
Artificial Intelligence in Drug Metabolism and Excretion Prediction: Recent Advances, Challenges, and Future Perspectives


Abstract
:1. Introduction
2. Evaluation Metrics
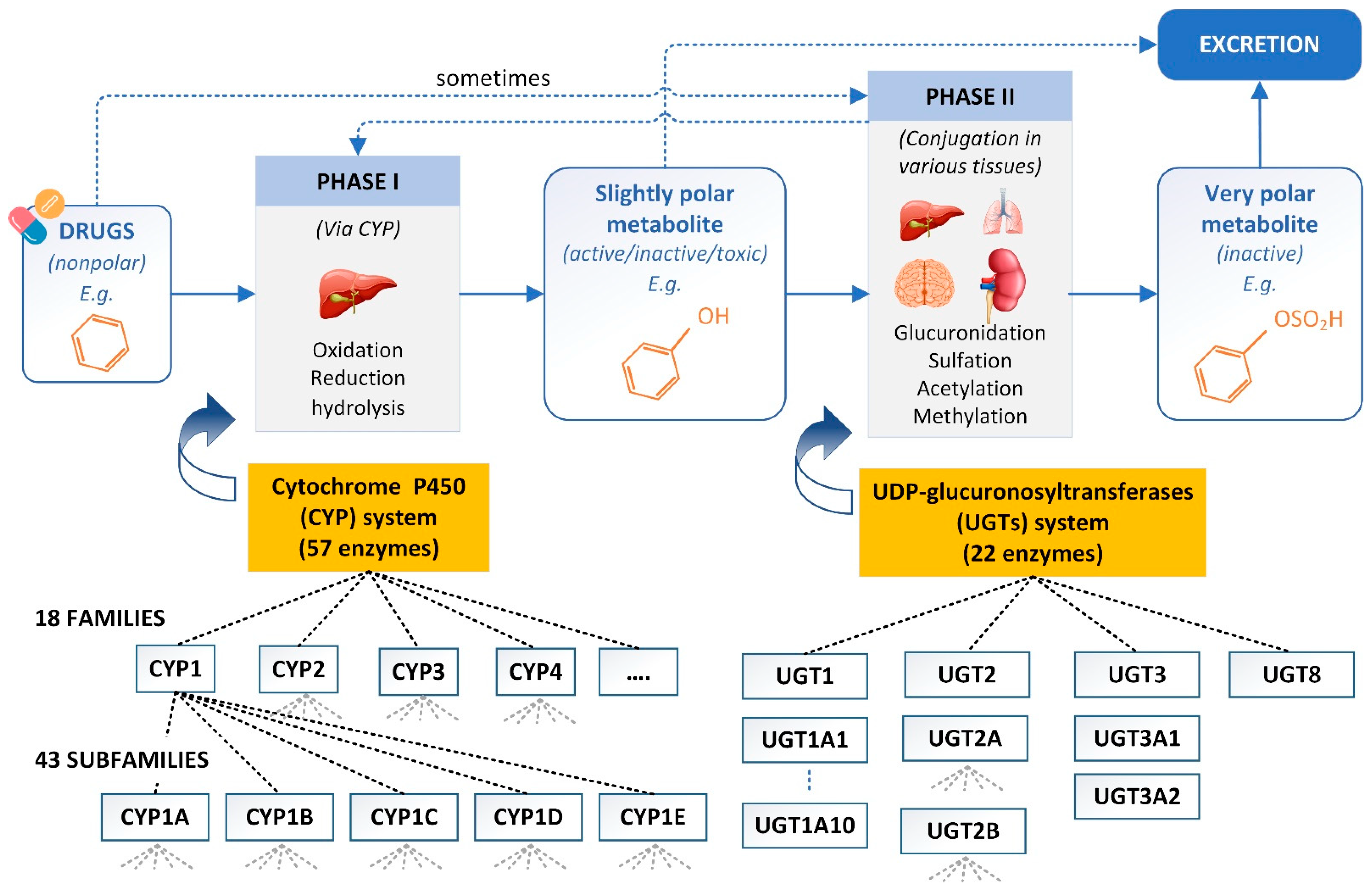
3. Drug Metabolism Prediction
3.1. SOMs and Metabolite Structure Predictions

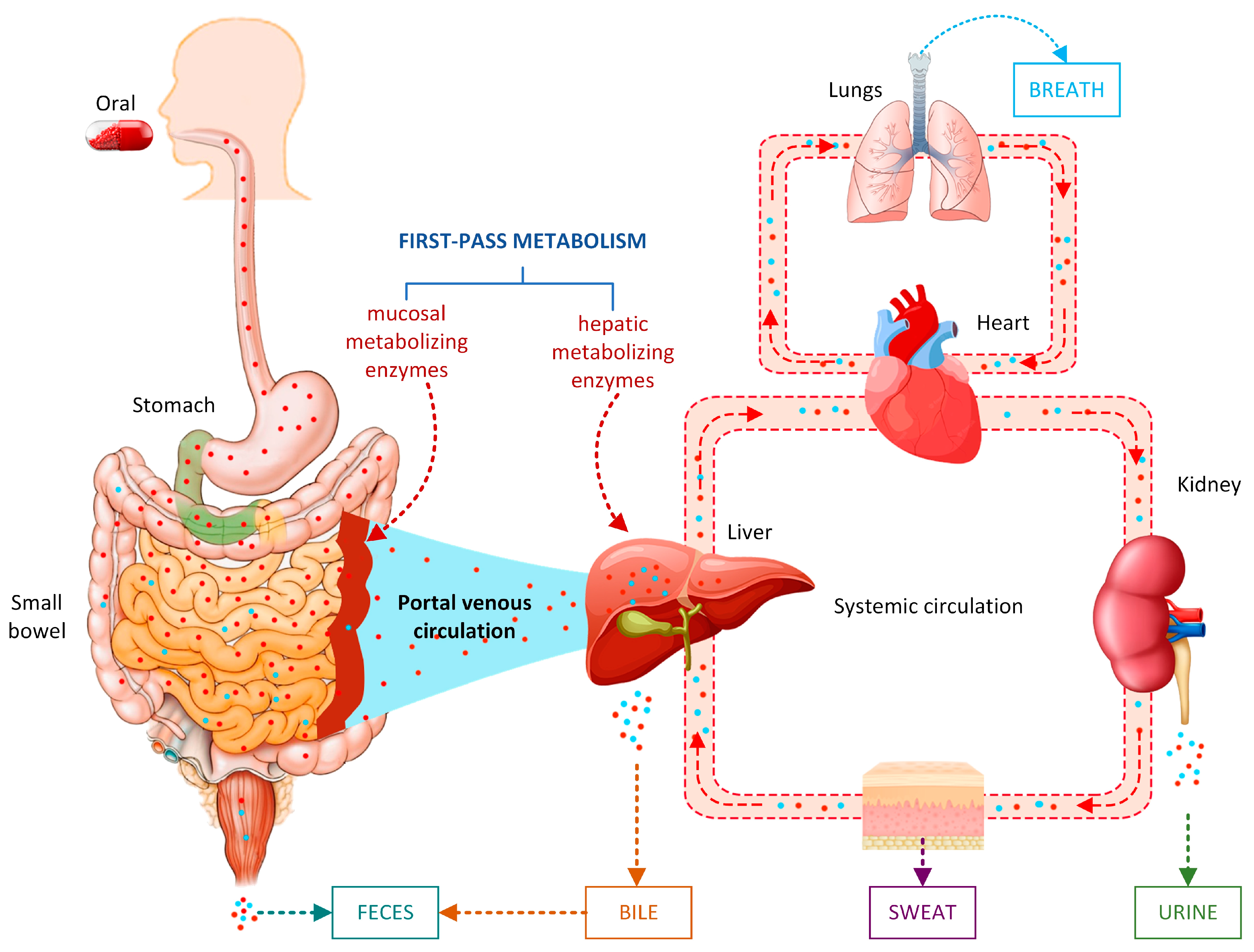{kind=link}
{kind=link}
| Name | Metabolism Prediction | Methods | Website * | Ref. |
|---|---|---|---|---|
| CyProduct (CypReact, CypBoM, MetaboGen) | Reactant, BoM for CYP, metabolite structure | ML | https://bitbucket.org/wishartlab/cyproduct/src/master/ | [29] |
| GLORYx | Metabolite structure | ML | https://nerdd.univie.ac.at/gloryx/ | [27] |
| FAME 3 | Phase 1 and 2 SOMs for CYP | ML | https://nerdd.univie.ac.at/fame3/ | [26] |
| BioTransformer 3.0 | Metabolic transformation | rule-based/knowledge-baseb, ML | http://biotransformer.ca/ | [28] |
| PreMetabo | Phase 1 and 2 SOMs for CYP, UGT, and SULT | Arrhenius equation and EaMEAD model | https://premetabo.bmdrc.kr/ | [30] |
| SMARTCyp 3.0 | SOMs for CYP | rule-based | http://smartcyp.sund.ku.d/ | [33] |
| HelixADMET | CYP inhibitors and substrates | GNN | https://paddlehelix.baidu.com/app/drug/admet/train | [34] |
| Interpretable-ADMET | CYP inhibitors and substrates | GAT, GCNN | http://cadd.pharmacy.nankai.edu.cn/interpretableadmet/ | [35] |
| FP-ADMET | CYP inhibitors and substrates | RF | https://gitlab.com/vishsoft/fpadmet | [36] |
| ADMETlab 2.0 | CYP inhibitors and substrates | GCNN | https://admetmesh.scbdd.com/ | [37] |
| AdmetSAR 2.0 | CYP inhibitors and substrates | RF, k-NN, SVM | http://lmmd.ecust.edu.cn/admetsar2/ | [38] |
| SwissADME | CYP inhibitors | MLR, RNN, SVM | http://www.swissadme.ch/ | [39] |
| ICDrug ADMET | CYP inhibitors and substrates | RF | www.icdrug.com/ICDrug/ADMET | [40] |
| Virtual Rat | CYP inhibitors | RF | https://virtualrat.cmdm.tw/ | [9] |
| DL-CYP | CYP inhibitors | DNN | http://www.pkumdl.cn/deepcyp/home.php | [41] |
| CYPstrate | CYP substrates | RF, SVM | https://nerdd.univie.ac.at/cypstrate/ | [42] |
| CYPlebrity | CYP inhibitors | RF | https://nerdd.univie.ac.at/cyplebrity/ | [43] |
| SuperCYPsPred | CYP inhibitors | RF | http://insilico-cyp.charite.de/SuperCYPsPred/ | [44] |
3.2. CYP Inhibitor and Substrate Prediction
| CYP Subtypes | Methods | Data Sources | Dataset Size (Compounds) | Best Performance | Ref. |
|---|---|---|---|---|---|
| 1A2, 2C19, 2D6, 2C9 and 3A4 inhibitors | RF | PubChem, SuperCYP | 18,313 | ACC = 0.97, AUC = 0.98 | [44] |
