1. Introduction
The development of nucleic acid vaccines began not only as an attempt to decrease the dependency on vaccines with live or attenuated vectors, but also as an investigation of the effects of the direct injection of DNA or RNA expression vectors [
1]. Plasmid DNA-based vaccines are still the most widely studied form of nucleic acid vaccine approaches, and immunization with purified circular plasmid DNA has already been shown to be successful in various tissues of small animals, even when deploying different routes of administration [
1,
2,
3,
4]. Despite being a flexible platform and successful in small animals tested in laboratories, DNA vaccines were not approved for human use until the pandemic scenario of SARS-CoV-2 in 2021, one year after the approval of mRNA-based vaccines in 2020. This delay has been ascribed to concerns regarding the potential oncogenic effects of DNA integration, and because DNA vaccines have shown lower potency in human clinical trials [
5]. Among the various types of RNA vaccines, only mRNA vaccines are currently approved; however, self-assembly/amplifying (sam) vaccines represent a promising immunogenic approach to nucleic acid vaccines. Replicase-containing RNA vectors have been shown to significantly more immunogenic than the conventional plasmid DNA, and recent results have shown that a single dose of 0.1 µg of these vectors injected intramuscularly was able to immunize mice [
6]. This methodology uses “replicons”: self-amplifying RNA vectors (samRNA) that lack a viral capsid and envelope, but contain the nonstructural protein genes that encode a viral replicase, 5′ and 3′ sequences important for replication, and a subgenomic promoter derived from alphavirus vectors [
7]. An increased number of RNA copies also leads to enhanced levels of transient gene expression, both in vitro and in vivo [
8]. Replicon particles derived from Venezuelan equine encephalitis (VEE) are an efficient platform for RNA vaccines, with robust effects on vaccine formulations when complexed with lipid formulations, promoting the protection of RNA from degradation and enhancing transfection efficiency [
9].
Liposomes are artificial round lipid-based nanoparticles consisting of one or more phospholipid bilayer membranes that encapsulate an internal aqueous compartment [
10]. They have played a major role as model systems for membrane studies; however, more recently, they have been successfully used as carriers for drugs and vaccines [
11]. Depending on the modification of the lipid moieties with cationic or anionic side groups, formed liposomes gain different affinities for charged or uncharged cargo molecules. These may be attached to the inner or outer surface of liposomes or confined inside the liposomes, protecting the cargo from degradation; this is important when the cargo molecules are nucleic acids (revised in [
12]). The DDAB/DC-Chol formulation used here is based on previously published studies on pDNA vaccines [
13,
14]. The high stability of DDAB/DC-Chol liposomes in association with pDNA has been previously reported for the development of antimalarial DNA vaccines [
14]; however, to date, the same approach has not been tested for mRNA delivery.
In this study, RNA replicons containing reporter genes that encode green fluorescent protein (GFP) and nanoluciferase (nLuc) incorporated into cationic liposomes were used for the in vitro transfection of Vero and HEK 293T cells, and for intradermal immunization of BALB/c and C57BL/6 mice by tattooing. The samRNA used in this study was derived from VEE vectors, and synthesized from a plasmid DNA template using the DNA-dependent SP6 RNA polymerase. Constructs containing the SP6 promoter, specifically SP6-VEE-GFP and SP6-VEE-nLuc, were used for in vitro transcription, resulting in samRNAs with a high degree of purity. These RNA replicons—expressing either GFP or nLuc—were incorporated into DDAB/DC-Chol liposomes by sonication, and the resulting lipoplexes were used for in vitro and in vivo assays. Lipoplex formulations were used for the intradermal immunization of BALB/c and C57BL/6 mice.
To analyze the efficiency of the immunization assays, an intravital imaging system was used to measure the relative fluorescence or bioluminescence in the tattooed areas. The lipoplex-samRNA approach was applied to develop a potential vaccine against
Plasmodium falciparum based on the vaccine candidate gene PfRH5, which is an essential and almost invariant ligand of the human blood cell receptor and a key for erythrocyte invasion of malarial parasites [
