
High-Throughput/High Content Imaging Screen Identifies Novel Small Molecule Inhibitors and Immunoproteasomes as Therapeutic Targets for Chordoma
 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Cells
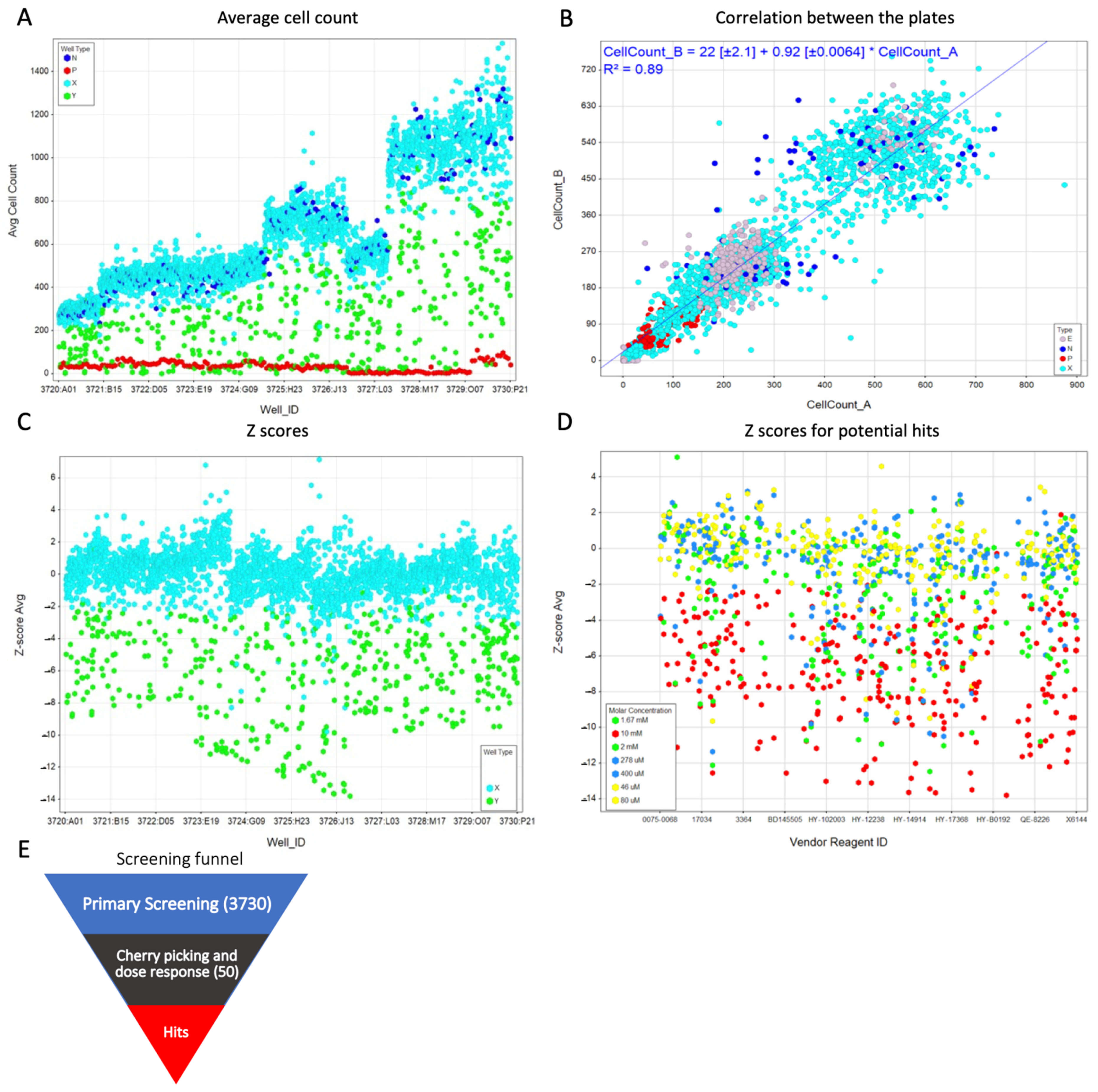
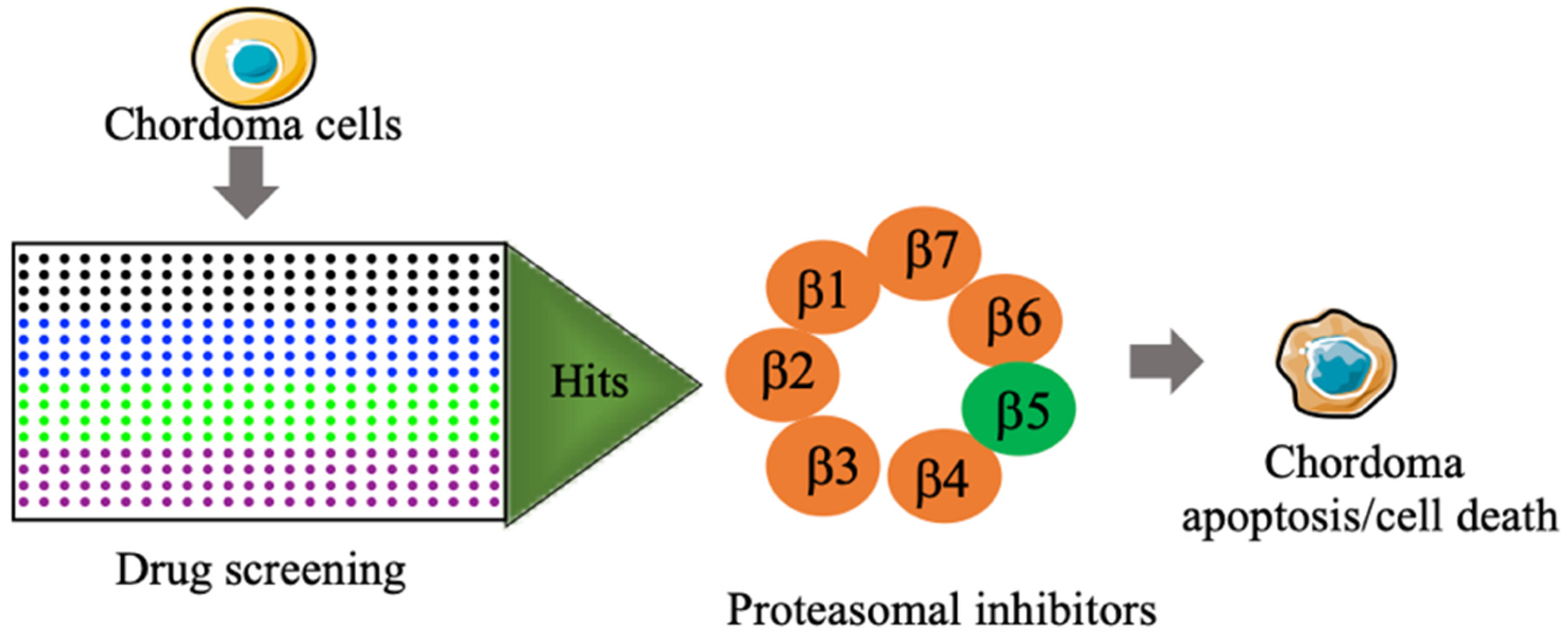
2.2. Primary Screening
2.3. Z′-Factor and Z-Score
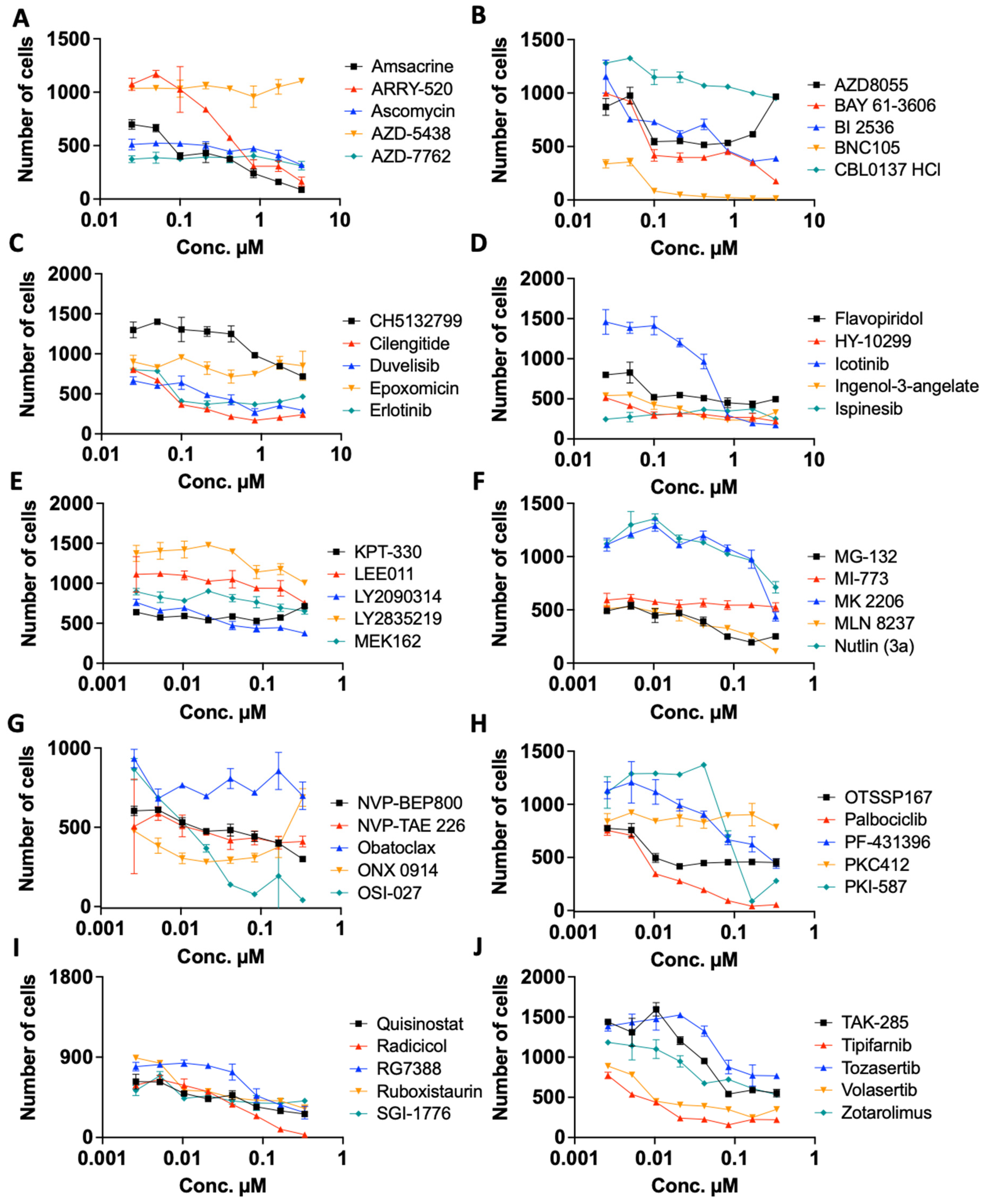
2.4. Re Confirmation and Dose Dependency Studies
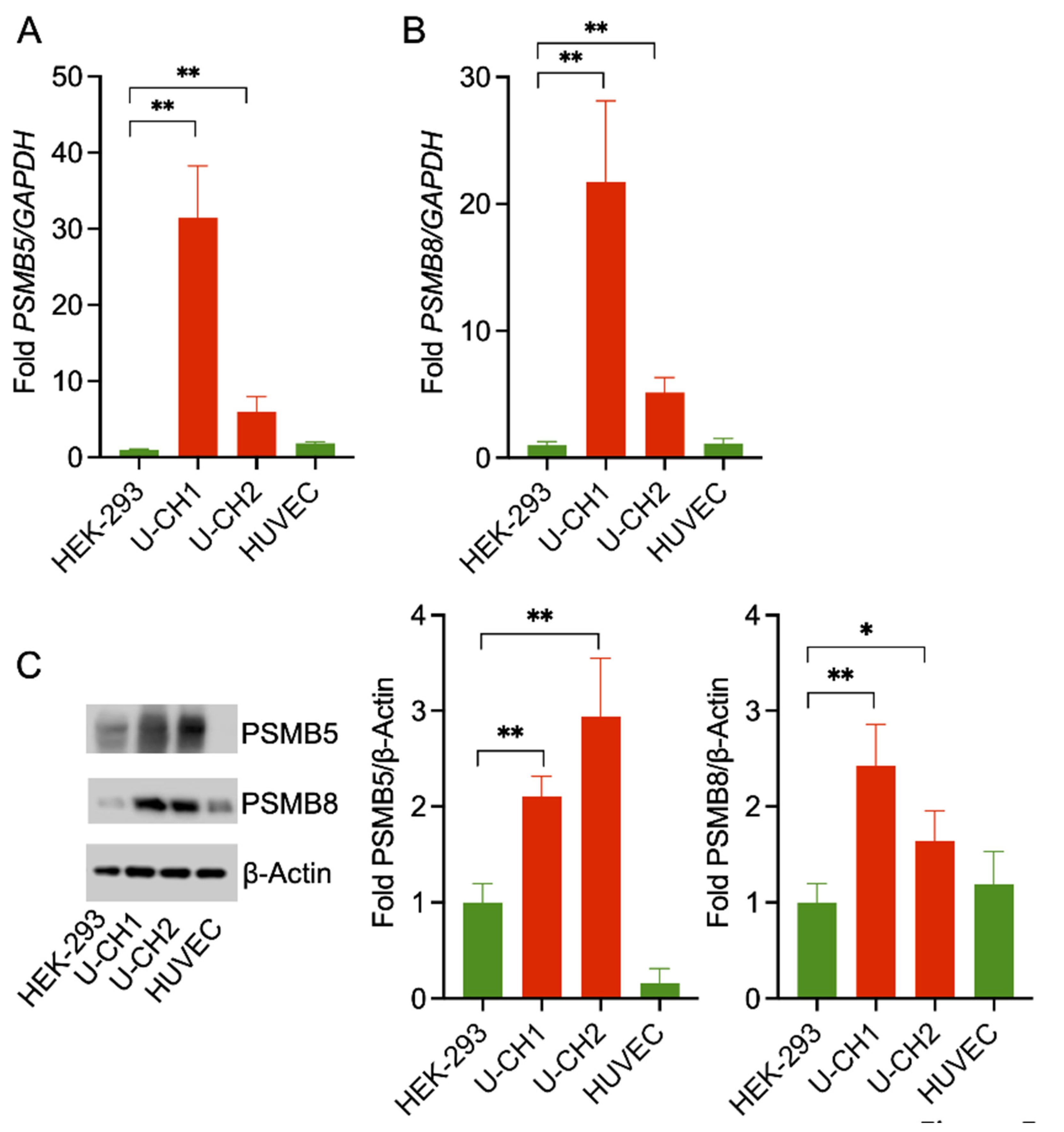
2.5. RNA Isolation, cDNA Synthesis, and Semi-Quantitative Real-Time (qRT) PCR
2.6. qRT-PCR
2.7. CellTiter 96® AQueous One Solution Cell Proliferation Assay
2.8. Western Blotting
2.9. Purity Statement
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, E.; Koffer, P.P.; DiPetrillo, T.A.; Kinsella, T.J. Incidence, treatment, and survival patterns for sacral chordoma in the United States, 1974–2011. Front. Oncol. 2016, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Chambers, K.J.; Lin, D.T.; Meier, J.; Remenschneider, A.; Herr, M.; Gray, S.T. Incidence and survival patterns of cranial chordoma in the United States. Laryngoscope 2014, 124, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- McMaster, M.L.; Goldstein, A.M.; Bromley, C.M.; Ishibe, N.; Parry, D.M. Chordoma: Incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control 2001, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Ng, D.; Alcorta, D.A.; Liebsch, N.J.; Sheridan, E.; Li, S.; Goldstein, A.M.; Parry, D.M.; Kelley, M.J. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat. Genet. 2009, 41, 1176–1178. [Google Scholar] [CrossRef]
- Nishino, A.; Satou, Y.; Morisawa, M.; Satoh, N. Brachyury (T) gene expression and notochord development in Oikopleura longicauda (Appendicularia, Urochordata). Dev. Genes Evol. 2001, 211, 219–231. [Google Scholar] [CrossRef]
- Oakley, G.J.; Fuhrer, K.; Seethala, R.R. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs. chondrosarcoma differential: A tissue microarray-based comparative analysis. Mod. Pathol. 2008, 21, 1461–1469. [Google Scholar] [CrossRef]
- Snyderman, C.H.; Gardner, P.A. Current opinion in otolaryngology and head and neck surgery: Clival chordoma and its management. Curr. Opin. Otolaryngol. Head Neck Surg. 2020, 28, 118–121. [Google Scholar] [CrossRef]
- Catton, C.; O’Sullivan, B.; Bell, R.; Laperriere, N.; Cummings, B.; Fornasier, V.; Wunder, J. Chordoma: Long-term follow-up after radical photon irradiation. Radiother. Oncol. 1996, 41, 67–72. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708–722. [Google Scholar] [CrossRef]
- Sun, S.; Jin, S.; Guo, R. Role of STAT3 in Resistance of Non-small Cell Lung Cancer. Zhongguo Fei Ai Za Zhi 2019, 22, 457–463. [Google Scholar] [CrossRef]