| 1A2, 2C9, 2C19, 2D6 and 3A4 inhibitors | RF | ChEMBL, PubChem, ADME | 134,844 | AUC = 0.92, ACC = 0.83 | [43] |
| 1A2, 2D6, 2C9, 2C8, 2C19, and 3A4 inhibitors | RF | [52,57] | 17,652 | ACC = 0.868, AUC = 0.741 | [36] |
| CYPs 1A2, 2C9, 2C19, 2D6 and 3A4 inhibitors | RF, SVM, k-NN | PubChem | 65,467 | AUC =0.93 | [46] |
| 1A2, 2D6, 2C9, 2C19, and 3A4 inhibitors 2D6, 2C9, and 3A4 substrate | RF, SVM, k-NN | [57] | 77,490 2018 | ACC = 0.855, AUC = 0.84 | [38] |
| 1A2, 2D6, 2C9, 2C19, and 3A4 inhibitors | DT | [58,59] | 64,129 | ACC = 0.93, Recall = 0.924 | [9] |
| 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4 substrates | Improved Bayesian method | SuperCYP [60], PubChem, DrugBank, CYP450 Engineering Database [61,62], Meta-CYP | 7114 | AUC = 0.92, ACC = 0.90 | [49] |
| 1A2, 2C19, 2C9, 2D6, and 3A4 inhibitors 1A2, 2C19, 2C9, 2D6, and 3A4 substrates | MGAF | ChEMBL, PubChem, OCHEM, literature | 62,771 (inhibitors) 3291 (substrates) | ACC = 0.886, AUC = 0.948 | [37] |
| 1A2, 2C19, 2C9, 2D6, and 3A4 inhibitors2C9, 2D6, and 3A4 substrates | GCNN, GAT | ChEMBL, PubChem, DrugBank, literature | 63,921 (inhibitors)2053 (substrates) | ACC = 0.85, AUC = 0.93 | [35] |
| 1A2, 2C19, 2C9, 2D6, and 3A4 inhibitors 1A2, 2C19, 2C9, 2D6, and 3A4 substrates | GNN | PubChem, CypReact [13], SuperCYP [44] | 64,801 (inhibitors) 9233 (substrates) | AUC = 0.967 | [34] |
| 1A2, 2D6, 2C9, and 2C19 inhibitors | RF, GBDT, XGB, DNN, CNN | [41] | 53,179 | ACC = 0.974, AUC = 0.991 | [63] |
| 1A2, 2C9, 2C19, 2D6 and 3A4 inhibitor | MT-DNN | PubChem | 153,484 | AUC = 0.937, ACC = 0.895 | [48] |
| 2C8 inhibitors | RF, SVM, k-NN, LR, ANN | PubChem and literature [64,65] | 514 | AUC = 0.90, ACC = 0.89 | [52] |
| 2C9 inhibitors | RF, SVM | ChEMBL | 8141 | ACC = 0. 843, MCC = 0.695 | [54] |
| 2C9 inhibitors | BT, multilayer feedforward of resilient backpropagation network | PubChem | >35,000 | AUC = 0.85 | [53] |
| 3A4 inhibitors | GCNN combined with the MT-DNN | ChEMBL and [66] | 3774 | R2 = 0.692 | [67] |
| 89,619 | R2 = 0.414 | ||||
| 3A4 inhibitors | SVM, XGB, and RF | In-house, public | 30,768 | ACC = 0.927, sensitivity = 0.788 | [56] |
| In-house | 26,138 | ACC = 0.90, AUC = 0.908 | |||
| 1B1 inhibitors | RF, SVM, ANN | ChEMBL, Pubchem, and [68,69,70] | 714 | MCC = 0.95 | [50] |
| 1A1 inhibitors | 658 | MCC = 0.96 | |||
| 1A2 inhibitors | CNN | PubChem | 21,721 | ACC = 0.722, AUC = 0.819 | [51] |
3.3. UGTs Prediction
| Methods | Data Sources | Dataset Size (Compounds) | Performance | Ref. |
|---|---|---|---|---|
| RF | MetaQSAR | 7962 | MCC = 0.76, AUC = 0.94 | [73] |
