
Microfluidic Liver-on-a-Chip for Preclinical Drug Discovery

Abstract
:1. Introduction
2. Liver-on-a-Chip
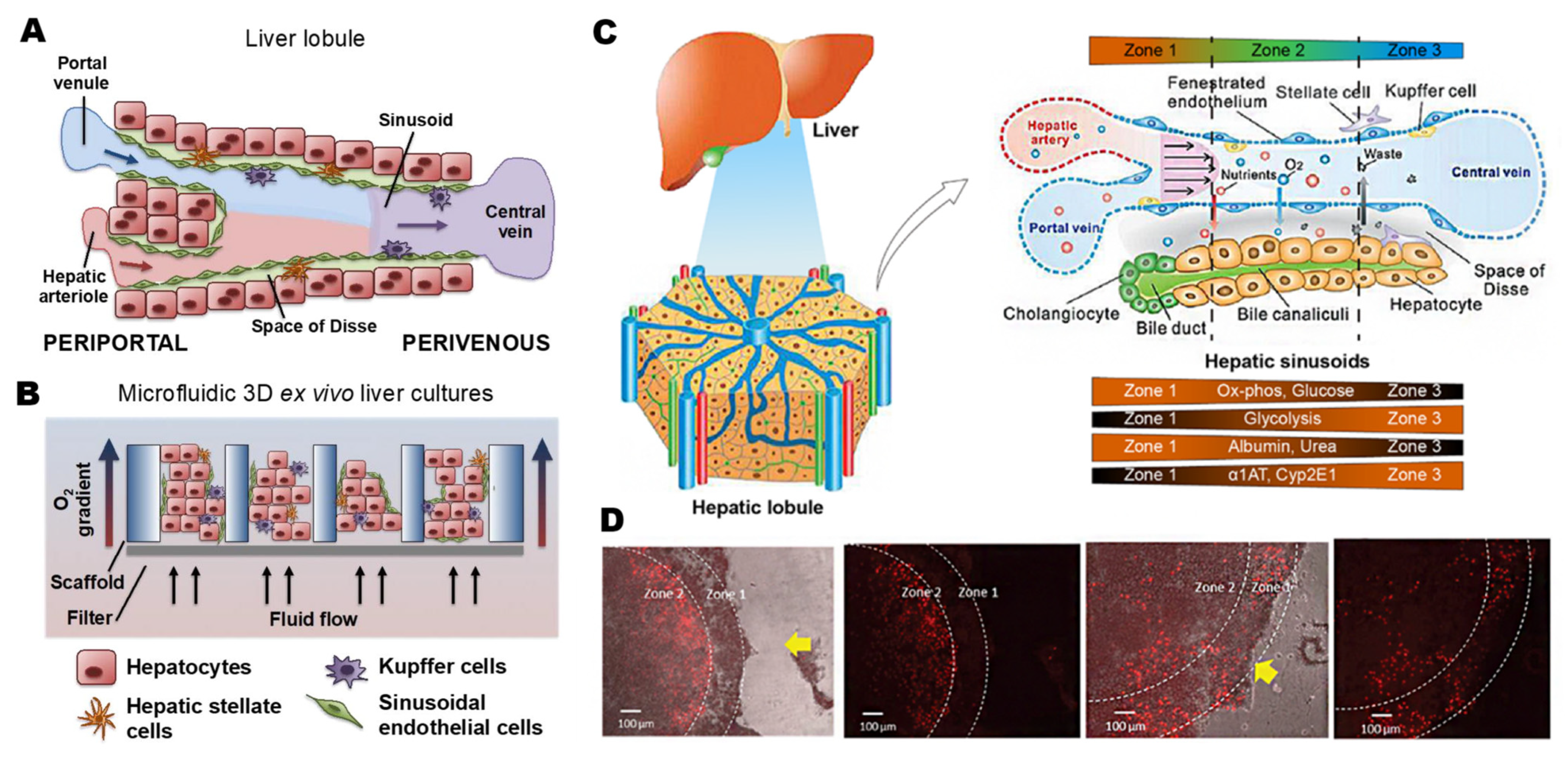
2.1. Physiological Microenvironment of the Liver
2.2. Liver-on-a-Chip Technology
2.2.1. Cell Sourcing
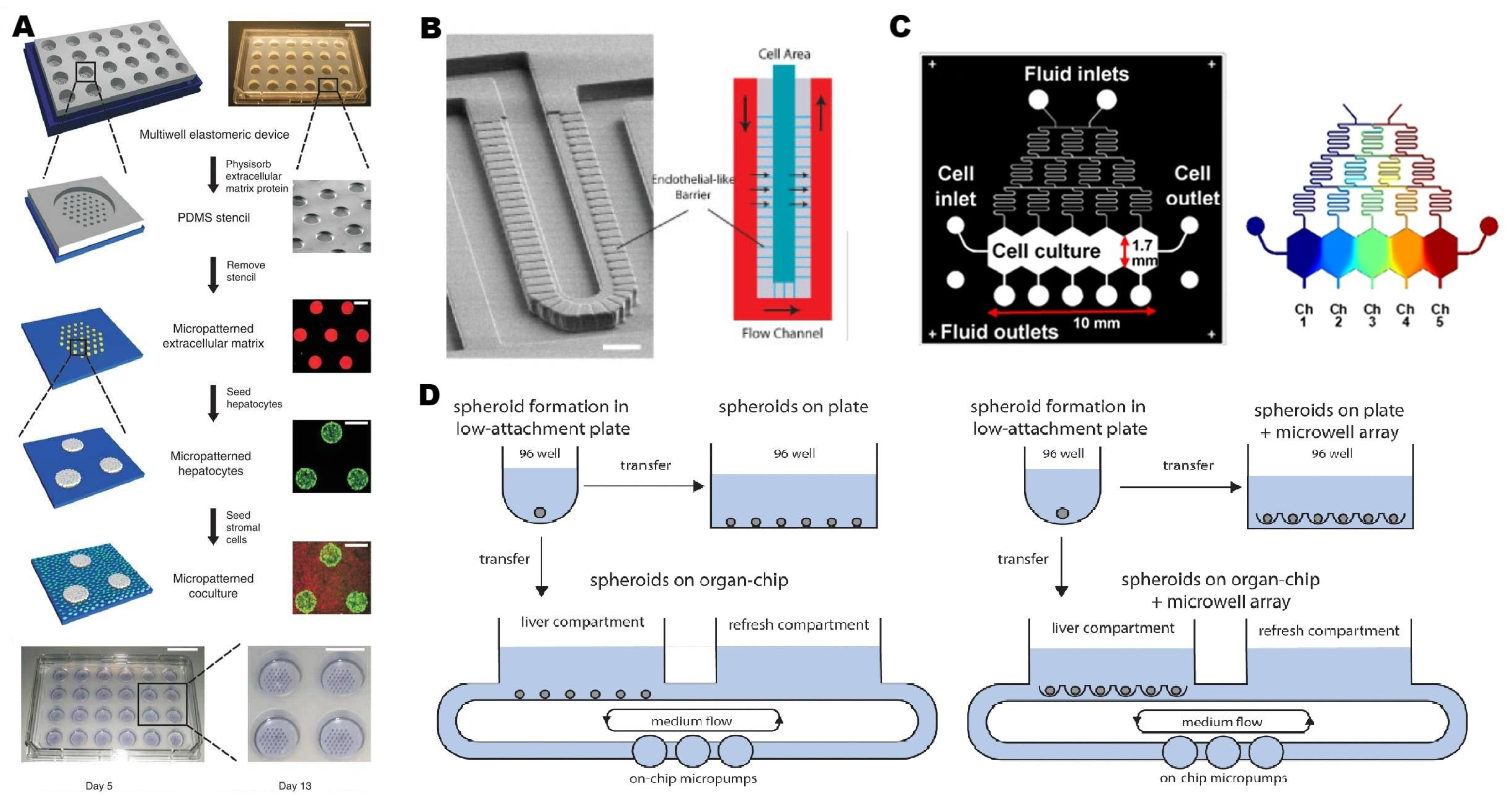
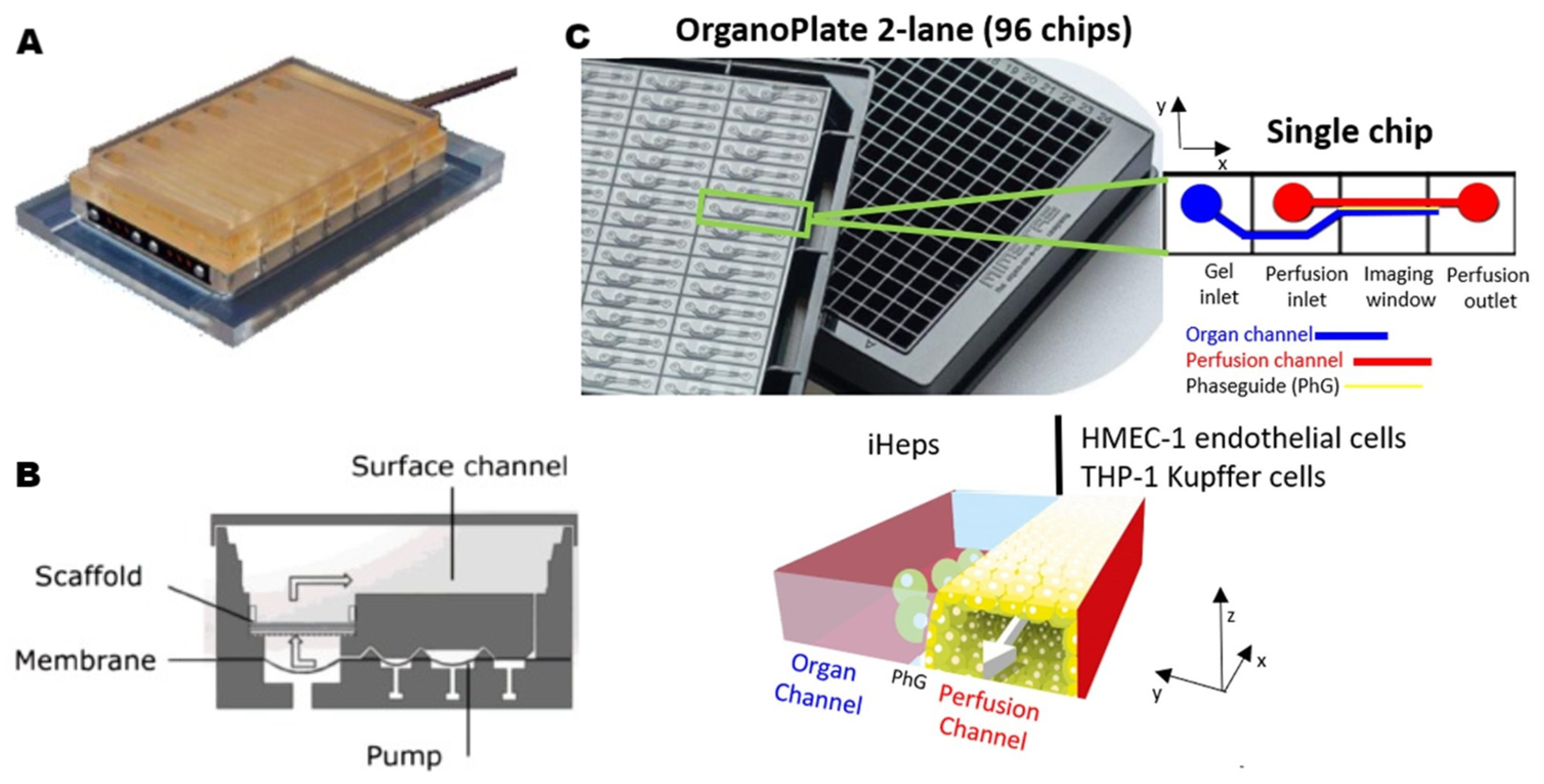
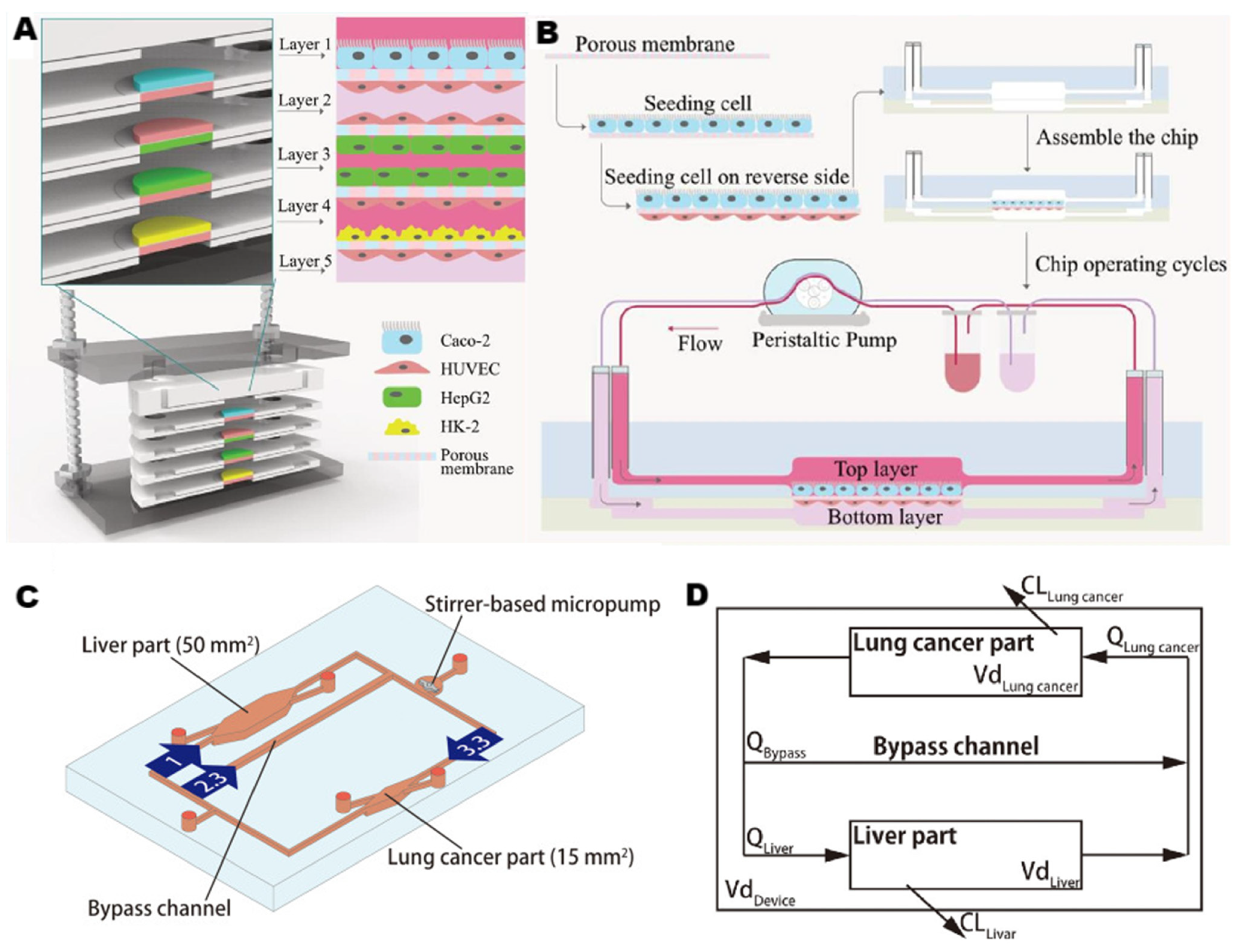
2.2.2. Microfluidic Strategies for Liver-on-a-Chip
3. Application in Preclinical Studies of Drug Discovery
3.1. Pharmacokinetics and Pharmacodynamics
3.2. Evaluation of Drug Efficiency and Safety
3.3. Multi-Organ Chips Systems
4. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hinkson, I.V.; Madej, B.; Stahlberg, E.A.; Consortium, A. Accelerating Therapeutics for Opportunities in Medicine: A Paradigm Shift in Drug Discovery. Front. Pharmacol. 2020, 11, 770. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 494–495. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Imai, R.; Ono, S. The Current Status of Drug Discovery and Development as Originated in United States Academia: The Influence of Industrial and Academic Collaboration on Drug Discovery and Development. Cts-Clin. Transl. Sci. 2018, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.X.; Gao, W.; Hu, H.X.; Zhou, S.M. Why 90 % of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef]
- Huang, R.L.; Xia, M.H.; Sakamuru, S.; Zhao, J.H.; Shahane, S.A.; Attene-Ramos, M.; Zhao, T.G.; Austin, C.P.; Simeonov, A. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat. Commun. 2016, 7, 10425. [Google Scholar] [CrossRef]
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H.K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 2019, 11, eaax5516. [Google Scholar] [CrossRef]
- Martic-Kehl, M.I.; Schibli, R.; Schubiger, P.A. Can animal data predict human outcome? Problems and pitfalls of translational animal research. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1492–1496. [Google Scholar] [CrossRef]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials Is it Time to Rethink Our Current Approach? JACC-Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.W.; Xie, J.X.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Treyer, A.; Musch, A. Hepatocyte Polarity. Compr. Physiol. 2013, 3, 243–287. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Berger, D.R.; Ballinger, K.R.; Davidson, M.D.; Lin, C.; Ware, B.R. Microengineered Liver Tissues for Drug Testing. Jala 2015, 20, 216–250. [Google Scholar] [CrossRef] [PubMed]
- Kulsharova, G.; Kurmangaliyeva, A. Liver microphysiological platforms for drug metabolism applications. Cell Prolif. 2021, 54, e13099. [Google Scholar] [CrossRef]
- Long, T.J.; Cosgrove, P.A.; Dunn, R.T.; Stolz, D.B.; Hamadeh, H.; Afshari, C.; McBride, H.; Griffith, L.G. Modeling Therapeutic Antibody-Small Molecule Drug-Drug Interactions Using a Three-Dimensional Perfusable Human Liver Coculture Platform. Drug Metab. Dispos. 2016, 44, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.; Shaeri, M.; Large, E.; Cornforth, T.; Robinson, A.; Kostrzewski, T.; Sison-Young, R.; Goldring, C.; Park, K.; Hughes, D. Perfused human hepatocyte microtissues identify reactive metabolite-forming and mitochondria-perturbing hepatotoxins. Toxicol. Vitr. 2018, 46, 29–38. [Google Scholar] [CrossRef]