- Sahu, N.; Chan, E.; Chu, F.; Pham, T.; Koeppen, H.; Forrest, W.; Merchant, M.; Settleman, J. Cotargeting of MEK and PDGFR/STAT3 Pathways to Treat Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1729–1738. [Google Scholar] [CrossRef]
- Xia, M.; Huang, R.; Sakamuru, S.; Alcorta, D.; Cho, M.H.; Lee, D.H.; Park, D.M.; Kelley, M.J.; Sommer, J.; Austin, C.P. Identification of repurposed small molecule drugs for chordoma therapy. Cancer Biol. Ther. 2013, 14, 638–647. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med. 2019, 25, 292–300. [Google Scholar] [CrossRef]
- Siu, I.M.; Ruzevick, J.; Zhao, Q.; Connis, N.; Jiao, Y.; Bettegowda, C.; Xia, X.; Burger, P.C.; Hann, C.L.; Gallia, G.L. Erlotinib inhibits growth of a patient-derived chordoma xenograft. PLoS ONE 2013, 8, e78895. [Google Scholar] [CrossRef]
- Macaulay, V.M.; Middleton, M.R.; Eckhardt, S.G.; Rudin, C.M.; Juergens, R.A.; Gedrich, R.; Gogov, S.; McCarthy, S.; Poondru, S.; Stephens, A.W.; et al. Phase I Dose-Escalation Study of Linsitinib (OSI-906) and Erlotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2897–2907. [Google Scholar] [CrossRef]
- Akhavan-Sigari, R.; Gaab, M.R.; Rohde, V.; Abili, M.; Ostertag, H. Expression of PDGFR-alpha, EGFR and c-MET in spinal chordoma: A series of 52 patients. Anticancer Res. 2014, 34, 623–630. [Google Scholar]
- Scheipl, S.; Barnard, M.; Cottone, L.; Jorgensen, M.; Drewry, D.H.; Zuercher, W.J.; Turlais, F.; Ye, H.; Leite, A.P.; Smith, J.A.; et al. EGFR inhibitors identified as a potential treatment for chordoma in a focused compound screen. J. Pathol. 2016, 239, 320–334. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Naegeli, K.M.; East, M.P.; Laitinen, T.; Havener, T.M.; Wells, C.I.; Johnson, G.L.; Drewry, D.H.; Zuercher, W.J.; Morris, D.C. Design of a Cyclin G Associated Kinase (GAK)/Epidermal Growth Factor Receptor (EGFR) Inhibitor Set to Interrogate the Relationship of EGFR and GAK in Chordoma. J. Med. Chem. 2019, 62, 4772–4778. [Google Scholar] [CrossRef]
- Magnaghi, P.; Salom, B.; Cozzi, L.; Amboldi, N.; Ballinari, D.; Tamborini, E.; Gasparri, F.; Montagnoli, A.; Raddrizzani, L.; Somaschini, A.; et al. Afatinib Is a New Therapeutic Approach in Chordoma with a Unique Ability to Target EGFR and Brachyury. Mol. Cancer Ther. 2018, 17, 603–613. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Maffuid, K.A.; Laitinen, T.; Torrice, C.D.; Tizzard, G.J.; Crona, D.J.; Zuercher, W.J. Targeting an EGFR Water Network with 4-Anilinoquin(az)oline Inhibitors for Chordoma. ChemMedChem 2019, 14, 1693–1700. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ajay, A.K.; Zhao, L.; Vig, S.; Fujiwara, M.; Thakurela, S.; Jadhav, S.; Cho, A.; Chiu, I.J.; Ding, Y.; Ramachandran, K.; et al. Deletion of STAT3 from Foxd1 cell population protects mice from kidney fibrosis by inhibiting pericytes trans-differentiation and migration. Cell Rep. 2022, 38, 110473. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Li, D.; Saha, P.; Wang, R.; Zhang, H.; Ajay, A.K.; Deban, C.; Sukenick, G.; Azzi, J.; Lin, G. Discovery of Highly Selective Inhibitors of the Human Constitutive Proteasome beta5c Chymotryptic Subunit. J. Med. Chem. 2023, 66, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Singh, P.K.; Ban, Y.; Qing, X.; Ah Kioon, M.D.; Fan, H.; Zhao, Q.; Wang, R.; Sukenick, G.; Salmon, J.; et al. Structure-Activity Relationships of Noncovalent Immunoproteasome beta5i-Selective Dipeptides. J. Med. Chem. 2020, 63, 13103–13123. [Google Scholar] [CrossRef]
- Birmingham, A.; Selfors, L.M.; Forster, T.; Wrobel, D.; Kennedy, C.J.; Shanks, E.; Santoyo-Lopez, J.; Dunican, D.J.; Long, A.; Kelleher, D.; et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods 2009, 6, 569–575. [Google Scholar] [CrossRef]
- Sui, Y.; Wu, Z. Alternative statistical parameter for high-throughput screening assay quality assessment. J. Biomol. Screen 2007, 12, 229–234. [Google Scholar] [CrossRef]
- Martin, P.; Ruan, J.; Furman, R.; Rutherford, S.; Allan, J.; Chen, Z.; Huang, X.; DiLiberto, M.; Chen-Kiang, S.; Leonard, J.P. A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2917–2921. [Google Scholar] [CrossRef]
- Han, Q.; Bai, H.; Xu, Y.; Zhou, M.; Zhou, H.; Dong, X.; Chen, B. Solamargine induces autophagy-mediated apoptosis and enhances bortezomib activity in multiple myeloma. Clin. Exp. Pharmacol. Physiol. 2022, 49, 674–685. [Google Scholar] [CrossRef]
- Trucco, M.M.; Awad, O.; Wilky, B.A.; Goldstein, S.D.; Huang, R.; Walker, R.L.; Shah, P.; Katuri, V.; Gul, N.; Zhu, Y.J.; et al. A novel chordoma xenograft allows in vivo drug testing and reveals the importance of NF-kappaB signaling in chordoma biology. PLoS ONE 2013, 8, e79950. [Google Scholar] [CrossRef]
- Scheipl, S.; Barnard, M.; Lohberger, B.; Zettl, R.; Brcic, I.; Liegl-Atzwanger, B.; Rinner, B.; Meindl, C.; Frohlich, E. Drug combination screening as a translational approach toward an improved drug therapy for chordoma. Cell Oncol. 2021, 44, 1231–1242. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Kisselev, A.F. Site-Specific Proteasome Inhibitors. Biomolecules 2021, 12, 54. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Wei, W.; Zou, Y.; Jiang, Q.; Zhou, Z.; Ding, H.; Yan, L.; Yang, S. PSMB5 is associated with proliferation and drug resistance in triple-negative breast cancer. Int. J. Biol. Markers 2018, 33, 102–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, W.; Liu, Q.; Ma, Z.; Hu, S.; Zhang, Z.; Jia, H.; He, X. Expression of immunoproteasome subunits in the brains of Toxoplasma gondii-infected mice. Exp. Mol. Pathol. 2021, 123, 104684. [Google Scholar] [CrossRef]
- Kiuchi, T.; Tomaru, U.; Ishizu, A.; Imagawa, M.; Iwasaki, S.; Suzuki, A.; Otsuka, N.; Ohhara, Y.; Kinoshita, I.; Matsuno, Y.; et al. Expression of the immunoproteasome subunit beta5i in non-small cell lung carcinomas. J. Clin. Pathol. 2021, 74, 300–306. [Google Scholar] [CrossRef]