15]. The results demonstrate that the samRNA-encoding PfRH5 represents a promising tool for the development of a safe and effective antimalarial vaccine delivered via the intradermal route.
2. Materials and Methods
2.1. Cloning of Plasmid Constructs
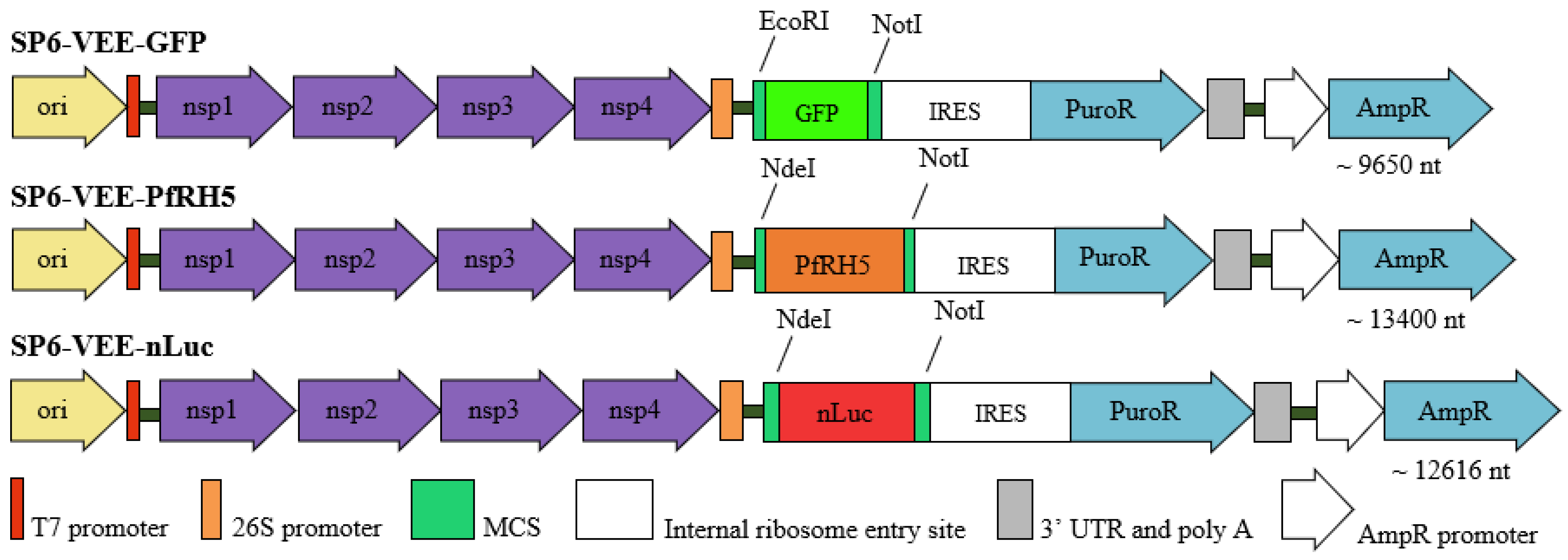
The plasmids used as templates for RNA replicon synthesis were SP6-VEE-IRES-Puro (ADDGENE: Plasmid #58971) [
16], modified by adding the protein of interest, nanoluciferase, as an amplified PCR product, and SP6-VEE-GFP (ADDGENE: Plasmid #58976). The self-amplifying RNA (samRNA) of SP6-VEE-nLuc and SP6-VEE-PfRH5, the products of subcloning with the enzymes NotI and NdeI, are shown in
Figure 1, along with the other constructs. For comparison, the following pcDNA3-derivates were used in the transfection and immunization assays: pcDNA3-GFP as a control for RNA replicons expressing GFP, and pcDNA3-Luc as a control for RNA replicons expressing nLuc. The plasmids were transformed into
E. coli DH10B competent cells using standard molecular techniques, and produced on a large scale using the Plasmidprep protocol for high purity [
17]. Additionally, for immunizations, stable, non-replicating mRNA encoding the PfRH5 antigen was used as a comparator. The PfRH5 gene was cloned into the vector TEV-MCS-101A kindly provided by Dr. Katalin Karikó (BioNTech, Mainz, Germany). In this vector, the mRNA for PfRH5 is flanked in the 5′ by the Tobacco etch virus (TEV) 5′ untranslated region (UTR), and in the 3′ by Xenopus beta-globin 3′UTR followed by 101 adenines.
2.2. RNA Replicon In Vitro Synthesis
The DNA plasmids SP6-VEE-GFP and SP6-VEE-nLuc were linearized downstream of the 3′ end of the RNA replicon template sequence by digestion with MluI. Linearized DNA templates were transcribed into RNA using a kit from RiboMAX™ Large Scale RNA Production Systems (Promega), following instructions from the manufacturer. The removal of SP6 polymerase was removed through phenol-chloroform extraction, and the RNA replicon was precipitated with 2.5 volumes of 95% ethanol and 0.1 volume of 3M sodium acetate (pH 5.2), according to the manufacturer’s instructions. The synthesized RNA replicons were stored at −80 °C until use. Alternatively, the plasmid TEV-MCS-100A was used to produce the mRNA encoding PfRH5. The RNA generated by the TEV vector did not replicate. We compared the efficiency of these two RNA forms and analyzed their immunological effects.
2.3. Liposome Preparation
The liposomes were produced, as previously described [
14]. All the lipids were purchased from Avanti Polar Lipids. In order to produce cationic liposomes, DDAB (dimethyldioctadecyl ammonium bromide) lipid vesicles were complexed with DC-Chol, a cationic cholesterol derivative (cholesteryl 3β-N-(di-methyl-amino-ethyl)-carbamate hydrochloride), at a 4:1 molar ratio. The DDAB/DC-Chol lipid vesicles were dissolved in chloroform, and the solution was evaporated under a nitrogen stream to yield a dry lipid film. The remaining chloroform was removed by drying the film under vacuum for 1 h, and the resulting dry lipid film was stored at 4 °C until use. Before use, the liposome preparation was rehydrated in 1 mL of 50 mM HEPES buffer (pH 7.4) to a final concentration of 1 mg/mL, and then maintained at 60 °C under vigorous shaking for 1 h (1000 rpm, Eppendorf tube benchtop mixer). The dispersed liposomes were then sonicated until the suspensions changed from an opaque/milky appearance, which indicated the presence of large particles, to a translucid solution with particles smaller than 100 nm in diameter. A concentration of 8 nM of lipids for each 1 µg of nucleic acid (RNA) was established for the lipoplexes in immunization with sPfRH5 RNA and samPfRH5 replicon. The mixture of liposomes and the nucleic acid of interest was sonicated in an ultrasonic bath for 2 min, and used for transfection and/or immunization immediately after this step.
2.4. Zeta Potential and Polydispersity
The charge and size parameters of the formulated liposomes were analyzed using a NanoPlusTM 2 (Zeta Potential Analyzer, Micromeritics, Norcross, GA, USA). The zeta potential describes the electrokinetic potential in colloidal dispersions, thus contributing to the evaluation of charge parameters. Dynamic light scattering (DLS) was used to determine the size (diameter in nm) of the particles in suspension. The polydispersity (PD) of a liposome formulation is a measure of the homogeneity or uniformity of a mixture.
