

In Silico and In Vivo Studies of a Tumor-Penetrating and Interfering Peptide with Antitumoral Effect on Xenograft Models of Breast Cancer
 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis and Sequence
2.2. Peptide NRP-1 Complex Structure Prediction
2.3. Molecular Dynamics Simulation
2.4. Mice
2.5. Breast Cancer Xenograft Models and Treatment
2.6. Analysis of Peptide Stability in Human Serum
2.7. Histological Samples Staining
2.8. Statistical Analysis
2.9. Ethical Considerations
3. Results
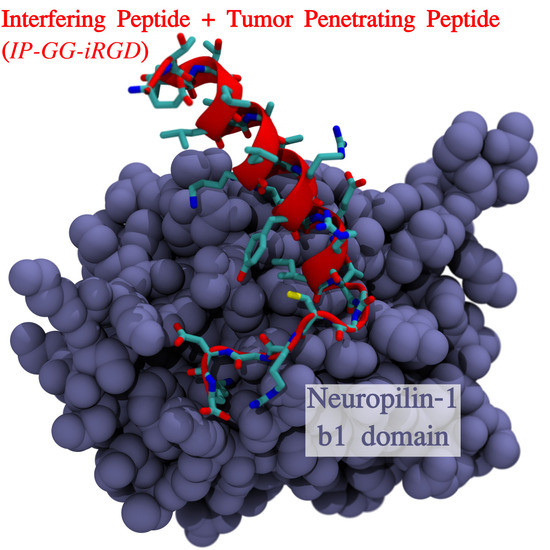
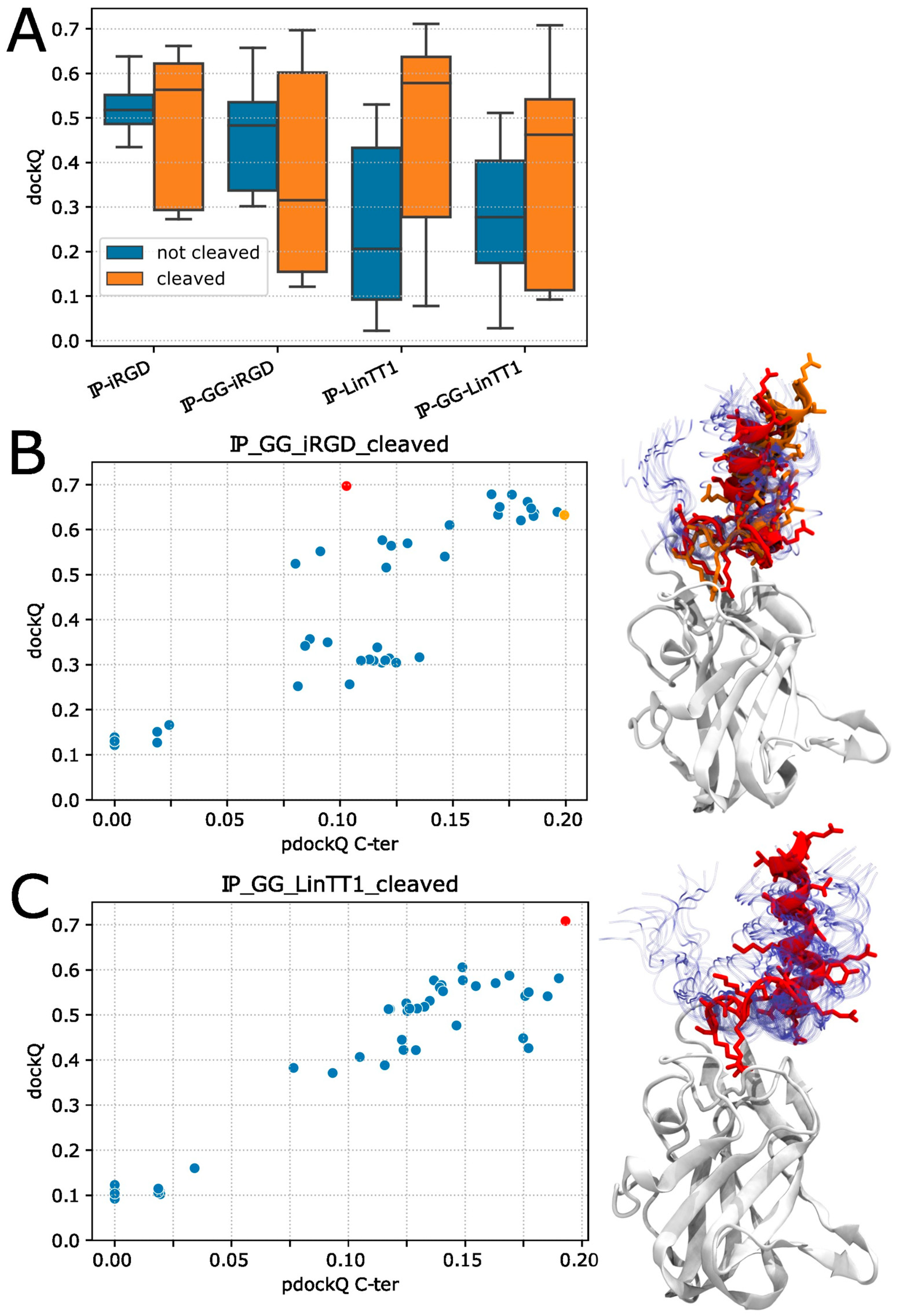
3.1. Neuropilin1–Peptide Interaction Models
3.2. Molecular Dynamics Simulation
3.3. Antitumoral Effect of Lin TT1 and iRGD Interfering Peptides on Breast Cancer Xenograft Models
3.4. Stability of iRGD-IP and Lin TT1-IP Peptides in Serum
3.5. TPP-IP Peptides Induce Apoptosis of Tumoral Graft
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Perl, A.; Leonard, D.; Sangodkar, J.; Narla, G. Therapeutic targeting of PP2A. Int. J. Biochem. Cell Biol. 2018, 96, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Pavan, V.; Ribaudo, G.; et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood 2015, 125, 3747–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibaldi, E.; Pagano, M.A.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Ribaudo, G.; Pavan, V.; Bordin, L.; Visentin, A.; et al. Targeted activation of the SHP-1/PP2A signaling axis elicits apoptosis of chronic lymphocytic leukemia cells. Haematologica 2017, 102, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Possemato, R.; Campbell, K.; Plattner, C.A.; Pallas, D.C.; Hahn, W.C. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 2004, 5, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V.; Goris, J.; Van Hoof, C. PP2A: The expected tumor suppressor. Curr. Opin. Genet. Dev. 2005, 15, 34–41. [Google Scholar] [CrossRef]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, e229–e238. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Makkinje, A.; Damuni, Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 1996, 271, 11059–11062. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cai, M.; Chen, J.; Liao, Y.; Mai, S.; Li, Y.; Huang, X.; Liu, Y.; Zhang, J.; Kung, H.; et al. α4 contributes to bladder urothelial carcinoma cell invasion and/or metastasis via regulation of E-cadherin and is a predictor of outcome in bladder urothelial carcinoma patients. Eur. J. Cancer 2014, 50, 840–851. [Google Scholar] [CrossRef]