- Lu, S.; Yang, J.; Chen, Z.; Gong, S.; Zhou, H.; Xu, X.; Wang, J. Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Exp. Hematol. 2009, 37, 831–837. [Google Scholar] [CrossRef]
- Kwon, C.H.; Park, H.J.; Choi, Y.R.; Kim, A.; Kim, H.W.; Choi, J.H.; Hwang, C.S.; Lee, S.J.; Choi, C.I.; Jeon, T.Y.; et al. PSMB8 and PBK as potential gastric cancer subtype-specific biomarkers associated with prognosis. Oncotarget 2016, 7, 21454–21468. [Google Scholar] [CrossRef]
- Yang, B.Y.; Song, J.W.; Sun, H.Z.; Xing, J.C.; Yang, Z.H.; Wei, C.Y.; Xu, T.Y.; Yu, Z.N.; Zhang, Y.N.; Wang, Y.F.; et al. PSMB8 regulates glioma cell migration, proliferation, and apoptosis through modulating ERK1/2 and PI3K/AKT signaling pathways. Biomed. Pharmacother. 2018, 100, 205–212. [Google Scholar] [CrossRef]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Schwind, S.; Tarighat, S.S.; Geyer, S.; Eisfeld, A.K.; Whitman, S.; Walker, A.; Klisovic, R.; Byrd, J.C.; Santhanam, R.; et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood 2012, 119, 6025–6031. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Leleu, X.; Arnulf, B.; Zamagni, E.; Cibeira, M.T.; Kwok, F.; Mollee, P.; Hajek, R.; Moreau, P.; Jaccard, A.; et al. Bortezomib, Melphalan, and Dexamethasone for Light-Chain Amyloidosis. J. Clin. Oncol. 2020, 38, 3252–3260. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Kastritis, E.; Wechalekar, A.D.; Schonland, S.O.; Kim, K.; Sanchorawala, V.; Landau, H.J.; Kwok, F.; Suzuki, K.; Comenzo, R.L.; et al. A randomized phase 3 study of ixazomib-dexamethasone versus physician’s choice in relapsed or refractory AL amyloidosis. Leukemia 2021, 36, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Attar, E.C.; Johnson, J.L.; Amrein, P.C.; Lozanski, G.; Wadleigh, M.; DeAngelo, D.J.; Kolitz, J.E.; Powell, B.L.; Voorhees, P.; Wang, E.S.; et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J. Clin. Oncol. 2013, 31, 923–929. [Google Scholar] [CrossRef]
- Attar, E.C.; Amrein, P.C.; Fraser, J.W.; Fathi, A.T.; McAfee, S.; Wadleigh, M.; Deangelo, D.J.; Steensma, D.P.; Stone, R.M.; Foster, J.; et al. Phase I dose escalation study of bortezomib in combination with lenalidomide in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Leuk. Res. 2013, 37, 1016–1020. [Google Scholar] [CrossRef]
- Messinger, Y.H.; Gaynon, P.S.; Sposto, R.; van der Giessen, J.; Eckroth, E.; Malvar, J.; Bostrom, B.C.; Therapeutic Advances in Childhood, L.; Lymphoma, C. Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 2012, 120, 285–290. [Google Scholar] [CrossRef]
- Piperdi, B.; Ling, Y.H.; Liebes, L.; Muggia, F.; Perez-Soler, R. Bortezomib: Understanding the mechanism of action. Mol. Cancer Ther. 2011, 10, 2029–2030. [Google Scholar] [CrossRef]
