescues MPP(+)/MPTP-Induced Ferroptosis by Increasing Glutathione Peroxidase 4. Mol. Neurobiol. 2021, 58, 3187–3197. [Google Scholar] [CrossRef]
- Xi, J.; Zhang, Z.; Wang, Z.; Wu, Q.; He, Y.; Xu, Y.; Ding, Z.; Zhao, H.; Da, H.; Zhang, F.; et al. Hinokitiol Functions as a Ferroptosis Inhibitor to Confer Neuroprotection. Free Radic. Biol. Med. 2022, 190, 202–215. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, W.; Li, J.; Qiu, Z.; Wang, X.; Xu, H.; Wang, H.; Lu, D.; Qi, R. Senegenin Rescues PC12 Cells with Oxidative Damage Through Inhibition of Ferroptosis. Mol. Neurobiol. 2022, 59, 6983–6992. [Google Scholar] [CrossRef]
- Huang, Z.; Han, J.; Wu, P.; Wu, C.; Fan, Y.; Zhao, L.; Hao, X.; Chen, D.; Zhu, M. Sorting Nexin 5 Plays an Important Role in Promoting Ferroptosis in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 5463134. [Google Scholar] [CrossRef]
- Tang, J.-J.; Huang, L.-F.; Deng, J.-L.; Wang, Y.-M.; Guo, C.; Peng, X.-N.; Liu, Z.; Gao, J.-M. Cognitive Enhancement and Neuroprotective Effects of OABL, a Sesquiterpene Lactone in 5xFAD Alzheimer’s Disease Mice Model. Redox Biol. 2022, 50, 102229. [Google Scholar] [CrossRef]
- Lotharius, J.; Falsig, J.; van Beek, J.; Payne, S.; Dringen, R.; Brundin, P.; Leist, M. Progressive Degeneration of Human Mesencephalic Neuron-Derived Cells Triggered by Dopamine-Dependent Oxidative Stress Is Dependent on the Mixed-Lineage Kinase Pathway. J. Neurosci. 2005, 25, 6329–6342. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Yin, M.; Zhang, M.-H. Cell-Based Assays for Parkinson’s Disease Using Differentiated Human LUHMES Cells. Acta Pharmacol. Sin. 2014, 35, 945–956. [Google Scholar] [CrossRef]
- Scholz, D.; Pöltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, Complete and Large-Scale Generation of Post-Mitotic Neurons from the Human LUHMES Cell Line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef]
- Gutbier, S.; Kyriakou, S.; Schildknecht, S.; Ückert, A.-K.; Brüll, M.; Lewis, F.; Dickens, D.; Pearson, L.; Elson, J.L.; Michel, S.; et al. Design and Evaluation of Bi-Functional Iron Chelators for Protection of Dopaminergic Neurons from Toxicants. Arch. Toxicol. 2020, 94, 3105–3123. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a Newly Characterized Form of Cell Death in Parkinson’s Disease That Is Regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.-B.; Kim, H.; El Touny, L.; Simeonov, A.; Gerhold, D. LUHMES Dopaminergic Neurons Are Uniquely Susceptible to Ferroptosis. Neurotox. Res. 2022, 40, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.; et al. Genome Dynamics of the Human Embryonic Kidney 293 Lineage in Response to Cell Biology Manipulations. Nat. Commun. 2014, 5, 4767. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Ye, C.; Sun, Y.; Peng, T.; Yang, S.; Wang, W.; Li, H. Mutant Huntingtin Induces Iron Overload via Up-Regulating IRP1 in Huntington’s Disease. Cell Biosci. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A. Toward a Cell Biology of Motor Neurons. In Handbook of Amyotrophic Lateral Sclerosis; Marcel Dekker Incorporated: New York, NY, USA, 1992. [Google Scholar]
- Kanjilal, B.; Keyser, B.M.; Andres, D.K.; Nealley, E.; Benton, B.; Melber, A.A.; Andres, J.F.; Letukas, V.A.; Clark, O.; Ray, R. Differentiated NSC-34 Cells as an in Vitro Cell Model for VX. Toxicol. Mech. Methods 2014, 24, 488–494. [Google Scholar] [CrossRef]