- Sarkar, U.; Rivera-Burgos, D.; Large, E.M.; Hughes, D.J.; Ravindra, K.C.; Dyer, R.L.; Ebrahimkhani, M.R.; Wishnok, J.S.; Griffith, L.G.; Tannenbaum, S.R. Metabolite Profiling and Pharmacokinetic Evaluation of Hydrocortisone in a Perfused Three-Dimensional Human Liver Bioreactor. Drug Metab. Dispos. 2015, 43, 1091–1099. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Kostrzewski, T.; Stokes, C.L.; Griffith, L.G.; Hughes, D.J.; Cirit, M. Quantitative Assessment of Population Variability in Hepatic Drug Metabolism Using a Perfused Three-Dimensional Human Liver Microphysiological Systems. J. Pharmacol. Exp. Ther. 2017, 360, 95–105. [Google Scholar] [CrossRef]
- Bircsak, K.M.; DeBiasio, R.; Miedel, M.; Alsebahi, A.; Reddinger, R.; Saleh, A.; Shun, T.Y.; Vernetti, L.A.; Gough, A. A 3D microfluidic liver model for high throughput compound toxicity screening in the OrganoPlate (R). Toxicology 2021, 450, 152667. [Google Scholar] [CrossRef]
- Toh, Y.C.; Lim, T.C.; Tai, D.; Xiao, G.F.; van Noort, D.; Yu, H.R. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.R.; Lv, T.; Tu, X.; Li, P.W.; Wang, T.T.; Dong, H.H.; Tu, P.F.; Ai, X.N. An integrated biomimetic array chip for establishment of collagen-based 3D primary human hepatocyte model for prediction of clinical drug-induced liver injury. Biotechnol. Bioeng. 2021, 118, 4687–4698. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.D.; Jiao, S.M.; Wei, J.H.; Zhang, X.B.; Pei, Y.X.; Pei, Z.C.; Li, J.J.; Du, Y.G. Investigation of absorption, metabolism and toxicity of ginsenosides compound K based on human organ chips. Int. J. Pharm. 2020, 587, 119669. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Hsieh, M.J.; Liao, Y.H.; Lin, Y.C.; Hou, Y.T. Liver-on-a-chip platform to study anticancer effect of statin and its metabolites. Biochem. Eng. J. 2021, 165, 107831. [Google Scholar] [CrossRef]
- Deguchi, S.; Takayama, K. State-of-the-art liver disease research using liver-on-a-chip. Inflamm. Regen. 2022, 42, 62. [Google Scholar] [CrossRef]
- Özkan, A.; Stolley, D.; Cressman, E.N.K.; McMillin, M.; DeMorrow, S.; Yankeelov, T.E.; Rylander, M.N. The Influence of Chronic Liver Diseases on Hepatic Vasculature: A Liver-on-a-chip Review. Micromachines 2020, 11, 487. [Google Scholar] [CrossRef]
- Abdel-Misih, S.R.Z.; Bloomston, M. Liver Anatomy. Surg. Clin. North Am. 2010, 90, 643–653. [Google Scholar] [CrossRef]
- Bale, S.S.; Vernetti, L.; Senutovitch, N.; Jindal, R.; Hegde, M.; Gough, A.; McCarty, W.J.; Bakan, A.; Bhushan, A.; Shun, T.Y.; et al. In vitro platforms for evaluating liver toxicity. Exp. Biol. Med. 2014, 239, 1180–1191. [Google Scholar] [CrossRef]
- Deng, J.; Wei, W.B.; Chen, Z.Z.; Lin, B.C.; Zhao, W.J.; Luo, Y.; Zhang, X.L. Engineered Liver-On-A-Chip Platform to Mimic Liver Functions and Its Biomedical Applications: A Review. Micromachines 2019, 10, 676. [Google Scholar] [CrossRef]
- Materne, E.M.; Tonevitsky, A.G.; Marx, U. Chip-based liver equivalents for toxicity testing—Organotypicalness versus cost-efficient high throughput. Lab Chip 2013, 13, 3481–3495. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, D.E.; Johnson, K.; Ryan, L.; Boorman, G.; Maronpot, R.R. New insights into functional aspects of liver morphology. Toxicol. Pathol. 2005, 33, 27–34. [Google Scholar] [CrossRef]