- Barry, J.J.; Jian, B.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Tihan, T.; Parsa, A.T. The next step: Innovative molecular targeted therapies for treatment of intracranial chordoma patients. Neurosurgery 2011, 68, 231–240. [Google Scholar] [CrossRef]
- Petrelli, A.; Giordano, S. From single- to multi-target drugs in cancer therapy: When aspecificity becomes an advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef]
- Nan, G.; Huang, L.; Li, Y.; Yang, Y.; Yang, Y.; Li, K.; Lai, F.; Chen, X.; Xiao, Z. Identification of N, C-capped di- and tripeptides as selective immunoproteasome inhibitors. Eur. J. Med. Chem. 2022, 234, 114252. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| List of Primers Used for RT-PCR | |
|---|---|
| PSMB5 F | 5′-AGGAACGCATCTCTGTAGCAG-3′ |
| PSMB5 R | 5′-AGGGCCTCTCTTATCCCAGC-3′ |
| PSMB8 F | 5′-CCTTACCTGCTTGGCACCATGT-3′ |
| PSMB8 R | 5′-TTGGAGGCTGCCGACACTGAAA-3′ |
| GAPDH F | 5′-ACAACTTTGGTATCGTGGAAGG-3′ |
| GAPDH R | 5′-GCCATCACGCCACAGTTTC-3′ |
| Compound Name | Reagent ID | Average Z-Score | Z-Score (SD) | Function | Vendor |
|---|---|---|---|---|---|
| Amsacrine | HY-13551 | −8.79 | 0.141 | A potent intercalating antineoplastic agent. It is effective in the treatment of acute leukemias and malignant lymphomas but has poor activity in the treatment of solid tumors. | Medchem Express |
| ARRY-520 | HY-15187 | −7.33 | 0.332 | Filanesib (ARRY-520) is a synthetic kinesin spindle protein (KSP) inhibitor | Medchem Express |
| Ascomycin | 11309 | −7.56 | 1.782 | Ascomycin inhibits the production of Th1 (interferon- and IL-2) and Th2 (IL-4 and IL-10) cytokines. | Cayman Chemical |
| AZD-5438 | HY-10012 | −7.19 | 0.148 | AZD-5438 is a potent inhibitor of CDK1/2/9 | Medchem Express |
| AZD-7762 | HY-10992 | −9.02 | 0.403 | AZD-7762 is a potent ATP-competitive checkpoint kinase (Chk) inhibitor | Medchem Express |
| AZD8055 | SS-4787 | −7.15 | 0.919 | AZD-8055 is a novel ATP-competitive inhibitor of mTOR kinase. AZD-8055 inhibits both mTORC1 and mTORC2. | Key Organics |
| BAY 61-3606 | HY-14985 | −9.85 | 4.172 | BAY-61-3606 is a potent and selective inhibitor of Syk kinase | Medchem Express |
| BI 2536 | HY-50698 | −7.88 | 1.902 | Bl2356 is a notable dual PLK1 and BRD4 inhibitor. BI-2536 suppresses IFNB (encoding IFN-β) gene transcription. | Medchem Express |
| BNC105 | HY-16114 | −6.33 | 0.346 | BNC105 is a tubulin polymerization inhibitor with potent antiproliferative and tumor vascular disrupting properties. | Medchem Express |
| CBL0137 HCl | HY-18935A | −6.77 | 0.792 | CBL0137(CBL-0137) activates p53 and inhibits NF-kB | Medchem Express |
| CH5132799 | HY-15466 | −5.87 | 0.354 | CH5132799 is a selective class I PI3K inhibitor | Medchem Express |
| Cilengitide | HY-16141 | −10.05 | 4.879 | Cilengitide is a cyclic RGD-containing peptide that binds cancer cells expressing high concentrations of αVβ3 and αVβ5 integrins. | Medchem Express |