| DT, RF, AdaBoost | Handbook of Metabolic Pathways of Xenobiotics [76], KEGG [77,78,79] | 586 | ACC = 0.867, AUC = 0.928 | [75] |
| LR, SVM | Fujitsu ADME database and MDL metabolism database | 200 | ACC = 0.81 | [30] |
4. Drug Excretion Prediction
4.1. Clearance Prediction
| Methods | Data Sources | Dataset Size (Compounds) | Performance | Ref. |
|---|---|---|---|---|
| RF | Human renal clearance [85] | 636 | R2 = 0.27, RMSE = 0.53 | [36] |
| Intrinsic clearance [84] | 244 | R2 = 0.29, RMSE = 1.02 | ||
| Metabolic intrinsic clearance [83] | 5278 | ACC = 0.74, AUC = 0.84 | ||
| Human liver microsomal clearance [66] | 5348 | R2 = 0.56, RMSE = 1.05 | ||
| RF, SVM, GBM, XGB | [92] | 1352 | R2 = 0.875, RMSE = 0.103 | [86] |
| RFR, RBF, PLS, GP2DS, GPFixed, GPFVS, GPRFVS, GPOPT | Takeda Pharmaceutical Company (Fujisawa, Japan) | 1114 | R2 = 0.61, RMSE = 0.31 | [87] |
| SVM | AstraZeneca in-house data | 73,620 | R2 = 0.356, RMSE = 0.377 | [89] |
| RF, NB, SVM, CT, k-NN, MLR, ANN | FDA drugs and [93,94,95,96] | 636 | R2 = 0.94, RMSE = 0.11 | [85] |
| RF, AdaBoost, Radial SVM, Linear SVM | ChEMBL v.23, KEGG DRUG [97] | 56,065 | ACC = 0.77, Kappa = 0.588 | [83] |
| RF, SVM, PLS, ANN | ChEMBL and Varma et al. [98] | 401 | R2= 0.92, RMSE = 0.12 | [88] |
| Combination conventional ML and DeepSnap-DL | in-house | 1545 | AUC = 0.943, ACC = 0.874 | [90] |
| ANN | Medivir in-house | 4794 | R2 = 0.717, RMSE = 0.327 | [91] |
| GCNN | ChEMBL, PubChem, OCHEM, literature | 831 | R2 = 0.692 | [37] |
| a molecular GCNN combined with the MT-DNN | [66] | 5348 | R2 = 0.62 | [67] |
| Amgen’s internal datasets | 86,470 | R2 = 0.445 | ||
| MT-DNN | ChEMBL v.23 | 5384 | R2 = 0.624 | [66] |
| MT-CNN | AstraZeneca | 139,907 | R2 = 0.59, RMSE = 0.35 | [99] |
4.2. Half-Life Prediction
| Methods | Data Sources | Dataset Size (Compounds) | Performance | Ref. |
|---|---|---|---|---|
| RF | [102] | 2127 | ACC = 0.76, AUC = 0.88 | [36] |
| SVM, RF, GBM, XGB | [92] | 1352 | R2 = 0.832, RMSE = 0.154 | [86] |
| MGAF | ChEMBL, PubChem, OCHEM, literature | 1219 | AUC = 0.822, ACC = 0.744 | [37] |
| GCNN, GAT | ChEMBL, PubChem, DrugBank, literature | 665 | ACC = 0.773, AUC = 0.766 | [35] |
5. Data Sources for Research Community
- HMDB 5.0 (https://hmdb.ca/ accessed on 22 January 2023): An extensive database of small molecule metabolites discovered in the human body, including information on their chemical and physical properties, metabolic pathways, and clinical biomarkers. Information on more than 220,000 metabolites and 8500 protein sequences can be found in HMDB. [103].