- Usta, O.B.; McCarty, W.J.; Bale, S.; Hegde, M.; Jindal, R.; Bhushan, A.; Golberg, I.; Yarmush, M.L. Microengineered cell and tissue systems for drug screening and toxicology applications: Evolution of in-vitro liver technologies. Technology 2015, 3, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip’. Exp. Cell Res. 2018, 363, 15–25. [Google Scholar] [CrossRef]
- Ma, L.D.; Wang, Y.T.; Wang, J.R.; Wu, J.L.; Meng, X.S.; Hu, P.; Mu, X.; Liang, Q.L.; Luo, G.A. Design and fabrication of a liver-on-a-chip platform for convenient, highly efficient, and safe in situ perfusion culture of 3D hepatic spheroids. Lab Chip 2018, 18, 2547–2562. [Google Scholar] [CrossRef]
- Moravcova, A.; Cervinkova, Z.; Kucera, O.; Mezera, V.; Rychtrmoc, D.; Lotkova, H. The Effect of Oleic and Palmitic Acid on Induction of Steatosis and Cytotoxicity on Rat Hepatocytes in Primary Culture. Physiol. Res. 2015, 64, S627–S636. [Google Scholar] [CrossRef]
- Tomlinson, L.; Hyndman, L.; Firman, J.W.; Bentley, R.; Kyffin, J.A.; Webb, S.D.; McGinty, S.; Sharma, P. In vitro Liver Zonation of Primary Rat Hepatocytes. Front. Bioeng. Biotechnol. 2019, 7, 17. [Google Scholar] [CrossRef]
- Kim, Y.; Kang, K.; Jeong, J.; Paik, S.S.; Kim, J.S.; Park, S.A.; Kim, W.D.; Park, J.; Choi, D. Three-dimensional (3D) printing of mouse primary hepatocytes to generate 3D hepatic structure. Ann. Surg. Treat. Res. 2017, 92, 67–72. [Google Scholar] [CrossRef]
- Lee, H.; Chae, S.; Kim, J.Y.; Han, W.; Kim, J.; Choi, Y.; Cho, D.W. Cell-printed 3D liver-on-a-chip possessing a liver microenvironment and biliary system. Biofabrication 2019, 11, 025001. [Google Scholar] [CrossRef]
- Madurska, M.J.; Poyade, M.; Eason, D.; Rea, P.; Watson, A.J.M. Development of a Patient-Specific 3D-Printed Liver Model for Preoperative Planning. Surg. Innov. 2017, 24, 145–150. [Google Scholar] [CrossRef]
- Witowski, J.S.; Pedziwiatr, M.; Major, P.; Budzynski, A. Cost-effective, personalized, 3D-printed liver model for preoperative planning before laparoscopic liver hemihepatectomy for colorectal cancer metastases. Int. J. Comput. Assist. Radiol. Surg. 2017, 12, 2047–2054. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Ueno, H.; Applegate, D.R.; Shuler, M.L. Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and 3D primary liver tissue. Lab Chip 2016, 16, 2719–2729. [Google Scholar] [CrossRef]
- Li, L.; Gokduman, K.; Gokaltun, A.; Yarmush, M.L.; Usta, O.B. A microfluidic 3D hepatocyte chip for hepatotoxicity testing of nanoparticles. Nanomedicine 2019, 14, 2209–2226. [Google Scholar] [CrossRef]
- Matsusaki, M.; Case, C.P.; Akashi, M. Three-dimensional cell culture technique and pathophysiology. Adv. Drug Deliv. Rev. 2014, 74, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.D.; Schirmer, K.; Munst, B.; Heinz, S.; Ghafoory, S.; Wolfl, S.; Simon-Keller, K.; Marx, A.; Oie, C.I.; Ebert, M.P.; et al. In Vitro Generation of Functional Liver Organoid-Like Structures Using Adult Human Cells. PLoS ONE 2015, 10, e0139345. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, X.; Cribbin, E.M.; Kim, A.M.; Li, J.J.; Yong, K.T. Liver-on-a-chip: Considerations, advances, and beyond. Biomicrofluidics 2022, 16, 061502. [Google Scholar] [CrossRef]