- Sobral, L.M.; Sousa, L.O.; Coletta, R.D.; Cabral, H.; Greene, L.J.; Tajara, E.H.; Gutkind, J.S.; Curti, C.; Leopoldino, A.M. Stable SET knockdown in head and neck squamous cell carcinoma promotes cell invasion and the mesenchymal-like phenotype in vitro, as well as necrosis, cisplatin sensitivity and lymph node metastasis in xenograft tumor models. Mol. Cancer 2014, 13, 32. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Tian, L.; Zhang, X.; Haesen, D.; Bravo, J.; Fominaya, J.; Choquet, S.; Zini, J.M.; Loisel, S.; Waelkens, E.; Janssens, V.; et al. Identification of PP2A/Set binding sites and design of interfering peptides with potential clinical applications. Int. J. Pept. Res. Ther. 2017, 24, 479–488. [Google Scholar] [CrossRef]
- Simón-Gracia, L.; Loisel, S.; Sidorenko, V.; Scodeller, P.; Parizot, C.; Savier, E.; Haute, T.; Teesalu, T.; Rebollo, A. Preclinical Validation of Tumor-Penetrating and Interfering Peptides against Chronic Lymphocytic Leukemia. Mol. Pharm. 2022, 19, 895–903. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.M.; Ruoslahti, E.M. Tumor-Penetrating Peptides. Front. Oncol. 2013, 3, 216. [Google Scholar] [CrossRef] [Green Version]
- Zanuy, D.; Kotla, R.; Nussinov, R.; Teesalu, T.; Sugahara, K.N.; Alemán, C.; Haspel, N. Sequence dependence of C-end rule peptides in binding and activation of neuropilin-1 receptor. J. Struct. Biol. 2013, 182, 78–86. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [Green Version]
- Braun, G.B.; Friman, T.; Pang, H.-B.; Pallaoro, A.; De Mendoza, T.H.; Willmore, A.-M.A.; Kotamraju, V.R.; Mann, A.P.; She, Z.-G.; Sugahara, K.N.; et al. Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nat. Mater. 2014, 13, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Braun, G.B.; de Mendoza, T.H.; Kotamraju, V.R.; French, R.P.; Lowy, A.M.; Teesalu, T.; Ruoslahti, E. Tumor-Penetrating iRGD Peptide Inhibits Metastasis. Mol. Cancer Ther. 2015, 14, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.N.; Scodeller, P.; Braun, G.B.; de Mendoza, T.H.; Yamazaki, C.M.; Kluger, M.D.; Kitayama, J.; Alvarez, E.; Howell, S.B.; Teesalu, T.; et al. A tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis. J. Control. Release 2015, 212, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Wonder, E.; Simón-Gracia, L.; Scodeller, P.; Majzoub, R.N.; Kotamraju, V.R.; Ewert, K.K.; Teesalu, T.; Safinya, C.R. Competition of charge-mediated and specific binding by peptide-tagged cationic liposome–DNA nanoparticles in vitro and in vivo. Biomaterials 2018, 166, 52–63. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paasonen, L.; Sharma, S.; Braun, G.B.; Kotamraju, V.R.; Chung, T.D.; She, Z.G.; Sugahara, K.N.; Yliperttula, M.; Wu, B.; Pellecchia, M.; et al. New p32/gC1qR Ligands for Targeted Tumor Drug Delivery. Chembiochem 2016, 17, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Simón-Gracia, L.; Hunt, H.; Scodeller, P.; Gaitzsch, J.; Kotamraju, V.R.; Sugahara, K.N.; Tammik, O.; Ruoslahti, E.; Battaglia, G.; Teesalu, T. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 2016, 104, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [Green Version]
- Fogal, V.; Babic, I.; Chao, Y.; Pastorino, S.; Mukthavaram, R.; Jiang, P.; Cho, Y.-J.; Pingle, S.C.; Crawford, J.R.; Piccioni, D.E.; et al. Mitochondrial p32 is upregulated in Myc expressing brain cancers and mediates glutamine addiction. Oncotarget 2015, 6, 1157–1170. [Google Scholar] [CrossRef] [Green Version]
- Fogal, V.; Zhang, L.; Krajewski, S.; Ruoslahti, E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008, 68, 7210–7218. [Google Scholar] [CrossRef] [Green Version]
- Simon-Gracia, L.; Savier, E.; Parizot, C.; Brossas, J.Y.; Loisel, S.; Teesalu, T.; Conti, F.; Charlotte, F.; Scatton, O.; Aoudjehane, L.; et al. Bifunctional therapeutic peptides for targeting malignant B cells and hepatocytes: Proof of concerpt in chronic lymphocytic leukemai. Adv. Ther. 2020, 3, 2000131. [Google Scholar] [CrossRef]
- Markowska, A.; Markowski, A.R.; Jarocka-Karpowicz, I. The Importance of 6-Aminohexanoic Acid as a Hydrophobic, Flexible Structural Element. Int. J. Mol. Sci. 2021, 22, 12122. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Johansson-Åkhe, I.; Mirabello, C.; Wallner, B. Predicting protein-peptide interaction sites using distant protein complexes as structural templates. Sci. Rep. 2019, 9, 4267. [Google Scholar] [CrossRef] [Green Version]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Berrocal, L.; Cirri, E.; Zhang, X.; Andrini, L.; Marin, G.H.; Lebel-Binay, S.; Rebollo, A. New Therapeutic Approach for Targeting Hippo Signalling Pathway. Sci. Rep. 2019, 9, 4771. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2009, 1795, 1–15. [Google Scholar] [CrossRef]
- Kalev, P.; Sablina, A.A. Protein phosphatase 2A as a potential target for anticancer therapy. Anticancer Agents Med. Chem. 2011, 11, 38–46. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nagahara, Y.; Ikekita, M.; Shinomiya, T. A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br. J. Pharmacol. 2003, 138, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, Q.; Lu, Y.; Li, X.; Huang, S. Reactivating PP2A by FTY720 as a Novel therapy for AML with C-KIT tyrosine kinase domain mutation. J. Cell. Biochem. 