- Martinez, A.M.; Mirkovic, J.; Stanisz, Z.A.; Patwari, F.S.; Yang, W.S. NSC-34 Motor Neuron-like Cells Are Sensitized to Ferroptosis upon Differentiation. FEBS Open Bio 2019, 9, 582–593. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Qian, X.; Song, H.; Ming, G.-L. Brain Organoids: Advances, Applications and Challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef]
- Kelava, I.; Lancaster, M.A. Stem Cell Models of Human Brain Development. Cell Stem Cell 2016, 18, 736–748. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Engineering of Human Brain Organoids with a Functional Vascular-like System. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Jang, J.; Yoo, J.-E.; Lee, J.-A.; Lee, D.R.; Kim, J.Y.; Huh, Y.J.; Kim, D.-S.; Park, C.-Y.; Hwang, D.-Y.; Kim, H.-S.; et al. Disease-Specific Induced Pluripotent Stem Cells: A Platform for Human Disease Modeling and Drug Discovery. Exp. Mol. Med. 2012, 44, 202–213. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha Synuclein Aggregation Drives Ferroptosis: An Interplay of Iron, Calcium and Lipid Peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with IPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Matsuo, T.; Adachi-Tominari, K.; Sano, O.; Kamei, T.; Nogami, M.; Ogi, K.; Okano, H.; Yano, M. Involvement of Ferroptosis in Human Motor Neuron Cell Death. Biochem. Biophys. Res. Commun. 2021, 566, 24–29. [Google Scholar] [CrossRef]
- Weinert, M.; Selvakumar, T.; Tierney, T.S.; Alavian, K.N. Isolation, Culture and Long-Term Maintenance of Primary Mesencephalic Dopaminergic Neurons from Embryonic Rodent Brains. J. Vis. Exp. 2015, 96, 52475. [Google Scholar] [CrossRef]
- Gaven, F.; Marin, P.; Claeysen, S. Primary Culture of Mouse Dopaminergic Neurons. J. Vis. Exp. 2014, 91, e51751. [Google Scholar] [CrossRef]
- Soto-Verdugo, J.; Ortega, A. Critical Involvement of Glial Cells in Manganese Neurotoxicity. Biomed Res. Int. 2021, 2021, 1596185. [Google Scholar] [CrossRef]
- Ravi, K.; Paidas, M.J.; Saad, A.; Jayakumar, A.R. Astrocytes in Rare Neurological Conditions: Morphological and Functional Considerations. J. Comp. Neurol. 2021, 529, 2676–2705. [Google Scholar] [CrossRef]
- Van der Star, B.J.; Vogel, D.Y.S.; Kipp, M.; Puentes, F.; Baker, D.; Amor, S. In Vitro and in Vivo Models of Multiple Sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 570–588. [Google Scholar] [CrossRef]
- Li, S.; Zhou, C.; Zhu, Y.; Chao, Z.; Sheng, Z.; Zhang, Y.; Zhao, Y. Ferrostatin-1 Alleviates Angiotensin II (Ang II)- Induced Inflammation and Ferroptosis in Astrocytes. Int. Immunopharmacol. 2021, 90, 107179. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial Phenotypes and Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Liang, X.; Liu, X.; Li, Y.; Wang, Y.; Kong, L.; Tang, M. Induction of Ferroptosis in Response to Graphene Quantum Dots through Mitochondrial Oxidative Stress in Microglia. Part. Fibre Toxicol. 2020, 17, 30. [Google Scholar] [CrossRef]
- Shen, D.; Wu, W.; Liu, J.; Lan, T.; Xiao, Z.; Gai, K.; Hu, L.; Luo, Z.; Wei, C.; Wang, X.; et al. Ferroptosis in Oligodendrocyte Progenitor Cells Mediates White Matter Injury after Hemorrhagic Stroke. Cell Death Dis. 2022, 13, 259. [Google Scholar] [CrossRef] [PubMed]
- Bert, A.; Gran, B.; Weissert, R. EAE: Imperfect but Useful Models of Multiple Sclerosis. Trends Mol. Med. 2011, 17, 119–125. [Google Scholar] [CrossRef]
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s Disease and Cancer: Two Wars, One Front. Nat. Rev. Cancer 2011, 11, 812–823. [Google Scholar] [CrossRef]