- Du, Y.Y.; Wang, J.L.; Jia, J.; Song, N.; Xiang, C.G.; Xu, J.; Hou, Z.Y.; Su, X.H.; Liu, B.; Jiang, T.; et al. Human Hepatocytes with Drug Metabolic Function Induced from Fibroblasts by Lineage Reprogramming. Cell Stem Cell 2014, 14, 394–403. [Google Scholar] [CrossRef]
- LeCluyse, E.L.; Alexandre, E.; Hamilton, G.A.; Viollon-Abadie, C.; Coon, D.J.; Jolley, S.; Richert, L. Isolation and culture of primary human hepatocytes. Methods Mol. Biol. 2005, 290, 207–229. [Google Scholar]
- Pichard, L.; Fabre, I.; Fabre, G.; Domergue, J.; Saint Aubert, B.; Mourad, G.; Maurel, P. Cyclosporin A drug interactions. Screening for inducers and inhibitors of cytochrome P-450 (cyclosporin A oxidase) in primary cultures of human hepatocytes and in liver microsomes. Drug Metab. Dispos. Biol. Fate Chem. 1990, 18, 595–606. [Google Scholar]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grun, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef] [PubMed]
- Korelova, K.; Jirouskova, M.; Sarnova, L.; Gregor, M. Isolation and 3D Collagen Sandwich Culture of Primary Mouse Hepatocytes to Study the Role of Cytoskeleton in Bile Canalicular Formation In Vitro. JOVE J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zeigerer, A.; Wuttke, A.; Marsico, G.; Seifert, S.; Kalaidzidis, Y.; Zerial, M. Functional properties of hepatocytes in vitro are correlated with cell polarity maintenance. Exp. Cell Res. 2017, 350, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.V.; Ioannides, C.; Parke, D.V. Cytochromes P450 and species differences in xenobiotic metabolism and activation of carcinogen. Environ. Health Perspect. 1998, 106, 633–641. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Chan, K.; Silber, P.M. Human and animal hepatocytes in vitro with extrapolation in vivo. Chem.-Biol. Interact. 2004, 150, 97–114. [Google Scholar] [CrossRef]
- Lin, J.H.; Chiba, M.; Balani, S.K.; Chen, I.W.; Kwei, G.Y.; Vastag, K.J.; Nishime, J.A. Species differences in the pharmacokinetics and metabolism of indinavir, a potent human immunodeficiency virus protease inhibitor. Drug Metab. Dispos. Biol. Fate Chem. 1996, 24, 1111–1120. [Google Scholar]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979, 282, 615–616. [Google Scholar] [CrossRef]
- Donato, M.T.; Lahoz, A.; Castell, J.V.; Gomez-Lechon, M.J. Cell lines: A tool for in vitro drug metabolism studies. Curr. Drug Metab. 2008, 9, 1–11. [Google Scholar]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef]
- Guo, L.; Dial, S.; Shi, L.M.; Branham, W.; Liu, J.; Fang, J.L.; Green, B.; Deng, H.; Kaput, J.; Ning, B.T. Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Kramer, L.; Hoffmann, E.; Klein, S.; Noor, F. 3D organotypic HepaRG cultures as in vitro model for acute and repeated dose toxicity studies. Toxicol. Vitr. 2014, 28, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Ramaiahgari, S.C.; den Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.M.; van de Water, B.; Price, L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.J.; Chouhan, B.; Regan, S.L.; Rollison, H.; Amberntsson, S.; Andersson, L.C.; Srivastava, A.; Darnell, M.; Cairns, J.; Lazic, S.E.; et al. Integrated in vitro models for hepatic safety and metabolism: Evaluation of a human Liver-Chip and liver spheroid. Arch. Toxicol. 2019, 93, 1021–1037. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Tolosa, L.; Conde, I.; Donato, M.T. Competency of different cell models to predict human hepatotoxic drugs. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1553–1568. [Google Scholar] [CrossRef]