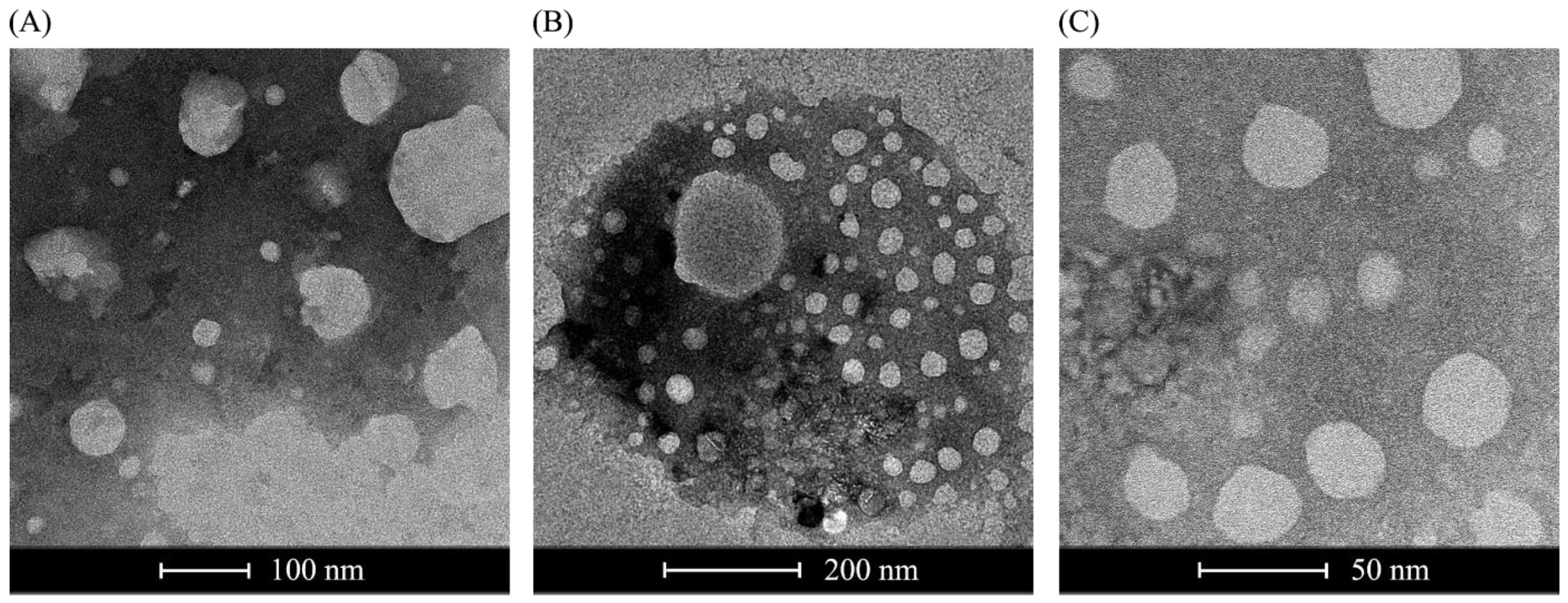
2.5. Negative Staining Transmission Electron Microscopy and Cryoelectron Microscopy
For negative staining transmission, the same liposome formulation used for transfection was diluted to a final volume of 1 mL, and a drop of the water-diluted suspension was stained with uranyl acetate for approximately 5 min. The samples were then dried at room temperature and analyzed using a transmission electron microscope (Jeol 100 CX II). For cryoelectron microscopy, 1 mL of plasmid RNA-loaded liposomes was applied over a holey carbon-film grid (Quantifoil Micro Tools, Jena, Germany), followed by flash freezing in liquid ethane using a Gatan Cryoplunge 3 (Gatan, Pleasanton, CA, USA). The visualization of the frozen, hydrated specimen was performed on a JEM2100 electron microscope (JEOL, Tokyo, Japan, operating at 200 kV), resulting in images of 2 μm scale. Images were recorded using a Gatan Ultrascan 4000 CCD camera at 40,000× magnification. Negative staining was performed at the ICB-USP (Institute of Biomedical Sciences, University of São Paulo).
2.6. Transfection of the RNA Replicons Stabilized by Liposomes
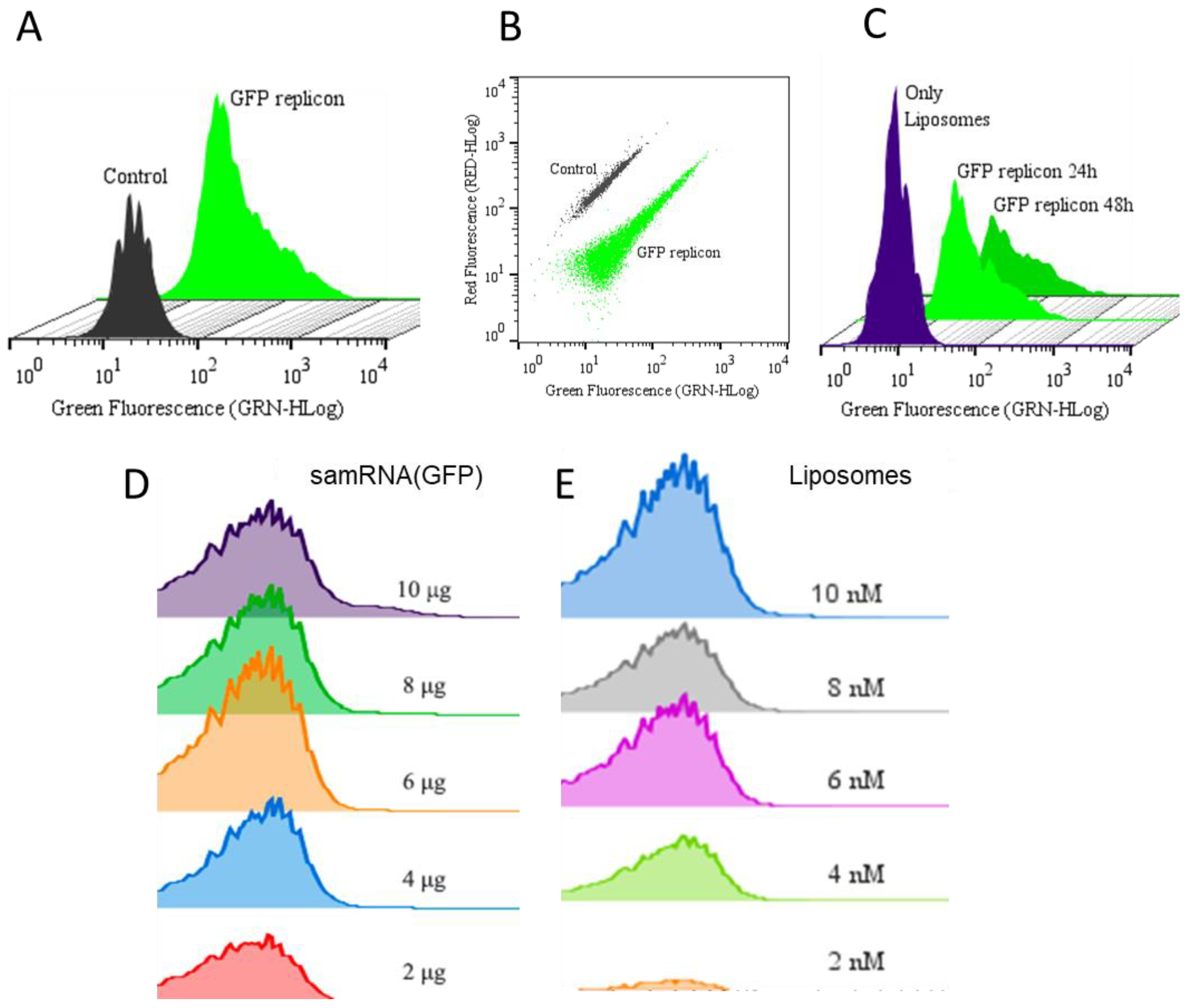
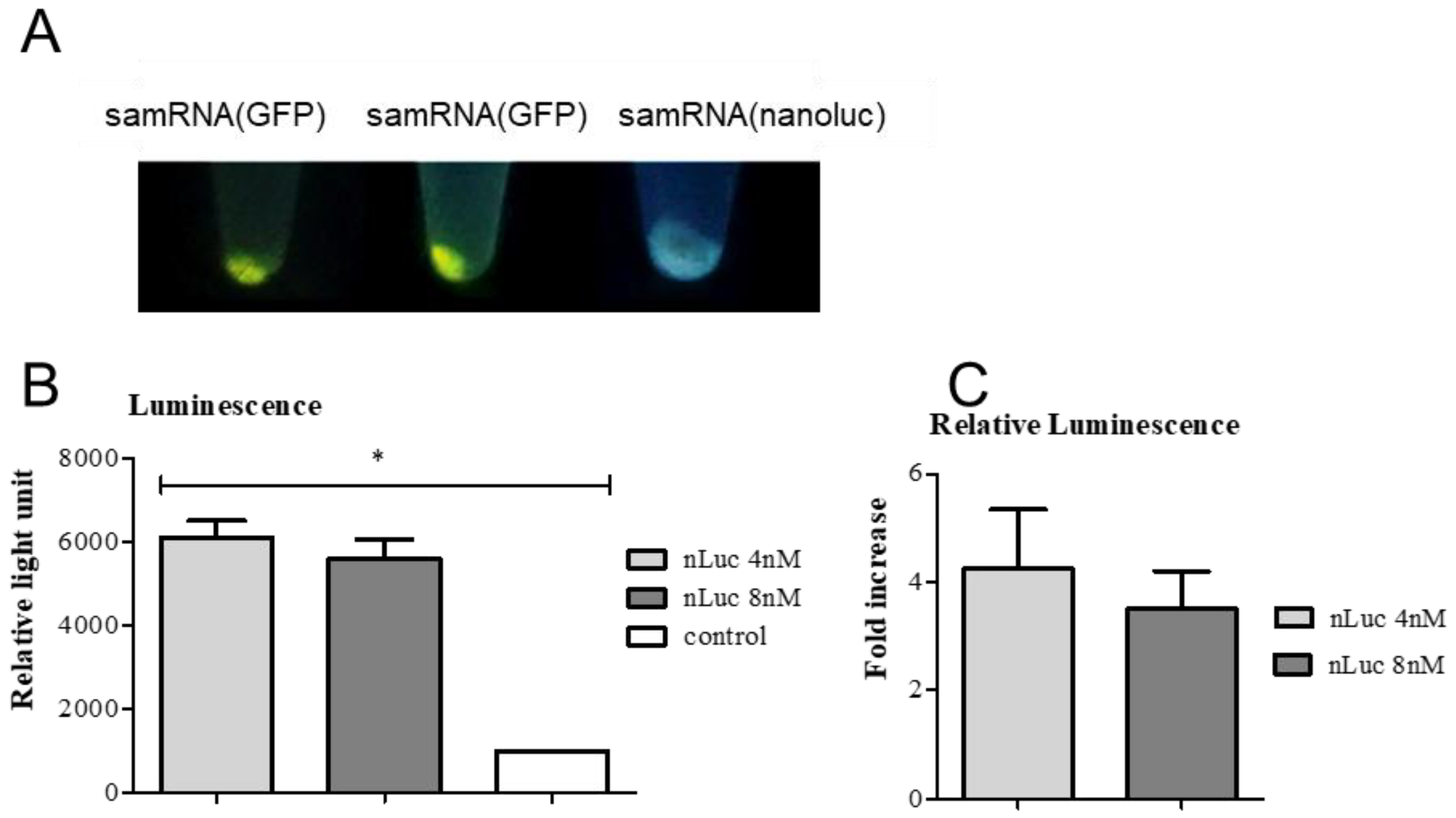