- Gamber, K.M. Animal Models of Parkinson’s Disease: New Models Provide Greater Translational and Predictive Value. Biotechniques 2016, 61, 210–211. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and New Animal Models of Parkinson’s Disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Klivenyi, P.; Vecsei, L. Pharmacological Models of Parkinson’s Disease in Rodents. Methods Mol. Biol. 2011, 793, 211–227. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Hare, D.J.; Duce, J.A.; George, J.L.; Adlard, P.A.; McLean, C.; Rogers, J.T.; Cherny, R.A.; Finkelstein, D.I.; et al. Parkinson’s Disease Iron Deposition Caused by Nitric Oxide-Induced Loss of β-Amyloid Precursor Protein. J. Neurosci. 2015, 35, 3591–3597. [Google Scholar] [CrossRef]
- Anandhan, A.; Chen, W.; Nguyen, N.; Madhavan, L.; Dodson, M.; Zhang, D.D. α-Syn Overexpression, NRF2 Suppression, and Enhanced Ferroptosis Create a Vicious Cycle of Neuronal Loss in Parkinson’s Disease. Free Radic. Biol. Med. 2022, 192, 130–140. [Google Scholar] [CrossRef]
- Puzzo, D.; Gulisano, W.; Palmeri, A.; Arancio, O. Rodent Models for Alzheimer’s Disease Drug Discovery. Expert Opin. Drug Discov. 2015, 10, 703–711. [Google Scholar] [CrossRef]
- De Bem, A.F.; Krolow, R.; Farias, H.R.; de Rezende, V.L.; Gelain, D.P.; Moreira, J.C.F.; Duarte, J.M.d.N.; de Oliveira, J. Animal Models of Metabolic Disorders in the Study of Neurodegenerative Diseases: An Overview. Front. Neurosci. 2021, 14, 1457. [Google Scholar] [CrossRef]
- Di Meco, A.; Lauretti, E.; Vagnozzi, A.N.; Praticò, D. Zileuton Restores Memory Impairments and Reverses Amyloid and Tau Pathology in Aged Alzheimer’s Disease Mice. Neurobiol. Aging 2014, 35, 2458–2464. [Google Scholar] [CrossRef]
- Li, L.; Li, W.-J.; Zheng, X.-R.; Liu, Q.-L.; Du, Q.; Lai, Y.-J.; Liu, S.-Q. Eriodictyol Ameliorates Cognitive Dysfunction in APP/PS1 Mice by Inhibiting Ferroptosis via Vitamin D Receptor-Mediated Nrf2 Activation. Mol. Med. 2022, 28, 11. [Google Scholar] [CrossRef]
- Yang, S.; Xie, Z.; Pei, T.; Zeng, Y.; Xiong, Q.; Wei, H.; Wang, Y.; Cheng, W. Salidroside Attenuates Neuronal Ferroptosis by Activating the Nrf2/HO1 Signaling Pathway in Aβ(1-42)-Induced Alzheimer’s Disease Mice and Glutamate-Injured HT22 Cells. Chin. Med. 2022, 17, 82. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Nitric Oxide Mechanism in the Protective Effect of Antidepressants against 3-Nitropropionic Acid-Induced Cognitive Deficit, Glutathione and Mitochondrial Alterations in Animal Model of Huntington’s Disease. Behav. Pharmacol. 2010, 21, 217–230. [Google Scholar] [CrossRef]
- Chen, L.; Na, R.; Danae McLane, K.; Thompson, C.S.; Gao, J.; Wang, X.; Ran, Q. Overexpression of Ferroptosis Defense Enzyme Gpx4 Retards Motor Neuron Disease of SOD1G93A Mice. Sci. Rep. 2021, 11, 12890. [Google Scholar] [CrossRef]
- Stamoula, E.; Siafis, S.; Dardalas, I.; Ainatzoglou, A.; Matsas, A.; Athanasiadis, T.; Sardeli, C.; Stamoulas, K.; Papazisis, G. Antidepressants on Multiple Sclerosis: A Review of In Vitro and In Vivo Models. Front. Immunol. 2021, 12, 677879. [Google Scholar] [CrossRef]
- Li, X.; Chu, Y.; Ma, R.; Dou, M.; Li, S.; Song, Y.; Lv, Y.; Zhu, L. Ferroptosis as a Mechanism of Oligodendrocyte Loss and Demyelination in Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2022, 373, 577995. [Google Scholar] [CrossRef]