- Tasnim, F.; Xing, J.W.; Huang, X.Z.; Mo, S.P.; Wei, X.N.; Tan, M.H.; Yu, H. Generation of mature kupffer cells from human induced pluripotent stem cells. Biomaterials 2019, 192, 377–391. [Google Scholar] [CrossRef]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdu, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113. [Google Scholar] [CrossRef]
- Koui, Y.; Kido, T.; Ito, T.; Oyama, H.; Chen, S.W.; Katou, Y.; Shirahige, K.; Miyajima, A. An In Vitro Human Liver Model by iPSC-Derived Parenchymal and Non-parenchymal Cells. Stem Cell Rep. 2017, 9, 490–498. [Google Scholar] [CrossRef]
- Ware, B.R.; Berger, D.R.; Khetani, S.R. Prediction of Drug-Induced Liver Injury in Micropatterned Co-cultures Containing iPSC-Derived Human Hepatocytes. Toxicol. Sci. 2015, 145, 252–262. [Google Scholar] [CrossRef]
- Li, Z.N.; Hui, J.A.; Yang, P.H.; Mao, H.J. Microfluidic Organ-on-a-Chip System for Disease Modeling and Drug Development. Biosensors 2022, 12, 370. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Microfluidics—Downsizing large-scale biology. Nat. Biotechnol. 2001, 19, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Atac, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef]
- Kim, D.; Lokuta, M.A.; Huttenlocher, A.; Beebe, D.J. Selective and tunable gradient device for cell culture and chemotaxis study. Lab Chip 2009, 9, 1797–1800. [Google Scholar] [CrossRef]
- Kothapalli, C.R.; van Veen, E.; de Valence, S.; Chung, S.; Zervantonakis, I.K.; Gertler, F.B.; Kamm, R.D. A high-throughput microfluidic assay to study neurite response to growth factor gradients. Lab Chip 2011, 11, 497–507. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef]
- Kang, Y.B.; Eo, J.; Mert, S.; Yarmush, M.L.; Usta, O.B. Metabolic Patterning on a Chip: Towards in vitro Liver Zonation of Primary Rat and Human Hepatocytes. Sci. Rep. 2018, 8, 8951. [Google Scholar] [CrossRef]
- Eckstrum, K.; Striz, A.; Ferguson, M.; Zhao, Y.; Sprando, R. Evaluation of the utility of the Beta Human Liver Emulation System (BHLES) for CFSAN’s regulatory toxicology program. Food Chem. Toxicol. 2022, 161, 112828. [Google Scholar] [CrossRef]
- Nawroth, J.C.; Petropolis, D.B.; Manatakis, D.V.; Maulana, T.I.; Burchett, G.; Schlunder, K.; Witt, A.; Shukla, A.; Kodella, K.; Ronxhi, J.; et al. Modeling alcohol-associated liver disease in a human Liver-Chip. Cell Rep. 2021, 36, 109393. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Barton, P.; Class, R.; Coxhead, H.; Delatour, C.; Gillent, E.; Henshall, J.; Isin, E.M.; King, L.; Valentin, J.-P. Setup of human liver-chips integrating 3D models, microwells and a standardized microfluidic platform as proof-of-concept study to support drug evaluation. Biomater. Biosyst. 2022, 7, 100054. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Linden, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed]