| Duvelisib | HY-17044 | −3.98 | 0.219 | Duvelisib is a Phosphoinositide 3-kinase inhibitor, specifically of the delta and gamma isoforms of PI3K | Medchem Express |
| Epoxomicin | HY-13821 | −9.65 | 0.134 | Epoxomicin (BU 4061T) is a naturally occurring selective proteasome inhibitor with anti-inflammatory activity. Epoxomicin covalently binds to the LMP7, X, MECL1, and Z catalytic subunits of the proteasome. | Medchem Express |
| Erlotinib | T0373 | −8.25 | 0.354 | Erlotinib is an epidermal growth factor receptor inhibitor (EGFR inhibitor). | Target Molecule |
| Flavopiridol | HY-10005 | −9.70 | 3.960 | Flavopiridol (Alvocidib) competes with ATP to inhibit CDKs including CDK1, CDK2, CDK4, CDK6, and CDK9 | Medchem Express |
| GSK-923295 | HY-10299 | −7.66 | 0.219 | Allosteric inhibitor of centromere-associated protein-E (CENP-E) kinesin motor ATPase activity | Medchem Express |
| Icotinib | HY-15164 | −5.31 | 0.686 | Icotinib Hydrochloride (BPI-2009) is a potent and specific EGFR inhibitor | Medchem Express |
| Ingenol-3-angelate | 16207 | −3.81 | 1.072 | Ingenol 3-Angelate is a protein kinase C activator | Cayman Chemical |
| Ispinesib | HY-50759 | −13.50 | 4.243 | Ispinesib selectively inhibits the mitotic motor protein, kinesin spindle protein (KSP), resulting in inhibition of mitotic spindle assembly, induction of cell cycle arrest during the mitotic phase, and cell death in tumor cells that are actively dividing. Because | Medchem Express |
| KPT-330 | S7252 | −10.72 | 0.205 | KPT-330, analog of KPT-185, is a selective inhibitor of CRM1. CRM1 is a nuclear export receptor involved in the active transport of transcription factors, cell-cycle regulators, tumor suppressors and RNA molecules. | Selleck Chemicals |
| LEE011 | HY-15777 | −3.76 | 0.035 | Ribociclib (LEE01) is a highly specific CDK4/6 inhibitor | Medchem Express |
| LY2090314 | HY-16294 | −5.87 | 1.478 | LY2090314 is a potent inhibitor of glycogen synthase kinase-3 | Medchem Express |
| LY2835219 | HY-16297 | −7.98 | 2.001 | Cyclophosphamide is a synthetic alkylating agent chemically related to the nitrogen mustards with antineoplastic activity, an immunosuppressant. | Medchem Express |
| MEK162 | HY-15202 | −4.32 | 0.792 | Binimetinib (MEK162) is an oral and selective MEK1/2 inhibitor. | Medchem Express |
| MG-132 | HY-13259 | −10.90 | 0.417 | MG132 is a potent cell-permeable proteasome and calpain inhibitor | Medchem Express |
| MI-773 (SAR405838) | S7649 | −8.13 | 0.488 | MI-773 is an Inhibitor of the MDM2-p53 interaction | Selleck Chemicals |
| MK 2206 | HY-10358 | −6.34 | 0.141 | MK-2206 dihydrochloride (MK-2206 (2HCl)) is an allosteric AKT inhibitor | Medchem Express |