- Morales Pantoja, I.E.; Hu, C.-L.; Perrone-Bizzozero, N.I.; Zheng, J.; Bizzozero, O.A. Nrf2-Dysregulation Correlates with Reduced Synthesis and Low Glutathione Levels in Experimental Autoimmune Encephalomyelitis. J. Neurochem. 2016, 139, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Luo, X.; Zeng, C.; Li, S.; Ma, M.; Wu, Y. Klotho Ameliorated Cognitive Deficits in a Temporal Lobe Epilepsy Rat Model by Inhibiting Ferroptosis. Brain Res. 2021, 1772, 147668. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Zhang, Z.; Zhang, R.; Sun, Q.; Zhang, Z.; Liu, Y.; Ma, B. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Delayed Neurocognitive Recovery in Aged Mice by Inhibiting Hippocampus Ferroptosis via Activating SIRT1/Nrf2/HO-1 Signaling Pathway. Oxid. Med. Cell. Longev. 2022, 2022, 3593294. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis Elegans as a Model to Study Alzheimer’s Disease and Other Neurodegenerative Diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef]
- Bayersdorfer, F.; Voigt, A.; Schneuwly, S.; Botella, J.A. Dopamine-Dependent Neurodegeneration in Drosophila Models of Familial and Sporadic Parkinson’s Disease. Neurobiol. Dis. 2010, 40, 113–119. [Google Scholar] [CrossRef]
- Meredith, G.E.; Sonsalla, P.K.; Chesselet, M.-F. Animal Models of Parkinson’s Disease Progression. Acta Neuropathol. 2008, 115, 385–398. [Google Scholar] [CrossRef]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cochemé, H.M.; et al. Direct Keap1-Nrf2 Disruption as a Potential Therapeutic Target for Alzheimer’s Disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The Role of TDP-43 Propagation in Neurodegenerative Diseases: Integrating Insights from Clinical and Experimental Studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Cha, S.J.; Han, Y.J.; Choi, H.-J.; Kim, H.-J.; Kim, K. Glutathione S-Transferase Rescues Motor Neuronal Toxicity in Fly Model of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 615. [Google Scholar] [CrossRef]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin Modulates Iron Toxicity in a Drosophila Model of Friedreich’s Ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in Ferrous Iron and Glutathione Promote Ferroptosis and Frailty in Aging Caenorhabditis Elegans. Elife 2020, 9, e56580. [Google Scholar] [CrossRef]
- Vázquez-Manrique, R.P.; González-Cabo, P.; Ros, S.; Aziz, H.; Baylis, H.A.; Palau, F. Reduction of Caenorhabditis Elegans Frataxin Increases Sensitivity to Oxidative Stress, Reduces Lifespan, and Causes Lethality in a Mitochondrial Complex II Mutant. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 172–174. [Google Scholar] [CrossRef]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e4. [Google Scholar] [CrossRef]
- Perez, M.A.; Clostio, A.J.; Houston, I.R.; Ruiz, J.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Ether Lipid Deficiency Disrupts Lipid Homeostasis Leading to Ferroptosis Sensitivity. PLoS Genet. 2022, 18, e1010436. [Google Scholar] [CrossRef]
- Bandmann, O.; Burton, E.A. Genetic Zebrafish Models of Neurodegenerative Diseases. Neurobiol. Dis. 2010, 40, 58–65. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 Mediated Pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Da Costa, M.M.J.; Allen, C.E.; Higginbottom, A.; Ramesh, T.; Shaw, P.J.; McDermott, C.J. A New Zebrafish Model Produced by TILLING of SOD1-Related Amyotrophic Lateral Sclerosis Replicates Key Features of the Disease and Represents a Tool for in Vivo Therapeutic Screening. Dis. Model. Mech. 2014, 7, 73–81. [Google Scholar] [CrossRef]
- Yang, W.; Chen, X.; Li, S.; Li, X.-J. Genetically Modified Large Animal Models for Investigating Neurodegenerative Diseases. Cell Biosci. 2021, 11, 218. [Google Scholar] [CrossRef]
- Yin, P.; Li, S.; Li, X.-J.; Yang, W. New Pathogenic Insights from Large Animal Models of Neurodegenerative Diseases. Protein Cell 2022, 13, 707–720. [Google Scholar] [CrossRef]