For transfection, Vero and HEK293T cells were seeded at a density of 104 cells/well, and grown overnight in a 24-well plate. Liposomes at either 4 nM with 2 µg of RNA replicons had their final volumes adjusted with 5% glucose to 50 µL per well, resulting in 1 µg of RNA replicon per mL in each well. Therefore, 50 µL of the formulation was pipetted over the cells, and DMEM medium was added to a final volume of 2 mL per well. After incubation in the presence of liposomes for 24 h, the cells were washed with incomplete RPMI medium, trypsinized, and resuspended in 100 µL of DMEM medium. Half of this volume was analyzed using a flow cytometer (Guava® EasyCyte Mini, Luminex, Austin, TX, USA), in the case of the GFP transfected wells, or using a luminometer (Berthold Lumat LB 9507, Bad Wildbad, Germany), in the case of nanoLuc transfected wells. DMEM was added to the cells, and the plates were incubated. Subsequent analyses were performed 48 h after transfection. The transfection efficiency of the GFP replicons and GFP was evaluated using flow cytometry, with excitation at 488 nm (FL1 filter) to detect the green fluorescent protein (GFP) in the transfected cells. The samples containing nanoLuc were treated with Brefeldin A (BD GolgiPlug™), according to the manufacturer’s instructions, 6–8 h before every analysis. The cells were pelleted at 18,800× g for 20 s (Eppendorf centrifuge) and resuspended in Nano-Glo® Luciferase Assay Substrate and Buffer, according to the manufacturer’s instructions, before analysis in the luminometer.