| MLN 8237 | 2003 | −6.69 | 0.424 | Alisertib (MLN 8237) induces apoptosis and autophagy through targeting the AKT/mTOR/AMPK/p38 pathway in leukemic cells. Antitumor activity. | Axon Medchem |
| Nutlin (3a) | HY-10029 | −7.81 | 0.021 | Nutlin 3a is an active enantiomer of Nutlin-3, acts as a murine double minute (MDM2) inhibitor that inhibits MDM2-p53 interactions and stabilizes the p53 protein, and thereby induces cell cycle arrest and apoptosis. | Medchem Express |
| NVP-BEP800 | 18383 | −5.33 | 1.287 | Ingenol-3-angelate causes inflammation due, at least in part, to activation of PKC, leading to antibody-dependent cellular cytotoxicity | Cayman Chemical |
| NVP-TAE 226 | HY-13203 | −7.35 | 1.407 | NVP-TAE 226 (TAE226) is a potent and ATP-competitive dual FAK and IGF-1R | Medchem Express |
| Obatoclax | HY-10969 | −13.05 | 4.313 | Obatoclax is an inhibitor of the Bcl-2 family of proteins. | Medchem Express |
| ONX 0914 | 16271 | −8.81 | 1.882 | ONX is a selective inhibitor of the β5i (LMP7) subunit of the immunoproteasome | Cayman Chemical |
| OSI-027 | HY-10423 | −5.39 | 0.085 | OSI-027 is an ATP-competitive mTOR kinase activity inhibitor. OSI-027 targets both mTORC1 and mTORC2 | Medchem Express |
| OTSSP167 (hydrochloride) | HY-15512A | −13.40 | 4.384 | OTSSP167, also known as OTS167, is an orally available inhibitor of maternal embryonic leucine zipper kinase (MELK) with potential antineoplastic activity. | Medchem Express |
| palbociclib | THR0011 | −5.70 | 0.849 | Selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6 | Pharmablock |
| PF-431396 | HY-10460 | −7.41 | 0.021 | PF-431396 is a potent and selective focal adhesion kinase (FAK) and proline-rich tyrosine kinase 2 (PYK2) inhibitor | Medchem Express |
| PKC412 | HY-10230 | −7.05 | 1.704 | Midostaurin (PKC412; CGP 41251) is a multi-targeted protein kinase inhibitor which inhibits PKCα/β/γ, Syk, Flk-1, Akt, PKA, c-Kit, c-Src, PDFRβ and VEGFR1/2 | Medchem Express |
| PKI-587 | HY-10681 | −8.36 | 0.035 | Gedatolisib, also known as PKI-587 and PF-05212384, is an agent targeting the phosphatidylinositol 3 kinase (PI3K) and mammalian target of rapamycin (mTOR) in the PI3K/mTOR signaling pathway, with potential antineoplastic activity | Medchem Express |
| Quisinostat | HY-15433 | −8.58 | 2.107 | Quisinostat is a "second generation" histone deacetylase inhibitor with antineoplastic activity.It is highly potent against class I and II HDACs | Medchem Express |
| Radicicol | BIR0140 | −7.70 | 0.495 | Radicicol inhibits the activities of Hsp90, Topo VI and PDK3 by blocking ATP binding to them. | Apollo Scientific |
| RG7388 | HY-15676 | −9.25 | 0.141 | Idasanutlin (RG7388) is a potent and selective MDM2 antagonist | Medchem Express |
| Ruboxistaurin | HY-10195B | −13.00 | 4.808 | LY333531 is a potent inhibitor of protein kinase Cβ | Medchem Express |