- Rofina, J.E.; Singh, K.; Skoumalova-Vesela, A.; van Ederen, A.M.; van Asten, A.J.A.M.; Wilhelm, J.; Gruys, E. Histochemical Accumulation of Oxidative Damage Products Is Associated with Alzheimer-like Pathology in the Canine. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2004, 11, 90–100. [Google Scholar] [CrossRef]
- Skoumalova, A.; Rofina, J.; Schwippelova, Z.; Gruys, E.; Wilhelm, J. The Role of Free Radicals in Canine Counterpart of Senile Dementia of the Alzheimer Type. Exp. Gerontol. 2003, 38, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Ghribi, O.; Herman, M.M.; Pramoonjago, P.; Savory, J. MPP+ Induces the Endoplasmic Reticulum Stress Response in Rabbit Brain Involving Activation of the ATF-6 and NF-KappaB Signaling Pathways. J. Neuropathol. Exp. Neurol. 2003, 62, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.M.; Bristot, I.J.; da Motta, L.L.; Parsons, R.B.; Klamt, F. Mimicking Parkinson’s Disease in a Dish: Merits and Pitfalls of the Most Commonly Used Dopaminergic In Vitro Models. Neuromolecular Med. 2017, 19, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, D.F.; Pavlou, M.A.S.; Outeiro, T.F. Cellular Models as Tools for the Study of the Role of Alpha-Synuclein in Parkinson’s Disease. Exp. Neurol. 2017, 298, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, J.C.M.; Saliba, S.W.; Oliveira, A.C.P. de Studying Neurodegenerative Diseases in Culture Models. Rev. Bras. Psiquiatr. 2013, 35 (Suppl. 2), S92–S100. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Schulz, J.B. Limitations of Cellular Models in Parkinson’s Disease Research. J. Neural Transm. Suppl. 2006, 70, 261–268. [Google Scholar] [CrossRef]
- Madji Hounoum, B.; Vourc’h, P.; Felix, R.; Corcia, P.; Patin, F.; Guéguinou, M.; Potier-Cartereau, M.; Vandier, C.; Raoul, C.; Andres, C.R.; et al. NSC-34 Motor Neuron-Like Cells Are Unsuitable as Experimental Model for Glutamate-Mediated Excitotoxicity. Front. Cell. Neurosci. 2016, 10, 118. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.; Tan, S.; Lin, T. Stem Cells for Modeling and Therapy of Parkinson’s Disease. Hum. Gene Ther. 2017, 28, 85–98. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced Pluripotent Stem Cells: Applications in Regenerative Medicine, Disease Modeling, and Drug Discovery. Front. cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef]
- Tsuda, L.; Lim, Y.-M. Alzheimer’s Disease Model System Using Drosophila. Adv. Exp. Med. Biol. 2018, 1076, 25–40. [Google Scholar] [CrossRef]
- Abolaji, A.O.; Kamdem, J.P.; Lugokenski, T.H.; Nascimento, T.K.; Waczuk, E.P.; Farombi, E.O.; Loreto, L.D.S.; Rocha, J.B.T. Involvement of Oxidative Stress in 4-Vinylcyclohexene-Induced Toxicity in Drosophila Melanogaster. Free Radic. Biol. Med. 2014, 71, 99–108. [Google Scholar] [CrossRef]
- Anet, A.; Olakkaran, S.; Kizhakke Purayil, A.; Hunasanahally Puttaswamygowda, G. Bisphenol A Induced Oxidative Stress Mediated Genotoxicity in Drosophila Melanogaster. J. Hazard. Mater. 2019, 370, 42–53. [Google Scholar] [CrossRef]
- Newman, M.; Ebrahimie, E.; Lardelli, M. Using the Zebrafish Model for Alzheimer’s Disease Research. Front. Genet. 2014, 5, 189. [Google Scholar] [CrossRef]
- Hannan, S.B.; Dräger, N.M.; Rasse, T.M.; Voigt, A.; Jahn, T.R. Cellular and Molecular Modifier Pathways in Tauopathies: The Big Picture from Screening Invertebrate Models. J. Neurochem. 2016, 137, 12–25. [Google Scholar] [CrossRef]