2.7. Liposome Toxicity Assay
Vero and HEK293T cells were seeded overnight in 24-well plates at a density of 104 cells/well and transfected with liposomes at various molar ratios (2 nM to 10 nM). After incubation for 24 h, the cells were washed with RPMI incomplete medium, trypsinized, and centrifuged for 5 min at 200× g. The pellets were resuspended in working solution from the LIVE/DEAD™ Cell Imaging Kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions, and incubated for 30 min at 25 °C. The samples were then washed twice with phosphate buffered saline (pH 7.4, PBS), and the percentages of live and dead cells were quantified using flow cytometry.
2.8. Fluorescence Microscopy of Transfected Cells
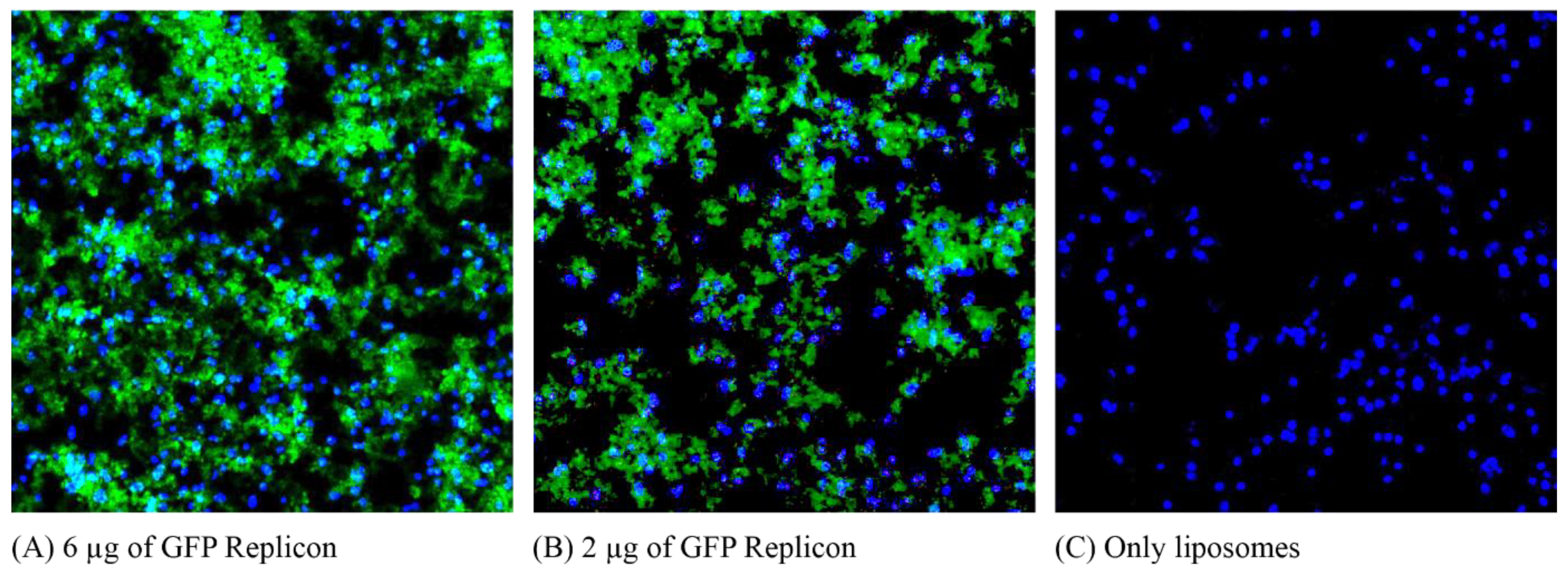
Vero cells were grown directly on slides in a Nunc Lab-Tek II Chamber Slide System to a confluence of 10
4 cells/well. The cells were transfected with liposomes at an 8 nM molar ratio with 6 µg and 2 µg of RNA replicons expressing GFP, and a control well was transfected only with liposomes. Transfection was performed in the same manner as in
Section 2.5 in this section, only in a smaller volume (0.5 mL instead of 2 mL per well). After 48 h of transfection, the sections were stained with 0.03 µg/mL DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich, St. Louis, MO, USA), washed with DMEM medium, and the slide was separated for microscopic examination. Figures were acquired using a DMRA2 fluorescence microscope (Leica, Wetzlar, Germany) and MetaMorph software (Molecular Devices Inc., San José, CA, USA), and edited using Figure J (Version 1.51n).
2.9. Confocal Immunofluorescence Analysis
Skin sections were embedded in cryoprotectant solution for 48 h, sliced (120 μm) using a Leica CM3050-S cryostat (Leica Biosystems), and mounted on gelatin-coated slides. The sections were then covered with ProLong Antifade reagent (Invitrogen, USA). Images of each section were obtained using a Nikon Eclipse Ti confocal laser microscope (Tokyo, Japan). A laser wavelength of 561 nm was used for GFP detection. The signal analysis was performed using NIS Elements AR4.00.04 (Nikon, Tokyo, Japan) software.