- METLIN (https://metlin.scripps.edu/ accessed on 22 January 2023): a metabolite database that contains information on more than 960,000 compounds [104]. It includes information on the chemical structure, molecular formula, and biological activities of metabolites. METLIN offers MS/MS data on various collision energy values in both positive and negative ionization modes. Additionally, it makes use of the elemental makeup, precise mass measurements, and the known structure of the metabolite to estimate the fragmented structure. The metabolomics-specific mobile interface METLIN Mobile allows you to see metabolite information from any cellular device.
- MetaCyc (https://metacyc.org/ accessed on 23 January 2023): A curated database of metabolic pathways and enzymes for a range of organisms. It includes information on 3085 pathways, 18,785 metabolites, and 18,391 reactions involved in metabolite biotransformation and can be used to construct metabolic models for specific organisms.
- MetaQSAR: A database for metabolites including information on the relationship between the chemical structure of a metabolite, its biological activity, the physicochemical properties of chemicals, as well as their predicted metabolic pathways and associated enzymes. It is a plug-in embedded in the VEGA ZZ programs (http://www.vegazz.net/ accessed on 23 January 2023) and contains 1890 substrates [74].
- MetXBioDB (https://bitbucket.org/djoumbou/biotransformerjar/src/master/ accessed on 23 January 2023): A database of metabolic pathways and enzymes for a range of organisms, including bacteria, archaea, and eukaryotes. MetXBioDB contains data on more than 2000 biotransformation including information on the structure and function of enzymes, as well as the reactions and pathways involved in metabolite biotransformation [28].
- Metabolights (https://www.ebi.ac.uk/metabolights/ accessed on 24 January 2023): A database of metabolomic data, which includes information on metabolites, metabolic pathways, and metabolic networks of more than 27,500 compounds. Metabolights also includes tools for data analysis and visualization, as well as resources for sharing and reusing metabolomic data [105].
- KEGG Pathway (https://www.genome.jp/kegg/pathway.html accessed on 24 January 2023): A database of metabolic pathways, including maps and diagrams of metabolic networks, as well as information on enzymes and metabolites. It includes information on more than 17,000 metabolic pathways and over 22,000 enzymes [106].
- HumanCyc (https://humancyc.org/ accessed on 24 January 2023): A curated database of metabolic pathways, enzymes for human metabolism, and the human genome. HumanCyc includes information on the reactions and pathways involved in metabolite biotransformation, as well as the enzymes and genes involved in these processes. Information on 28,783 genes, their products, and the metabolic processes and pathways they catalyze is contained in the pathway/genome database that was created as a consequence [107].
- BiGG (http://bigg.ucsd.edu/ accessed on 24 January 2023): In order to simulate systems biology and predict metabolic flux balance, the BiGG database reconstructs human metabolism metabolically. The 1496 ORFs, 2004 protein complexes, 2766 metabolites, and 3311 metabolic and transport processes are all included in this thorough literature-based genome-scale metabolic reconstruction. It was put together from building 35 of the human genome [108].
- DrugBank (http://www.drugbank.ca/ accessed on 24 January 2023): A comprehensive database of drug and drug target information including information on drug metabolism and pharmacokinetics, as well as the enzymes involved in drug biotransformation. It contains information on more than 500,000 drugs and their associated targets, pathways, and metabolic pathways [109].
- ChEMBL (www.ebi.ac.uk/chembl/ accessed on 24 January 2023): A database of bioactive molecules, including drugs and drug candidates, with information on their activities, targets, and metabolic pathways. It contains data on more than 2.3 million compounds and their associated activities and targets [110].
- ChemSpider (http://www.chemspider.com/ accessed on 24 January 2023): A chemical structure database that includes information on more than 115 million compounds including information on chemical structures, properties, and associated metadata, such as chemical identifiers and references [111].
- PubChem (https://pubchem.ncbi.nlm.nih.gov/ accessed on 25 January 2023): A public database of chemical structures and their associated biological activities including information on more than 114 million compounds, as well as tools for data analysis and visualization [112].
- ZINC20 (https://zinc20.docking.org/ accessed on 25 January 2023): A database of commercially available compounds for drug discovery including information on more than 750 million purchasable compounds, as well as tools for searching and filtering compounds based on various criteria, such as molecular weight, bioavailability, and toxicity [113].
- OCHEM (https://ochem.eu/home/show.do accessed on 25 January 2023): A platform for the development and validation of predictive models for chemical and biological data. OCHEM includes tools for data preprocessing, feature selection, and model training, as well as a library of pre-trained models. OCHEM contains more than 3.7 million records for 689 properties [114].
- Therapeutics Data Commons (TDC) (https://tdcommons.ai/ accessed on 25 January 2023): A database of clinical trial data for FDA-approved drugs including information on drug pharmacokinetics, pharmacodynamics, and adverse events, as well as data on drug metabolism and excretion. TDC contains data on more than 4.2 million compounds, 34,000 genes, and approximately 2 million reactions [115].
- openFDA (https://open.fda.gov/ accessed on 25 January 2023): A database of FDA-approved drugs, including information on drug labeling, adverse events, and clinical trial data. OpenFDA includes tools for data analysis and visualization, as well as an API for accessing FDA data [116].