- Bouleau, S.; Tricoire, H. Drosophila Models of Alzheimer’s Disease: Advances, Limits, and Perspectives. J. Alzheimer’s Dis. 2015, 45, 1015–1038. [Google Scholar] [CrossRef]
- Shaye, D.D.; Greenwald, I. OrthoList: A Compendium of C. Elegans Genes with Human Orthologs. PLoS ONE 2011, 6, e20085. [Google Scholar] [CrossRef]
- Gerlai, R. Zebra Fish: An Uncharted Behavior Genetic Model. Behav. Genet. 2003, 33, 461–468. [Google Scholar] [CrossRef]
- Blaser, R.; Gerlai, R. Behavioral Phenotyping in Zebrafish: Comparison of Three Behavioral Quantification Methods. Behav. Res. Methods 2006, 38, 456–469. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Echevarria, D.J.; Stewart, A.M. Gaining Translational Momentum: More Zebrafish Models for Neuroscience Research. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 55, 1–6. [Google Scholar] [CrossRef]
- Jeong, J.-Y.; Kwon, H.-B.; Ahn, J.-C.; Kang, D.; Kwon, S.-H.; Park, J.A.; Kim, K.-W. Functional and Developmental Analysis of the Blood-Brain Barrier in Zebrafish. Brain Res. Bull. 2008, 75, 619–628. [Google Scholar] [CrossRef]
- Musa, A.; Lehrach, H.; Russo, V.A. Distinct Expression Patterns of Two Zebrafish Homologues of the Human APP Gene during Embryonic Development. Dev. Genes Evol. 2001, 211, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Levy, N. The Use of Animal as Models: Ethical Considerations. Int. J. Stroke Off. J. Int. Stroke Soc. 2012, 7, 440–442. [Google Scholar] [CrossRef] [PubMed]





| Model | Main Conclusions | Reference | |
|---|---|---|---|
| Rodents | Model: APP knockout in mouse models (APP-/-) Ferroptosis inhibitor: DFP (50 mg/kg/day in drinking water for 3 months) | APP-/- mouse model presented an iron elevation in the CNS, but DFP ameliorated the SN neuronal loss. | [152] |
| Model: 3 and 6 month old Nrf2+/+ and Nrf2-/- mice that overexpress human α-syn (hα-Syn+:Nrf2+/+ and hα-Syn+:Nrf2-/-) | Nrf2 is a critical anti-ferroptotic mediator of neuronal survival, and the vicious cycle of α-syn overexpression and Nrf2 suppression leads to an enhanced neuronal ferroptotic cell death. | [153] | |
| Model: triple transgenic mice (3xTg mice, harboring the APP Swedish mutation (KM670/671NL), the M146V human mutant PS1, and the Tau P301L) Ferroptosis inhibitor: Zileuton (200 mg/L of drinking water for three months) | Aβ1-42 levels were significantly lower in the brain of the zileuton-treated animals; besides, a significant reduction in the amount of Aβ peptides deposited in the brain of zileuton-treated-animals was also observed. | [156] | |
| Model: APPswe/PS1E9 transgenic mice Ferroptosis inhibitor: Eriodictyol (50 mg/kg, i.p. for 3 months) | Eriodictyol reduced Aβ aggregation, decreased the levels of Aβ peptide and p-tau, ferrous iron, total iron, TfR and FTH in the cortex and hippocampus and decreased MDA content, and also an increased GPX4 expression levels. | [157] | |
| Model: Aβ1-42 AD mice Ferroptosis inhibitor: Salidroside (50 mg/kg/day) | Salidroside attenuated mitochondrial changes and alleviated the decrease in GPX4 levels caused by Aβ1-42 in the hippocampus and cortex. | [158] | |
| Model: Cuprizone model of MS | Increased NCOA4, TfR1 and COX2 activity and augmented 4-HNE levels. GPX4 levels were also significantly reduced, as well as the expression of system Xc-. | [86] | |
| Model: Rat model of Temporal lobe epilepsy induced by LiCl-Pilo Ferroptosis inhibitor: klotho (overexpressed using an adeno-associated viral vector) | Klotho was mainly located in the neurons, rather than in the astrocytes. FPN levels were increase in animals exposed to klotho, and the opposite occurred in DMT-1 levels. Also, the levels of GSH and GPX4 in the hippocampus were significantly elevated, while ROS levels were suppressed. | [165] | |