- Baran, S.W.; Brown, P.C.; Baudy, A.R.; Fitzpatrick, S.C.; Frantz, C.; Fullerton, A.; Gan, J.; Hardwick, R.N.; Hillgren, K.M.; Kopec, A.K.; et al. Perspectives on the evaluation and adoption of complex in vitro models in drug development: Workshop with the FDA and the pharmaceutical industry (IQ MPS Affiliate). Altex 2022, 39, 297–314. [Google Scholar] [CrossRef]
- Docci, L.; Klammers, F.; Ekiciler, A.; Molitor, B.; Umehara, K.; Walter, I.; Krähenbühl, S.; Parrott, N.; Fowler, S. In Vitro to In Vivo Extrapolation of Metabolic Clearance for UGT Substrates Using Short-Term Suspension and Long-Term Co-cultured Human Hepatocytes. AAPS J. 2020, 22, 131. [Google Scholar] [CrossRef]
- Dixit, V.; Moore, A.; Tsao, H.; Hariparsad, N. Application of Micropatterned Cocultured Hepatocytes to Evaluate the Inductive Potential and Degradation Rate of Major Xenobiotic Metabolizing Enzymes. Drug Metab. Dispos. 2016, 44, 250–261. [Google Scholar] [CrossRef]
- Docci, L.; Milani, N.; Ramp, T.; Romeo, A.A.; Godoy, P.; Franyuti, D.O.; Krähenbühl, S.; Gertz, M.; Galetin, A.; Parrott, N.; et al. Exploration and application of a liver-on-a-chip device in combination with modelling and simulation for quantitative drug metabolism studies. Lab Chip 2022, 22, 1187–1205. [Google Scholar] [CrossRef]
- Han, W.J.; Wu, Q.; Zhang, X.H.; Duan, Z.P. Innovation for hepatotoxicity in vitro research models: A review. J. Appl. Toxicol. 2019, 39, 146–162. [Google Scholar] [CrossRef]
- Van Ness, K.P.; Cesar, F.; Yeung, C.K.; Himmelfarb, J.; Kelly, E.J. Microphysiological systems in adsorption, distribution, metabolism, and elimination sciences. Cts-Clin. Transl. Sci. 2022, 15, 9–42. [Google Scholar] [CrossRef]
- Kimura, H.; Ikeda, T.; Nakayama, H.; Sakai, Y.; Fujii, T. An On-Chip Small Intestine-Liver Model for Pharmacokinetic Studies. Jala 2015, 20, 265–273. [Google Scholar] [CrossRef]
- Shinha, K.; Nihei, W.; Ono, T.; Nakazato, R.; Kimura, H. A pharmacokinetic-pharmacodynamic model based on multi-organ-on-a-chip for drug-drug interaction studies. Biomicrofluidics 2020, 14, 044108. [Google Scholar] [CrossRef] [PubMed]
- Lohasz, C.; Bonanini, F.; Hoelting, L.; Renggli, K.; Frey, O.; Hierlemann, A. Predicting Metabolism-Related Drug-Drug Interactions Using a Microphysiological Multitissue System. Adv. Biosyst. 2020, 4, 2000079. [Google Scholar] [CrossRef] [PubMed]
- Barrile, R.; van der Meer, A.D.; Park, H.; Fraser, J.P.; Simic, D.; Teng, F.; Conegliano, D.; Nguyen, J.; Jain, A.; Zhou, M.; et al. Organ-on-Chip Recapitulates Thrombosis Induced by an anti-CD154 Monoclonal Antibody: Translational Potential of Advanced Microengineered Systems. Clin. Pharmacol. Ther. 2018, 104, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.C.; Bagnaninchi, P.; Ye, H.; Sancho-Bru, P.; Falcon-Perez, J.M.; Royo, F.; Garcia-Ruiz, C.; Konu, O.; Miranda, J.; Lunov, O.; et al. Advanced preclinical models for evaluation of drug-induced liver injury—Consensus statement by the European Drug-Induced Liver Injury Network PRO-EURO-DILI-NET. J. Hepatol. 2021, 75, 935–959. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Sebastian, S.; Maharjan, S.; Lesha, A.; Carpenter, A.M.; Liu, X.L.; Xie, X.; Livermore, C.; Zhang, Y.S.; Zarrinpar, A. Liver-on-a-Chip Models of Fatty Liver Disease. Hepatology 2020, 71, 733–740. [Google Scholar] [CrossRef]