| SGI-1776 | HY-13287 | −8.44 | 1.768 | SGI-1776 is an inhibitor of Pim kinases | Medchem Express |
| TAK-285 | HY-15196 | −5.74 | 0.955 | TAK-285 is a potent, selective, ATP-competitive and orally active HER2 and EGFR(HER1) inhibitor | Medchem Express |
| Tipifarnib | HY-10502 | −9.51 | 0.120 | Tipifarnib, is a farnesyltransferase inhibitor. It inhibits the Ras kinase in a post translational modification step before the kinase pathway becomes hyperactive. | Medchem Express |
| Tozasertib | HY-10161 | −6.29 | 0.339 | Tozasertib (VX 680; MK-0457) is an inhibitor of Aurora A/B/C kinases | Medchem Express |
| Volasertib | HY-12137 | −12.15 | 0.071 | BI6727 (Volasertib) is a selective inhibitor of Plk1, Plk2, and Plk3 | Vitas M Labs |
| Zotarolimus | HY-12424 | −4.31 | 0.587 | Zotarolimus is mechanistically similar to sirolimus in having high-affinity binding to the immunophilin FKBP12 and comparable potency for inhibiting in vitro proliferation of both human and rat T cells. | Medchem Express |
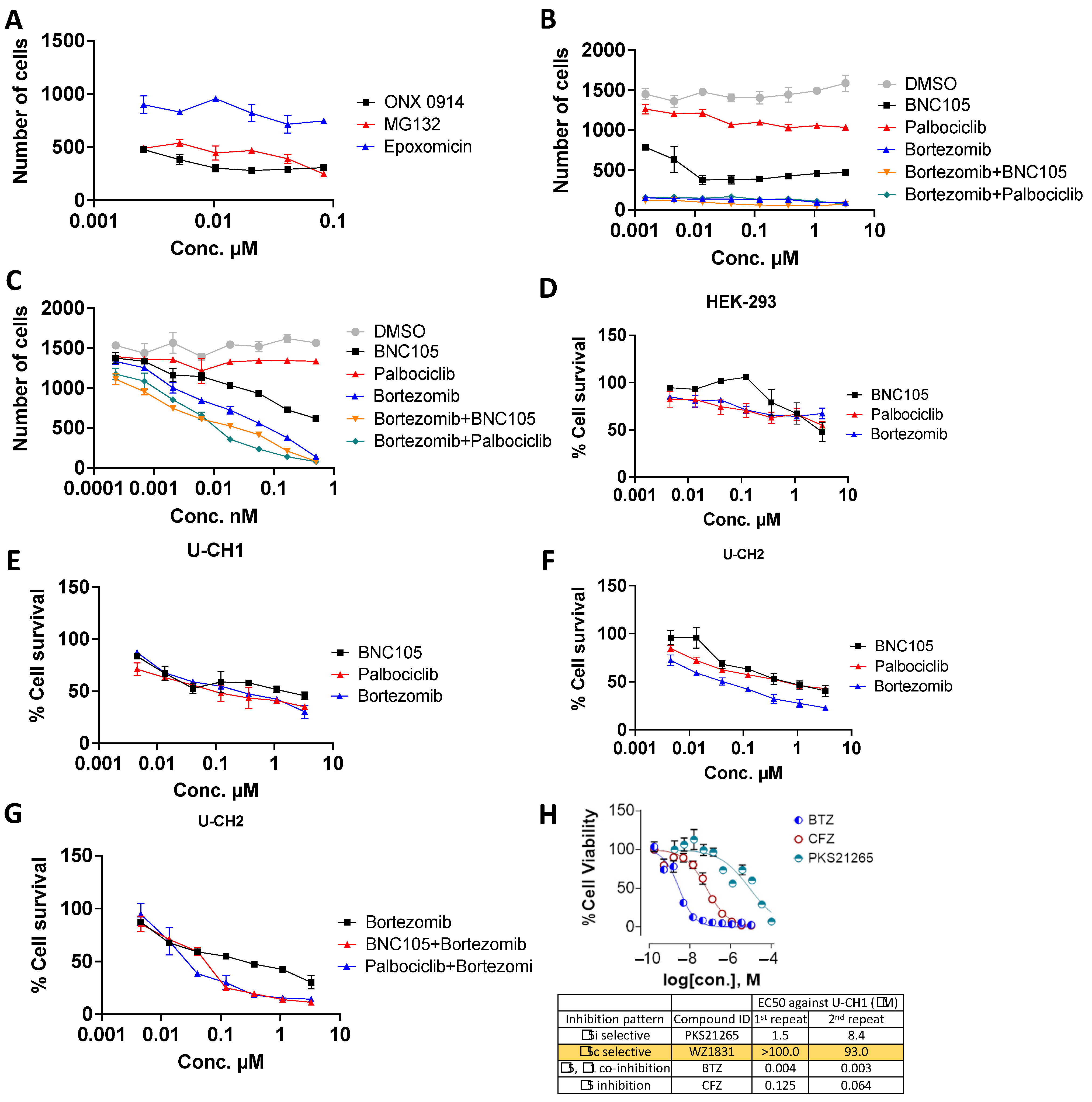
| Compound Name | IC50 (μM) |
|---|---|
| ARRY-520 | 1.086 |
| BAY 61-3606 (dihydrochloride) | 1.229 |
| BNC105 | 0.9265 |
| Bortezomib | 2.31 |
| Icotinib (Hydrochloride) | 1.129 |
| MG-132 | 0.03263 |
| MLN 8237 | 0.02419 |
| OSI-027 | 0.9978 |
| Palbociclib | 0.1067 |
| Tipifarnib | 0.9954 |
| Compound Name | IC50 (μM) |
|---|---|
| HEK-293 | |
| BNC105 | 0.8489 |
| Bortezomib | 0.8969 |
| Palbociclib | 0.9903 |
| U-CH1 | |
| BNC105 | 0.9539 |
| Bortezomib | 0.6041 |
| Palbociclib | 0.5906 |
| U-CH2 | |
| BNC105 | 0.2939 |
| Bortezomib | 0.5761 |
| Palbociclib | 0.7048 |
| U-CH2 | |
| Bortezomib | 0.6041 |
| BNC105+Bortezomib | 0.3805 |
| Palbociclib+Bortezomib | 0.4477 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajay, A.K.; Chu, P.; Patel, P.; Deban, C.; Roychowdhury, C.; Heda, R.; Halawi, A.; Saad, A.; Younis, N.; Zhang, H.; et al. High-Throughput/High Content Imaging Screen Identifies Novel Small Molecule Inhibitors and Immunoproteasomes as Therapeutic Targets for Chordoma. Pharmaceutics 2023, 15, 1274. https://doi.org/10.3390/pharmaceutics15041274
Ajay AK, Chu P, Patel P, Deban C, Roychowdhury C, Heda R, Halawi A, Saad A, Younis N, Zhang H, et al. High-Throughput/High Content Imaging Screen Identifies Novel Small Molecule Inhibitors and Immunoproteasomes as Therapeutic Targets for Chordoma. Pharmaceutics. 2023; 15(4):1274. https://doi.org/10.3390/pharmaceutics15041274
Chicago/Turabian StyleAjay, Amrendra K., Philip Chu, Poojan Patel, Christa Deban, Chitran Roychowdhury, Radhika Heda, Ahmad Halawi, Anis Saad, Nour Younis, Hao Zhang, and et al. 2023. "High-Throughput/High Content Imaging Screen Identifies Novel Small Molecule Inhibitors and Immunoproteasomes as Therapeutic Targets for Chordoma" Pharmaceutics 15, no. 4: 1274. https://doi.org/10.3390/pharmaceutics15041274