2.10. Intradermal Immunization
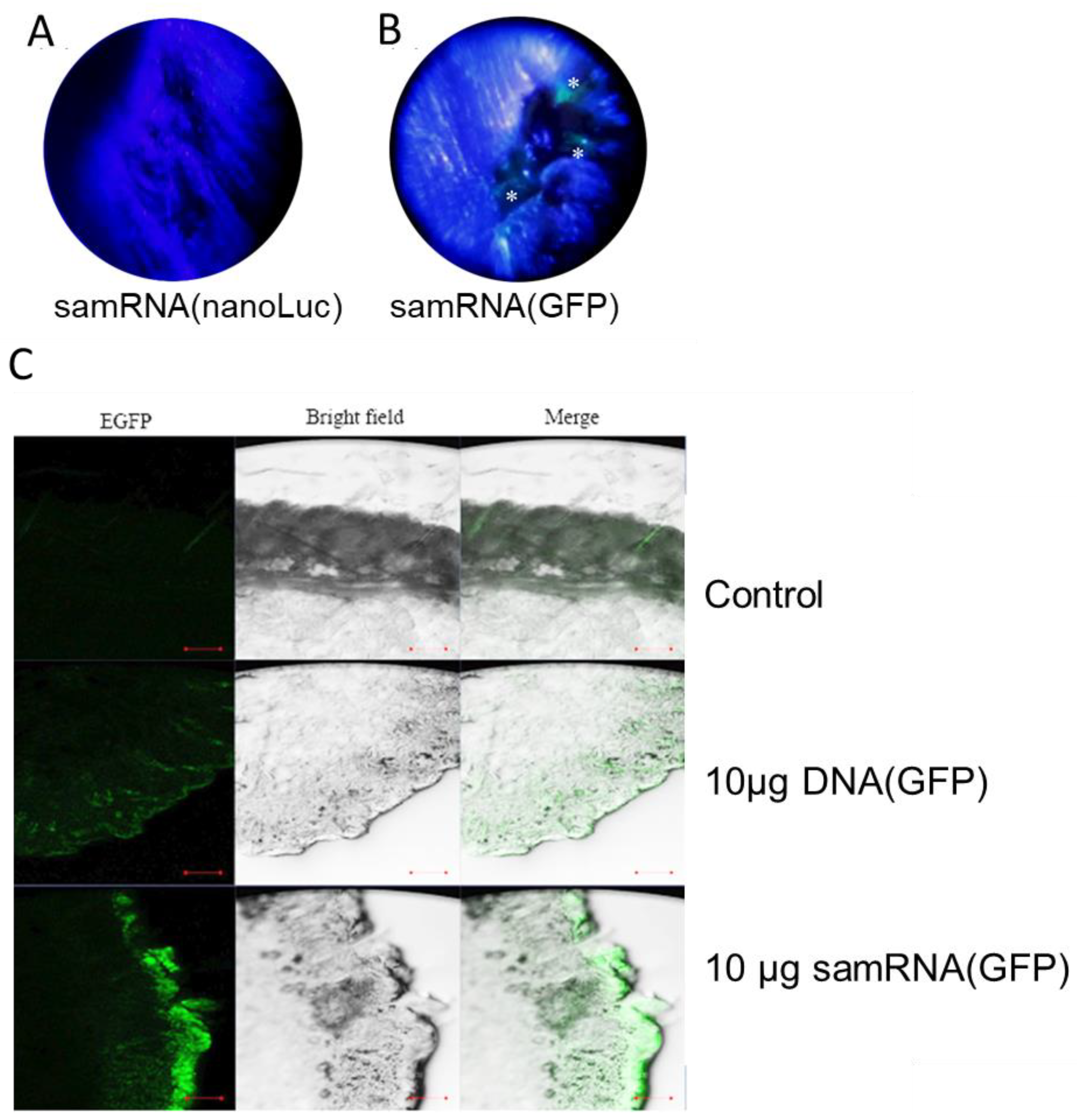
BALB/c mice of age 5–12 weeks were obtained from the breeding facility for isogenic mice of the Department of Parasitology (ICB/USP), and maintained under pathogen-free conditions during the course of the experiment. Ethical clearance was obtained from the local Ethics Committee at the Institute for Biomedical Sciences/USP (Protocol No. 15/3/3). Groups of five BALB/c mice were sedated with ketamine/xylazine, and had hair at the hindlimb removed using a commercial hair removal cream; the underlying skin was sterilized with 70% ethanol. For the delivery of mRNA/samRNA or DNA as control, 30 µL of liposome/pDNA or RNA mixture at 8 nM concentration was administered in two drops at the hairless skin in the tibia anterior muscle, followed by tattooing of a 2 × 1 cm skin area using a commercial tattoo machine. The tattoo device was adjusted to expose no more than 2 mm of the needle, and was used twice in the muscle for no more than 15 s at a voltage of 16 V set on the power supply. Under these conditions, every mouse received, in the course of one immunization, 10 µg of RNA in 50 µL of liposome solution. Given that some trauma had been caused to the skin, a silicone cream was applied to the tattooed area. Additionally, other groups of mice were immunized with an RNA replicon/liposome mixture. The treated animals were then exposed to an IVIS Spectrum CT (Caliper Life Sciences, Hopkinton, MA, USA) located in the CEFAP-ICB/USP for bioimaging. The tattooed area was analyzed for relative fluorescence (for GFP) or bioluminescence (for nLuc), according to the manufacturer’s protocol. When applicable, euthanasia using a nitrogen chamber was performed in mice to remove treated tissues, which were used for further histological analysis to evaluate the possible effects of the lipoplexes in the dermal tissue and potentially affected cells.