- Ingber, D.E. Developmentally inspired human ‘organs on chips’. Development 2018, 145, dev156125. [Google Scholar] [CrossRef]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 265. [Google Scholar] [CrossRef]
- Gurevich, I.; Burton, S.A.; Munn, C.; Ohshima, M.; Goedland, M.E.; Czysz, K.; Rajesh, D. iPSC-derived hepatocytes generated from NASH donors provide a valuable platform for disease modeling and drug discovery. Biol. Open 2020, 9, bio055087. [Google Scholar] [CrossRef]
- Sances, S.; Ho, R.; Vatine, G.; West, D.; Laperle, A.; Meyer, A.; Godoy, M.; Kay, P.S.; Mandefro, B.; Hatata, S.; et al. Human iPSC-Derived Endothelial Cells and Microengineered Organ-Chip Enhance Neuronal Development. Stem Cell Rep. 2018, 10, 1222–1236. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, H.; Deng, P.W.; Chen, W.W.; Guo, Y.Q.; Tao, T.T.; Qin, J.H. In situ differentiation and generation of functional liver organoids from human iPSCs in a 3D perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef]
- Yi, F.; Liu, G.H.; Belmonte, J.C.I. Human induced pluripotent stem cells derived hepatocytes: Rising promise for disease modeling, drug development and cell therapy. Protein Cell 2012, 3, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.Y.; Chao, B.S.; Wu, J.C. Strategies for Improving the Maturity of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2018, 123, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, O.; Cascione, S.; Michielin, F.; Elvassore, N. The emergence of the circadian clock network in hiPSC-derived hepatocytes on chip. Biochem. Biophys. Res. Commun. 2022, 601, 109–115. [Google Scholar] [CrossRef]
- Lohasz, C.; Loretan, J.; Sterker, D.; Goerlach, E.; Renggli, K.; Argast, P.; Frey, O.; Wiesmann, M.; Wartmann, M.; Rausch, M.; et al. A Microphysiological Cell-Culturing System for Pharmacokinetic Drug Exposure and High-Resolution Imaging of Arrays of 3D Microtissues. Front. Pharmacol. 2021, 12, 3798. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhuo, S.M.; Qu, Y.H.; Choudhury, D.; Wang, Z.P.; Iliescu, C.; Yu, H. On chip two-photon metabolic imaging for drug toxicity testing. Biomicrofluidics 2017, 11, 034108. [Google Scholar] [CrossRef]
- Peel, S.; Corrigan, A.M.; Ehrhardt, B.; Jang, K.J.; Caetano-Pinto, P.; Boeckeler, M.; Rubins, J.E.; Kodella, K.; Petropolis, D.B.; Ronxhi, J.; et al. Introducing an automated high content confocal imaging approach for Organs-on-Chips. Lab Chip 2019, 19, 410–421. [Google Scholar] [CrossRef]
- Kane, K.I.W.; Moreno, E.L.; Hachi, S.; Walter, M.; Jarazo, J.; Oliveira, M.A.P.; Hankemeier, T.; Vulto, P.; Schwamborn, J.C.; Thoma, M.; et al. Automated microfluidic cell culture of stem cell derived dopaminergic neurons. Sci. Rep. 2019, 9, 1796. [Google Scholar] [CrossRef]
- Buchanan, B.C.; Yoon, J.Y. Microscopic Imaging Methods for Organ-on-a-Chip Platforms. Micromachines 2022, 13, 328. [Google Scholar] [CrossRef]
- Tetsuka, K.; Ohbuchi, M.; Kawabe, T.; Goto, T.; Kiyonaga, F.; Takama, K.; Yamazaki, S.; Fujimori, A. Reconstituted Human Organ Models as a Translational Tool for Human Organ Response: Definition, Expectations, Cases, and Strategies for Implementation in Drug Discovery and Development. Biol. Pharm. Bull. 2020, 43, 375–383. [Google Scholar] [CrossRef]