6. Challenges in Drug Metabolism and Excretion Prediction Based on AI
7. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Talevi, A.; Bellera, C.L. Drug excretion. In ADME Processes in Pharmaceutical Sciences; Talevi, A., Quiroga, P., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Ha, C.-E.; Bhagavan, N.V. Chapter 35—Drug metabolism. In Essentials of Medical Biochemistry, 3rd ed.; Ha, C.-E., Bhagavan, N.V., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 795–805. [Google Scholar]
- Susa, S.T.; Preuss, C.V. Drug Metabolism; Statpearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Barreto, E.F.; Larson, T.R.; Koubek, E.J. Drug Excretion; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Gad, S.C.; Spainhour, C.B. Nonclinical Drug Administration: Formulations, Routes and Regimens for Solving Drug Delivery Problems in Animal Model Systems; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Mackenzie, P.I.; Somogyi, A.A.; Miners, J.O. Advances in drug metabolism and pharmacogenetics research in Australia. Pharmacol. Res. 2017, 116, 7–19. [Google Scholar] [CrossRef]
- Lai, Y.; Chu, X.; Di, L.; Gao, W.; Guo, Y.; Liu, X.; Lu, C.; Mao, J.; Shen, H.; Tang, H. Recent advances in the translation of drug metabolism and pharmacokinetics science for drug discovery and development. Acta Pharm. Sin. B 2022. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.; Su, B.H.; Tseng, Y.J. Current development of integrated web servers for preclinical safety and pharmacokinetics assessments in drug development. Brief Bioinform. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, S.R.; Jun, R.; Yu, M.-S.; Jung, C.; Na, D. In silico approaches and tools for the prediction of drug metabolism and fate: A review. Comput. Biol. Med. 2019, 106, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.F. Artificial intelligence in drug safety and metabolism. Artif. Intell. Drug Des. 2022, 483–501. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Tian, S.; Djoumbou-Feunang, Y.; Greiner, R.; Wishart, D.S. Cypreact: A Software Tool for in Silico Reactant Prediction for Human Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2018, 58, 1282–1291. [Google Scholar] [CrossRef]
- Phang-Lyn, S.; Llerena, V.A. Biochemistry, biotransformation. In Statpearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Zheng, M.; Luo, X.; Shen, Q.; Wang, Y.; Du, Y.; Zhu, W.; Jiang, H. Site of metabolism prediction for six biotransformations mediated by cytochromes P450. Bioinformatics 2009, 25, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.; Melton, R.; Thompson, D. Drug metabolism in drug discovery and preclinical development. In Drug Metabolism; IntechOpen: London, UK, 2021. [Google Scholar]
- Litsa, E.E.; Das, P.; Kavraki, L.E. Machine learning models in the prediction of drug metabolism: Challenges and future perspectives. Expert Opin. Drug Met. 2021, 17, 1245–1247. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed]
- de Bruyn Kops, C.; Stork, C.; Sicho, M.; Kochev, N.; Svozil, D.; Jeliazkova, N.; Kirchmair, J. GLORY: Generator of the Structures of Likely Cytochrome P450 Metabolites Based on Predicted Sites of Metabolism. Front Chem. 2019, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Williamson, M.J.; Afzal, A.M.; Tyzack, J.D.; Choy, A.P.; Howlett, A.; Rydberg, P.; Glen, R.C. FAst MEtabolizer (FAME): A rapid and accurate predictor of sites of metabolism in multiple species by endogenous enzymes. J. Chem. Inf. Model. 2013, 53, 2896–2907. [Google Scholar] [CrossRef]
- Sicho, M.; de Bruyn Kops, C.; Stork, C.; Svozil, D.; Kirchmair, J. FAME 2: Simple and effective machine learning model of cytochrome P450 regioselectivity. J. Chem. Inf. Model. 2017, 57, 1832–1846. [Google Scholar] [CrossRef]
- Sicho, M.; Stork, C.; Mazzolari, A.; Kops, C.D.; Pedretti, A.; Testa, B.; Vistoli, G.; Svozil, D.; Kirchmair, J. FAME 3: Predicting the Sites of Metabolism in Synthetic Compounds and Natural Products for Phase 1 and Phase 2 Metabolic Enzymes. J. Chem. Inf. Model. 2019, 59, 3400–3412. [Google Scholar] [CrossRef]
- de Bruyn Kops, C.; Sicho, M.; Mazzolari, A.; Kirchmair, J. GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics. Chem. Res. Toxicol. 2021, 34, 286–299. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la-Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. Biotransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Cao, X.; Greiner, R.; Li, C.; Guo, A.; Wishart, D.S. Cyproduct: A Software Tool for Accurately Predicting the Byproducts of Human Cytochrome P450 Metabolism. J. Chem. Inf. Model. 2021, 61, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Shin, H.K.; Shin, S.E.; Seo, M.; Jeon, H.N.; Yim, D.E.; Kim, D.H.; No, K.T. PreMetabo: An in silico phase I and II drug metabolism prediction platform. Drug Metab. Pharm. 2020, 35, 361–367. [Google Scholar] [CrossRef]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Gloriam, D.E.; Zaretzki, J.; Breneman, C.; Olsen, L. SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug Metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.; Montefiori, M.; Tran, K.P.; Jorgensen, F.S. SMARTCyp 3.0: Enhanced cytochrome P450 site-of-metabolism prediction server. Bioinformatics 2019, 35, 3174–3175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, Z.; Huang, Y.; Liu, L.; He, D.; Wang, W.; Fang, X.; Zhang, X.; Wang, F.; Wu, H.; et al. HelixADMET: A robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer. Bioinformatics 2022, 38, 3444–3453. [Google Scholar] [CrossRef]
- Wei, Y.; Li, S.; Li, Z.; Wan, Z.; Lin, J. Interpretable-ADMET: A Web Service for ADMET Prediction and Optimization based on Deep Neural Representation. Bioinformatics 2022, 38, 2863–2871. [Google Scholar] [CrossRef]