2.11. ELISA and Western Blots
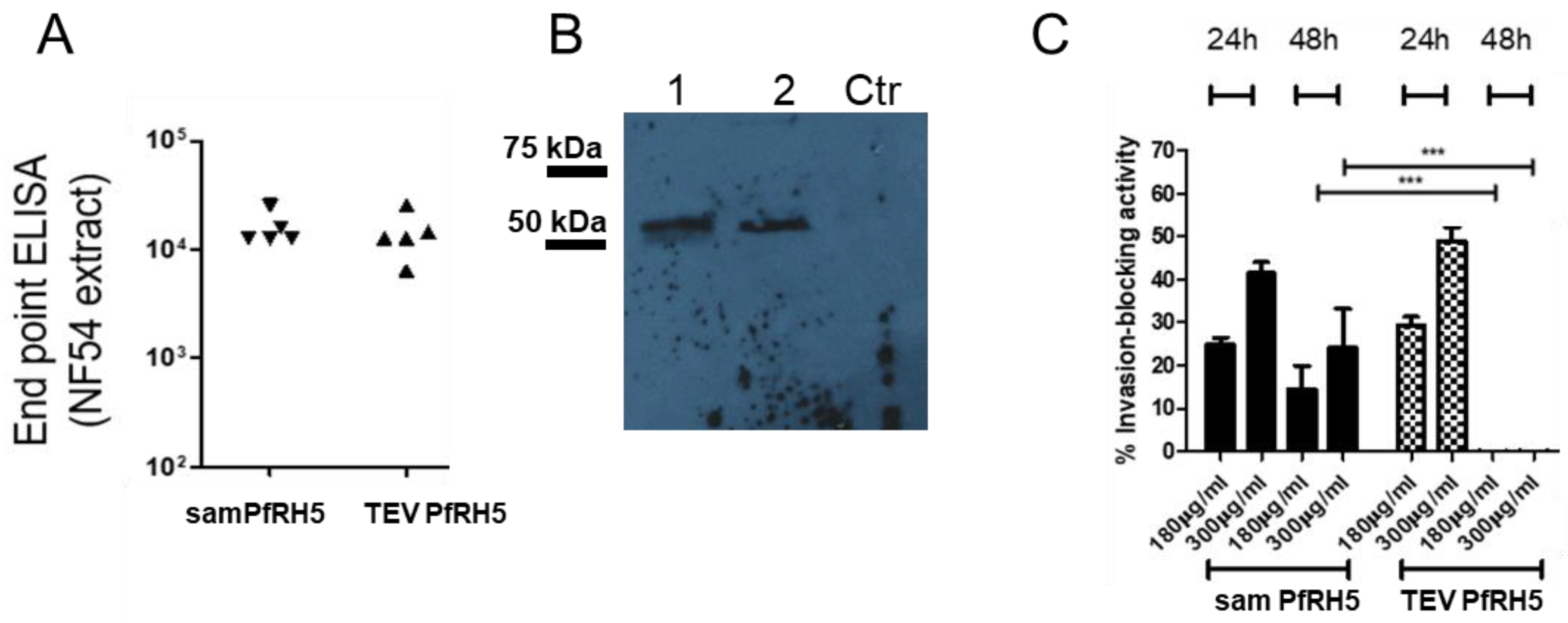
Each 96-well ELISA medium binding plate (Jet Biofil, Guangzhou, China) was coated with schizont extract at 200 ng/well and at 4 °C overnight. On the next day, the plates were washed 3 times with PBS/Tween 0.05% (PBS/T) and blocked with 2% skim milk/PBS for at least 1 h at 25 °C. The plates were washed and incubated with antisera generated from mice that were immunized with mRNA and samRNA encoding PfRH5 antigen in a solution of 1% skim milk/PBS. The antisera were endpoint-diluted and incubated for 2 h at room temperature. After four washes with PBS/T, anti-murine IgG coupled to horseradish peroxidase (KPL-Seracare, Milford, MA, USA) was applied for 1 h at 25 °C (1:2500). After repeated washing, all of the wells were developed with TMB substrate (Pierce/Thermo Fisher Scientific), stopped after 5 min with 1 M HCl, and the colorimetric reaction was analyzed using a BioTek plate reader (BioTek, Winooski, VT, USA) at 450 nm/595 nm.
For Western blots, 10 µg of schizont extract or 2 µg of recombinant PfRH5GPI [
18] was electrophoresed under non-reducing conditions in SDS-polyacrylamide gels (10%). Afterward, proteins in the gels were transferred onto nitrocellulose membranes (Hybond C; GE Healthcare), and these were blocked with 4% milk in PBS/T for 1 h at room temperature. After four washing steps with PBS/T, the primary antibodies (serum pool of plasmid-liposome immunized animals) were incubated overnight at room temperature at a dilution of 1:500 in 1% milk PBS/T. After five washes with PBS/T, the membranes were incubated with horseradish peroxidase-conjugated anti-mouse secondary antibody diluted 1:3000 for one hour at room temperature. After five washes with PBS/T, the membranes were briefly soaked in ECL reagent (GE Healthcare), and chemiluminescence was documented on Hypermax X-ray films (Kodak) or photographed using a GE Image Quant 3000 apparatus.
2.12. P. falciparum Culture and Invasion Inhibition Assays
For the assays with schizont extracts or invasion inhibition, the NF54 strain of
P. falciparum was maintained in human B
+ red blood cells (hematocrit 3–5%, ethical clearance for the use of human blood was granted by the local Committee for Ethics in Research involving Humans at ICB/USP, protocol number 803/2016) in RPMI medium supplemented with 0.5% Albumax 1 (Invitrogen/ThermoFisher Scientific). The cultures were kept in airtight boxes (candle jars) at 37 °C, with daily medium changes [
19]. Parasitemia was monitored by microscopy using thin blood smears stained with a modified Giemsa stain (Panótico Quick kit; LaborClin, Pinhais, Brazil). The schizont-stage parasites were collected for ELISA and Western blot analyses. The cultures were synchronized with intermittent plasma flotation [
20] (Voluven 6%; Fresenius Kabi, Campinas, Brazil), followed by sorbitol lysis [
21]. For growth inhibition tests, the cultures were plated in 96-well plates at an initial parasitemia of 1%. Protein A-purified IgG fractions from immunized mice were added at different concentrations (180 μg/mL and 300 μg/mL), and the volumes were matched with RPMI medium for the control IgG (300 µg/mL) from pre-immune sera. Measurements were taken after 24 and 48 h. In order to measure parasitemia, aliquots were removed from wells and stained with ethidium bromide (0.1 ug/mL), and analyzed by flow cytometry (Guava Easycyte Mini), as described previously [
22].