- Deguchi, S.; Tsuda, M.; Kosugi, K.; Sakamoto, A.; Mimura, N.; Negoro, R.; Sano, E.; Nobe, T.; Maeda, K.; Kusuhara, H.; et al. Usability of Polydimethylsiloxane-Based Microfluidic Devices in Pharmaceutical Research Using Human Hepatocytes. Acs Biomater. Sci. Eng. 2021, 7, 3648–3657. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Characteristic | Cell Types | Drugs Involved | Analysis | Ref. |
|---|---|---|---|---|---|
| Drug metabolism | 3D perfusion, co-culture | hepatocytes, Kupffer cells | hydrocortisone | the anti-inflammatory effect of glucocorticoids on liver cultures | [18] |
| Drug metabolism | 3D perfusion, co-culture | PHHs, Kupffer cells | tocilizumab | dual regulation of inflammatory factors and therapeutic antibodies | [16] |
| Drug metabolism | 3D perfusion, co-culture, liver–small intestine | Caco-2, HepG2, A549 | epirubicin (EPI), irinotecan (CPT-11), and cyclophosphamide (CPA) | reproduce both blood circulation and organs volume relation in vitro | [90] |
| Drug metabolism | 3D perfusion, co-culture, liver–small intestine–kidney | Caco-2, HepG2, HUVEC, HK-2 | ginsenosides compound K (CK) | the pharmacological investigation of carbohydrate drugs | [23] |
| Population variability of liver drug metabolism | 3D perfusion | PHHs from five different donors | phenacetin, diclofenac, lidocaine, ibuprofen, propranolol, and prednisolone | in vitro assessment of population variability in drug metabolism | [19] |
| Drug toxicity | 3D microfluidics | hepatocytes (rats) | acetaminophen, diclofenac, quinidine, rifampin and ketoconazole | multiplexed testing | [21] |
| Drug toxicity | 3D perfusion, co-culture | iPSC-derived hepatocytes, endothelial cells, Kupffer-like immune cells | troglitazone, a library of 159 compounds | high throughput screening | [20] |
| Drug toxicity | collagen-based 3D model, integrated biomimetic array chip, cell–extracellular matrix interaction | PHHs | 122 clinical drugs evaluated for liver toxicity | large-scale hepatotoxicity screening | [22] |
| Drug toxicity | 3D microfluidics, co-culture, cell–extracellular matrix interaction | hepatocytes, sinusoidal endothelial cells, Kupffer cells, hepatic stellate cells (humans, rats, and dogs) | bosentan, analgesic acetaminophen (APAP), methotrexate (MTX), fialuridine (FIAU), a Janssen proprietary compound (JNJ-2) | human and cross-species drug toxicities | [7] |
| Drug-drug interactions | gravity-driven microfluidic, liver–intestines | PHHs, HCT116 | cyclophosphamide, ifosfamide, ritonavir | quantization of the impact of other drugs on the efficacy of anticancer drugs | [92] |
| Drug-drug interactions | 3D microfluidics, liver-lung | HepG2, A549 | simvastatin, ritonavir, CPT-11 | quantization of the impact of other drugs on the metabolism of anticancer drugs | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, J.; Qiu, H.; Tan, C.S. Microfluidic Liver-on-a-Chip for Preclinical Drug Discovery. Pharmaceutics 2023, 15, 1300. https://doi.org/10.3390/pharmaceutics15041300
Fu J, Qiu H, Tan CS. Microfluidic Liver-on-a-Chip for Preclinical Drug Discovery. Pharmaceutics. 2023; 15(4):1300. https://doi.org/10.3390/pharmaceutics15041300
Chicago/Turabian StyleFu, Jingyu, Hailong Qiu, and Cherie S. Tan. 2023. "Microfluidic Liver-on-a-Chip for Preclinical Drug Discovery" Pharmaceutics 15, no. 4: 1300. https://doi.org/10.3390/pharmaceutics15041300