- Venkatraman, V. FP-ADMET: A compendium of fingerprint-based ADMET prediction models. J. Cheminform. 2021, 13. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zhang, X.; Pan, X.; Wang, B.; Ji, C.; Qi, Y.; Zhang, J.Z. HobPre: Accurate prediction of human oral bioavailability for small molecules. J. Cheminform. 2022, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Y.J.; Lai, L.H.; Pei, J.F. Prediction of human cytochrome P450 inhibition using a multitask deep autoencoder neural network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef]
- Stork, C.; Embruch, G.; Šícho, M.; de Bruyn Kops, C.; Chen, Y.; Svozil, D.; Kirchmair, J. NERDD: A web portal providing access to in silico tools for drug discovery. Bioinformatics 2020, 36, 1291–1292. [Google Scholar] [CrossRef]
- Plonka, W.; Stork, C.; Sicho, M.; Kirchmair, J. CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes. Bioorg. Med. Chem. 2021, 46, 116388. [Google Scholar] [CrossRef]
- Banerjee, P.; Dunkel, M.; Kemmler, E.; Preissner, R. SuperCYPsPred-a web server for the prediction of cytochrome activity. Nucleic Acids Res. 2020, 48, W580–W585. [Google Scholar] [CrossRef]
- Ridder, L.; Wagener, M. SyGMa: Combining expert knowledge and empirical scoring in the prediction of metabolites. ChemMedChem Chem. Enabling Drug Discov. 2008, 3, 821–832. [Google Scholar] [CrossRef]
- Nguyen-Vo, T.-H.; Trinh, Q.H.; Nguyen, L.; Nguyen-Hoang, P.-U.; Nguyen, T.-N.; Nguyen, D.T.; Nguyen, B.P.; Le, L. iCYP-MFE: Identifying human cytochrome P450 inhibitors using multitask learning and molecular fingerprint-embedded encoding. J. Chem. Inf. Model. 2021, 62, 5059–5068. [Google Scholar] [CrossRef]
- Shan, X.; Wang, X.; Li, C.-D.; Chu, Y.; Zhang, Y.; Xiong, Y.; Wei, D.-Q. Prediction of CYP450 enzyme–substrate selectivity based on the network-based label space division method. J. Chem. Inf. Model. 2019, 59, 4577–4586. [Google Scholar] [CrossRef]
- Park, H.; Brahma, R.; Shin, J.M.; Cho, K.H. Prediction of human cytochrome P450 inhibition using bio-selectivity induced deep neural network. Bulletin Korean Chem. Soc. 2022, 43, 261–269. [Google Scholar] [CrossRef]
- Dai, H.; Zheng, Y.-X.; Shan, X.-Q.; Chu, Y.-Y.; Wang, W.; Xiong, Y.; Wei, D.-Q. Computational prediction of the isoform specificity of cytochrome P450 substrates by an improved bayesian method. Res. Sq. 2019. [Google Scholar] [CrossRef]
- Raju, B.; Verma, H.; Narendra, G.; Sapra, B.; Silakari, O. Multiple machine learning, molecular docking, and ADMET screening approach for identification of selective inhibitors of CYP1B1. J. Biomol. Struct. Dyn. 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.T.; Yang, Y.W.; Huang, S.H.; Chen, L.X.; Kuang, Z.Y.; Heng, Y.; Mei, H. Molecular image-based convolutional neural network for the prediction of ADMET properties. Chemom. Intell. Lab. Syst. 2019, 194, 103853. [Google Scholar] [CrossRef]
- Zhang, X.X.; Zhao, P.A.; Wang, Z.Y.; Xu, X.; Liu, G.X.; Tang, Y.; Li, W.H. In silico prediction of CYP2C8 inhibition with machine-learning methods. Chem. Res. Toxicol. 2021, 34, 1850–1859. [Google Scholar] [CrossRef]
- Racz, A.; Keseru, G.M. Large-scale evaluation of cytochrome P450 2C9 mediated drug interaction potential with machine learning-based consensus modeling. J. Comput.—Aided. Mol. Des. 2020, 34, 831–839. [Google Scholar] [CrossRef]
- Goldwaser, E.; Laurent, C.; Lagarde, N.; Fabrega, S.; Nay, L.; Villoutreix, B.O.; Jelsch, C.; Nicot, A.B.; Loriot, M.A.; Miteva, M.A. Machine learning-driven identification of drugs inhibiting cytochrome P450 2C9. PLoS Comput. Biol. 2022, 18, e1009820. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, Y. Classification and prediction model of compound pharmacokinetic properties based on ensemble learning method. In Proceedings of the 2nd International Symposium on Artificial Intelligence for Medicine Sciences, Zhengzhou, China, 29–31 October 2021; pp. 526–531. [Google Scholar]
- Sasahara, K.; Shibata, M.; Sasabe, H.; Suzuki, T.; Takeuchi, K.; Umehara, K.; Kashiyama, E. Predicting drug metabolism and pharmacokinetics features of in-house compounds by a hybrid machine-learning model. Drug Metab. Pharmacokinet. 2021, 39. [Google Scholar] [CrossRef]
- Veith, H.; Southall, N.; Huang, R.; James, T.; Fayne, D.; Artemenko, N.; Shen, M.; Inglese, J.; Austin, C.P.; Lloyd, D.G. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [Google Scholar] [CrossRef]
- Shao, C.-Y.; Su, B.-H.; Tu, Y.-S.; Lin, C.; Lin, O.A.; Tseng, Y.J. Cyprules: A rule-based P450 inhibition prediction server. Bioinformatics 2015, 31, 1869–1871. [Google Scholar] [CrossRef]
- Su, B.-H.; Tu, Y.-S.; Lin, C.; Shao, C.-Y.; Lin, O.A.; Tseng, Y.J. Rule-based prediction models of cytochrome P450 inhibition. J. Chem. Inf. Model. 2015, 55, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Preissner, S.; Kroll, K.; Dunkel, M.; Senger, C.; Goldsobel, G.; Kuzman, D.; Guenther, S.; Winnenburg, R.; Schroeder, M.; Preissner, R. SuperCYP: A comprehensive database on cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010, 38, D237–D243. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Knoll, M.; Sirim, D.; Wagner, F.; Funke, S.; Pleiss, J. The cytochrome P450 engineering database: A navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 2007, 23, 2015–2017. [Google Scholar] [CrossRef] [PubMed]
- Sirim, D.; Wagner, F.; Lisitsa, A.; Pleiss, J. The cytochrome P450 engineering database: Integration of biochemical properties. BMC Biochem. 2009, 10, 1–4. [Google Scholar] [CrossRef]