2.13. Statistical Analyses
All of the experiments were performed at least in triplicate. GraphPad Prism 5.03 (Graph Pad Software, San Diego, CA, USA) and Origin 8 (OriginLab Corporation, Northampton, MA, USA) were used for all of the statistical analyses. Student’s t-test was used to compare normally distributed values between groups, and one-way ANOVA or the Kruskal–Wallis Test with Bonferroni or Dunnett’s post hoc test was used to compare three or more groups (p < 0.05, considered statistically significant). An analysis using nonlinear regression as dose-response curves was performed to determine the levels of liposome toxicity and compare the transfection efficiency between groups.
4. Discussion
The feasibility of using mRNA as an immunogen has long been discussed, and mRNA immunization has become pivotal in combating the SARS-CoV-2 pandemic. A breakthrough in mRNA immunization became possible after the discovery that nucleotides in the delivered mRNA must be modified to avoid unwanted inflammatory responses against the RNA itself [
25]. Since the production of synthetic mRNA and encapsulation does not depend on the sequence of the genes encoded, the combination of modified nucleotides and self-amplifying RNA, such as from the engineered alphavirus genomes used here, theoretically represents a platform for a swift response to rapidly emerging global health threats, such as the SARS-CoV-2 pandemic. In this study, we demonstrated the feasibility of samRNA vaccines packaged in cationic liposomes delivered intradermally with a tattooing device by detecting GFP expression in tattooed skin and generating anti-
P. falciparum inhibitory antibodies against the blood stage antigen PfRH5.
Skin tattooing is an intradermal immunization approach that has been applied to DNA vaccinations to induce stronger immune responses than intramuscular needle injections combined with adjuvants [
26]. Some key advantages make skin tattooing a good choice for intradermal immunization: (i) it covers a large skin area, which could potentially elicit a stronger immune response, bearing in mind that it is a region abundant in antigen-presenting cells; (ii) it is inexpensive; (iii) it produces low DNA damage and (iv) it recruits immune cells, such as macrophages and dendritic cells, enhancing the immune process [
27]. The level of expression suggests that tattooing RNA robustly promotes antigen production in the dermis. Surprisingly, tattoo-transfection of RNA showed a stronger induction of reporter proteins than DNA transfection (see
Figure 6C). In contrast to other groups that attempted to decrease the inflammatory effects of unmodified RNA during immunization [
25], a large production of GFP was observed following the delivery of unmodified samRNA. Although a large amount of RNA (10 µg) was initially used, it can be assumed that only a small portion was effectively transferred to the skin. This probably occurred because a conventional tattooing machine was used, in contrast to previous studies in which specific multilayer tattooing delivery was more effective [
28]. Notably, the mRNA and samRNA used in this study were not produced using a capping reagent or pseudo-uridine, which can improve the translation of RNA molecules [
25]. This highlights the efficiency of the method applied, but also simplifies the application of RNA transfection or immunization when sophisticated devices are not available. Additionally, the advantage of samRNA is that it can at least partially overcome the suppression of the foreign unmodified RNA immunization effect because the self-replicating RNA inside the transfected cell is modified by the cell itself.
Liposomes have advantages over other drug delivery systems because of their ability to solubilize hydrophilic molecules in the aqueous phase, preventing their degradation and precise release until the target is reached. They can also carry lipophilic molecules attached to their lipid bilayers when drugs show low solubility in water. Liposomes can be classified according to their size and number of lipid bilayers. While small unilamellar vesicles (SUVs) are typically smaller than 50 nm, large unilamellar vesicles (LUVs) range between 50 and 500 nm in size, and giant unilamellar vesicles (GUVs) are capable of reaching up to 100 µm [
29]. The particle size is especially important when designing synthetic vaccines because it affects their dispersion and diffusion speeds, drainage to the lymph nodes, and antigen concentration [
30]; these ultimately regulate intracellular uptake by antigen-presenting cells. In the present study, the encapsulation method was tested with the successful incorporation of a DNA vaccine encoding PfRH5 [
31]. As performed previously with plasmid DNA, tests to determine the ideal lipid/cargo ratio were conducted, and it was possible to obtain cationic particles with an ideal size of 50–100 nm, which protected their cargo and were easily taken up by antigen-presenting cells [
32].
The obtained antiPfRH5 titers and their inhibitory effects were perfectly comparable in efficiency to previous results obtained by us [
31] and others [
33]; we are currently not aware of any other published study that employed samRNA immunization of PfRH5. In terms of inhibitory activity, the samRNA performed less well than when native proteins were incorporated into liposomes [
34,
35], but performed better than liposomes with recombinant PfRH5 displayed on their surface [
18]. This may be because merozoite protein-loaded particles elicit antibodies against multiple antigens, including antiPfRH5, which then function synergistically to slow down erythrocyte invasion [
36]. It must be noted that the non-polymorphic antigen PfRH5 alone is not sufficiently effective as an anti-blood stage vaccine, because only specific inhibitory antibodies against determined epitopes provide protection [
37]; even when delivered in PfRH5′s natural complex with CYRPA and RIPR, invasion inhibition is not greatly augmented [
38], indicating that more antigens are needed to block merozoite invasion. In this sense, samRNA encoding a number of different antigens against which antibodies exert invasion-inhibitory activity, seems to be an attractive avenue to follow in terms of vaccine development. An additional advantage of mRNA-encoded antigens lies in the stimulation of cellular immune responses, as proteins synthesized after translation from delivered mRNA are internally processed and presented as MHC1 molecules, thus calling for the inclusion of liver-stage antigens in a samRNA-based approach.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}