| Model: transgenic mice expressing 5 familial AD mutation (5xFAD mice) Ferroptosis inhibitor: OABL (20 mg/kg/day i.p.) | OABL inhibited mitochondrial changes, increased GSH levels and reduced MDA content, and decreased the levels of Aβ1-42 and phosphorylated Tau observed in 5xFAD mice. | [116] | |
| Model: Delayed neurocognitive recovery aged mice Ferroptosis inhibitor: MSCs-Exo and Fer-1 (1.5 mg/kg) | MSCs-Exo inhibited ferroptosis in dNCR aged mice, as same as Fer-1 (positive control). | [166] | |
| Model: Rat model of PD (mice exposed to 25 μg 6-OHDA in the medial forebrain bundle) | 6-OHDA decreased, in vivo, the levels of TH and GPX4, decreased the GSH/GSSG ratio and increased MDA levels, suggesting increased lipid peroxidation. | [115] | |
| Drosophila melanogaster | Model: Drosophila melanogaster model of AD | Aβ42 was able to inhibit the activity of Nrf2 in neurons and loss-of-function mutations in Keap1 significantly protected against Ab42-induced toxicity. | [170] |
| Model: Drosophila melanogaster expressing hTDP-43-expressing Ferroptosis inhibitor: GstO2 | GstO2 was proposed as a key regulator of hTDP-43-related ALS pathogenesis and highlighted as a potential target in the treatment of ALS. | [172] | |
| Model: Drosophila melanogaster with frataxin deficiency | This deficiency induced iron hypersensitivity, a reduction life span and a reduction in the activity of aconitase and complex II, leading to mitochondria damage. | [173] | |
| Caenorhabditis elegans | Ferroptosis inducer: DEM (1 mM) Ferroptosis inhibitor: Lip-1 (200 μM) | DEM induced death of 4-day old adult worms, but the ferroptosis inhibitor reversed this death. DEM promoted an acute GSH depletion that triggered a marked increase in the levels of MDA and 4-HNE, which was ameliorated by Lip-1. | [174] |
| Model: C. elegans with a reduction of the frataxin gene | Shortening the frataxin gene increased the animal sensitivity to oxidative stress. | [175] | |
| Ferroptosis inducer: DGLA Model: Ether lipid-deficient mutant strains | Dietary lipids may greatly influence germ cells’ sensitivity to ferroptosis. | [176] | |
| Zebrafish | Model: Embryonic zebrafish Compound: Hydrogen peroxide (5 mM) | Mutant SOD1 significantly induced oxidative stress (which was demonstrated by a reduction in survival of embryos upon incubation with hydrogen peroxide), potentially promoting ferroptosis (as oxidative stress induces the depletion of antioxidant defenses such as GSH, being GSH depletion a hallmark of ferroptosis). | [180] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, I.; Barbosa, D.J.; Silva, V.; Benfeito, S.; Borges, F.; Remião, F.; Silva, R. Research Models to Study Ferroptosis’s Impact in Neurodegenerative Diseases. Pharmaceutics 2023, 15, 1369. https://doi.org/10.3390/pharmaceutics15051369
Costa I, Barbosa DJ, Silva V, Benfeito S, Borges F, Remião F, Silva R. Research Models to Study Ferroptosis’s Impact in Neurodegenerative Diseases. Pharmaceutics. 2023; 15(5):1369. https://doi.org/10.3390/pharmaceutics15051369
Chicago/Turabian StyleCosta, Inês, Daniel José Barbosa, Vera Silva, Sofia Benfeito, Fernanda Borges, Fernando Remião, and Renata Silva. 2023. "Research Models to Study Ferroptosis’s Impact in Neurodegenerative Diseases" Pharmaceutics 15, no. 5: 1369. https://doi.org/10.3390/pharmaceutics15051369