- Wu, Z.X.; Lei, T.L.; Shen, C.; Wang, Z.; Cao, D.S.; Hou, T.J. Admet evaluation in drug discovery. 19. Reliable prediction of human cytochrome P450 inhibition using artificial intelligence approaches. J. Chem. Inf. Model. 2019, 59, 4587–4601. [Google Scholar] [CrossRef]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef]
- Walsky, R.L.; Gaman, E.A.; Obach, R.S. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J. Clin. Pharmacol. 2005, 45, 68–78. [Google Scholar] [CrossRef]
- Wenzel, J.; Matter, H.; Schmidt, F. Predictive multitask deep neural network models for adme-tox properties: Learning from large data sets. J. Chem. Inf. Model. 2019, 59, 1253–1268. [Google Scholar] [CrossRef]
- Liu, K.; Sun, X.; Jia, L.; Ma, J.; Xing, H.; Wu, J.; Gao, H.; Sun, Y.; Boulnois, F.; Fan, J. Chemi-net: A molecular graph convolutional network for accurate drug property prediction. Int. J. Mol. Sci. 2019, 20, 3389. [Google Scholar] [CrossRef]
- Dong, J.; Wang, Z.; Cui, J.; Meng, Q.; Li, S. Synthesis and structure-activity relationship studies of α-naphthoflavone derivatives as CYP1b1 inhibitors. Eur. J. Med. Chem. 2020, 187, 111938. [Google Scholar] [CrossRef]
- Kubo, M.; Yamamoto, K.; Itoh, T. Design and synthesis of selective CYP1B1 inhibitor via dearomatization of α-naphthoflavone. Bioorganic Med. Chemistry. 2019, 27, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Wang, Z.; Cui, J.; Cui, Q.; Dong, J.; Zhang, Q.; Li, S. Design, synthesis, and biological evaluation of cytochrome P450 1b1 targeted molecular imaging probes for colorectal tumor detection. J. Med. Chem. 2018, 61, 10901–10909. [Google Scholar] [CrossRef] [PubMed]
- Czechtizky, W.; Su, W.; Ripa, L.; Schiesser, S.; Höijer, A.; Cox, R.J. Advances in the design of new types of inhaled medicines. Prog. Med. Chem. 2022, 61, 93–162. [Google Scholar] [PubMed]
- Smith, P.; Sorich, M.; Low, L.; McKinnon, R.; Miners, J. Towards integrated ADME prediction: Past, present and future directions for modelling metabolism by UDP-glucuronosyltransferases. J. Mol. Graph. Model. 2004, 22, 507–517. [Google Scholar] [CrossRef]
- Mazzolari, A.; Afzal, A.M.; Pedretti, A.; Testa, B.; Vistoli, G.; Bender, A. Prediction of UGT-mediated metabolism using the manually curated metaqsar database. ACS Med. Chem. Lett. 2019, 10, 633–638. [Google Scholar] [CrossRef]
- Pedretti, A.; Mazzolari, A.; Vistoli, G.; Testa, B. Metaqsar: An integrated database engine to manage and analyze metabolic data. J. Med. Chem. 2018, 61, 1019–1030. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, H.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. Computational prediction of site of metabolism for UGT-catalyzed reactions. J. Chem. Inf. Model. 2019, 59, 1085–1095. [Google Scholar] [CrossRef]
- Lee, P.W.; Aizawa, H.; Gan, L.; Prakash, C.; Zhong, D. Handbook of Metabolic Pathways of Xenobiotics; Wiley Online Library: Hoboken, NJ, USA, 2014. [Google Scholar]
- Peng, J.; Lu, J.; Shen, Q.; Zheng, M.; Luo, X.; Zhu, W.; Jiang, H.; Chen, K. In silico site of metabolism prediction for human UGT-catalyzed reactions. Bioinformatics 2014, 30, 398–405. [Google Scholar] [CrossRef]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. SOMP: Web server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics 2015, 31, 2046–2048. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Horde, G.W.; Gupta, V. Drug clearance. In Statpearls; Treasure Island: FL, USA, 2022. [Google Scholar]
- Wade, K.C. Pharmacokinetics in neonatal medicine. In Fanaroff and Martin’s Neonatal-Perinatal Medicine; Elsevier: Philadelphia, PA, USA, 2020; pp. 722–734. [Google Scholar]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Clearance in drug design. J. Med. Chem. 2019, 62, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Esaki, T.; Watanabe, R.; Kawashima, H.; Ohashi, R.; Natsume-Kitatani, Y.; Nagao, C.; Mizuguchi, K. Data curation can improve the prediction accuracy of metabolic intrinsic clearance. Mol. Inform. 2019, 38, 1800086. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.W.; Fagerholm, U.; Norinder, U. In silico categorization of in vivo intrinsic clearance using machine learning. Mol. Pharm. 2013, 10, 1318–1321. [Google Scholar] [CrossRef]
- Chen, J.H.; Yang, H.B.; Zhu, L.; Wu, Z.R.; Li, W.H.; Tang, Y.; Liu, G.X. In silico prediction of human renal clearance of compounds using quantitative structure-pharmacokinetic relationship models. Chem. Res. Toxicol. 2020, 33, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Liu, H.C.; Fan, Y.R.; Chen, X.Y.; Yang, Y.; Zhu, L.; Zhao, J.N.; Chen, Y.D.; Zhang, Y.M. In silico prediction of human intravenous pharmacokinetic parameters with improved accuracy. J. Chem. Inf. Model. 2019, 59, 3968–3980. [Google Scholar] [CrossRef]
- Kosugi, Y.; Hosea, N. Direct comparison of total clearance prediction: Computational machine learning model versus bottom-up approach using in vitro assay. Mol. Pharm. 2020, 17, 2299–2309. [Google Scholar] [CrossRef]
- Watanabe, R.; Ohashi, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Mizuguchi, K. Development of an in silico prediction system of human renal excretion and clearance from chemical structure information incorporating fraction unbound in plasma as a descriptor. Sci. Rep. 2019, 9, 18782. [Google Scholar] [CrossRef] [PubMed]
- Oprisiu, I.; Winiwarter, S. Silico ADME Modeling; Academic Press: Cambridge, MA, USA, 2021; pp. 208–222. [Google Scholar]
- Mamada, H.; Nomura, Y.; Uesawa, Y. Prediction model of clearance by a novel quantitative structure-activity relationship approach, combination deepsnap-deep learning and conventional machine learning. ACS Omega 2021, 6, 23570–23577. [Google Scholar] [CrossRef]
- Sohlenius-Sternbeck, A.-K.; Terelius, Y. Evaluation of ADMET predictor in early discovery drug metabolism and pharmacokinetics project work. Drug Metab. Dispos. 2022, 50, 95–104. [Google Scholar] [CrossRef]
- Lombardo, F.; Berellini, G.; Obach, R.S. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 1352 drug compounds. Drug Metab. Dispos. 2018, 46, 1466–1477. [Google Scholar] [CrossRef]
- Paine, S.W.; Barton, P.; Bird, J.; Denton, R.; Menochet, K.; Smith, A.; Tomkinson, N.P.; Chohan, K.K. A rapid computational filter for predicting the rate of human renal clearance. J. Mol. Graph. Model. 2010, 29, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Feng, B.; Obach, R.S.; Troutman, M.D.; Chupka, J.; Miller, H.R.; El-Kattan, A. Physicochemical determinants of human renal clearance. J. Med. Chem. 2009, 52, 4844–4852. [Google Scholar] [CrossRef]
- Lombardo, F.; Obach, R.S.; Varma, M.V.; Stringer, R.; Berellini, G. Clearance mechanism assignment and total clearance prediction in human based upon in silico models. J. Med. Chem. 2014, 57, 4397–4405. [Google Scholar] [CrossRef] [PubMed]
- Scotcher, D.; Jones, C.; Rostami-Hodjegan, A.; Galetin, A. Novel minimal physiologically-based model for the prediction of passive tubular reabsorption and renal excretion clearance. Eur. J. Pharm. Sci. 2016, 94, 59–71. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Obach, R.S.; Rotter, C.; Miller, H.R.; Chang, G.; Steyn, S.J.; El-Kattan, A.; Troutman, M.D. Physicochemical space for optimum oral bioavailability: Contribution of human intestinal absorption and first-pass elimination. J. Med. Chem. 2010, 53, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.M.; Subramanian, V.; Miljković, F. Multi-task convolutional neural networks for predicting in vitro clearance endpoints from molecular images. J. Comput.—Aided Mol. Des. 2022, 36, 443–457. [Google Scholar] [CrossRef]
- Hallare, J.; Gerriets, V. Half Life; Statpearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Relevance of half-life in drug design. J. Med. Chem. 2018, 61, 4273–4282. [Google Scholar] [CrossRef]
- Podlewska, S.; Kafel, R. Metstabon-online platform for metabolic stability predictions. Int. J. Mol. Sci. 2018, 19, 1040. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The human metabolome database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Montenegro-Burke, J.R.; Guijas, C.; Siuzdak, G. Metlin: A tandem mass spectral library of standards. Comput. Methods Data Anal. Metab. 2020, 2104, 149–163. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. ‘In Silico‘ Simulation of Biological Processes: Novartis Foundation Symposium 247. In The Kegg Database; Wiley Online Library: New York, NY, USA, 2002; pp. 91–103. [Google Scholar]
- Trupp, M.; Altman, T.; Fulcher, C.A.; Caspi, R.; Krummenacker, M.; Paley, S.; Karp, P.D. Beyond the genome (btg) is a (pgdb) pathway genome database: HumanCyc. Genome Biol. 2010, 11, 1. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. Bigg models 2020: Multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 2020, 48, D402–D406. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the drugbank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput.—Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C.; Sun, J.; Zitnik, M. Artificial intelligence foundation for therapeutic science. Nat. Chem. Biol. 2022, 18, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Kass-Hout, T.A.; Xu, Z.; Mohebbi, M.; Nelsen, H.; Baker, A.; Levine, J.; Johanson, E.; Bright, R.A. OpenFDA: An innovative platform providing access to a wealth of fda’s publicly available data. J. Am. Med. Inform. Assoc. 2016, 23, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, K.M.; Brennan, L.; Segurado, R.; Barron, R.E.; Gibney, E.R.; Ryan, M.F.; Gibney, M.J.; O’Sullivan, A.M. Genetic and environmental contributions to variation in the stable urinary NMR metabolome over time: A classic twin study. J. Proteome Res. 2021, 20, 3992–4000. [Google Scholar] [CrossRef]
- Pearson, P.G.; Wienkers, L.C. Handbook of Drug Metabolism; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Arabi, A.A. Artificial intelligence in drug design: Algorithms, applications, challenges and ethics. Future Drug Discov. 2021, 3, FDD59. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Aittokallio, T. What are the current challenges for machine learning in drug discovery and repurposing? Expert Opin. Drug Discov. 2022, 17, 423–425. [Google Scholar] [CrossRef]
- Sheridan, R.P. Interpretation of QSAR models by coloring atoms according to changes in predicted activity: How robust is it? J. Chem. Inf. Model. 2019, 59, 1324–1337. [Google Scholar] [CrossRef] [PubMed]
- Azram, N.A.; Atan, R.; Mustafa, S.; Desa, M.N.M. A review on integration of scientific experimental data through metadata. Recent Trends Adv. Wirel. IoT-Enabled Netw. 2019, 63–72. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.T.V.; Tayara, H.; Chong, K.T. Artificial Intelligence in Drug Metabolism and Excretion Prediction: Recent Advances, Challenges, and Future Perspectives. Pharmaceutics 2023, 15, 1260. https://doi.org/10.3390/pharmaceutics15041260
Tran TTV, Tayara H, Chong KT. Artificial Intelligence in Drug Metabolism and Excretion Prediction: Recent Advances, Challenges, and Future Perspectives. Pharmaceutics. 2023; 15(4):1260. https://doi.org/10.3390/pharmaceutics15041260
Chicago/Turabian StyleTran, Thi Tuyet Van, Hilal Tayara, and Kil To Chong. 2023. "Artificial Intelligence in Drug Metabolism and Excretion Prediction: Recent Advances, Challenges, and Future Perspectives" Pharmaceutics 15, no. 4: 1260. https://doi.org/10.3390/pharmaceutics15041260