2012, 113, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.J.; Chen, Y.; Oddo, J.; Matta, K.M.; Neil, J.; Davis, E.D.; Volkheimer, A.D.; Lanasa, M.C.; Friedman, D.R.; Goodman, B.K.; et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: A predictor of aggressive disease and a new treatment target. Blood 2011, 118, 4150–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Guo, H.; Damuni, Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 1995, 34, 1988–1996. [Google Scholar] [CrossRef]
- Neviani, P.; Perrotti, D. SETting OP449 into the PP2A-Activating Drug Family. Clin. Cancer Res. 2014, 20, 2026–2028. [Google Scholar] [CrossRef] [Green Version]
- Campello, R.J.G.B.; Moulavi, D.; Sander, J. Density-Based Clustering Based on Hierarchical Density Estimates. In Proceedings of the Advances in Knowledge Discovery and Data Mining, Gold Coast, Australia, 14–17 April 2013; Pei, J., Tseng, V.S., Cao, L., Motoda, H., Xu, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 160–172. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iRGD-IP | ETVTLLVALKVRYRERIT-Ahx-CRGDKGPDC-CONH2 (C-C disulfide bond) |
| LinTT1-IP | ETVTLLVALKVRYRERIT-Ahx-AKRGARSTA-CONH2 |
| IP-iRGD (no linker, full iRGD) | ETVTLLVALKVRYRERITCRGDKGPDC |
| IP-GG-iRGD (GG linker, full iRGD) | ETVTLLVALKVRYRERITGGCRGDKGPDC |
| IP-iRGD-cl (no linker, cleaved iRGD) | ETVTLLVALKVRYRERITCRGDK |
| IP-GG-iRGD-cl (GG linker, cleaved iRGD) | ETVTLLVALKVRYRERITGGCRGDK |
| IP-LinTT1 (no linker, full LinTT1) | ETVTLLVALKVRYRERITAKRGARSTA |
| IP-GG-LinTT1(GG linker, full LinTT1) | ETVTLLVALKVRYRERITGGAKRGARSTA |
| IP-LinTT1-cl (no linker, cleaved LinTT1) | ETVTLLVALKVRYRERITAKRGAR |
| IP-GG-LinTT1-cl (GG linker, cleaved LInTT1) | ETVTLLVALKVRYRERITGGAKRGAR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin, G.H.; Murail, S.; Andrini, L.; Garcia, M.; Loisel, S.; Tuffery, P.; Rebollo, A. In Silico and In Vivo Studies of a Tumor-Penetrating and Interfering Peptide with Antitumoral Effect on Xenograft Models of Breast Cancer. Pharmaceutics 2023, 15, 1180. https://doi.org/10.3390/pharmaceutics15041180
Marin GH, Murail S, Andrini L, Garcia M, Loisel S, Tuffery P, Rebollo A. In Silico and In Vivo Studies of a Tumor-Penetrating and Interfering Peptide with Antitumoral Effect on Xenograft Models of Breast Cancer. Pharmaceutics. 2023; 15(4):1180. https://doi.org/10.3390/pharmaceutics15041180
Chicago/Turabian StyleMarin, Gustavo H., Samuel Murail, Laura Andrini, Marcela Garcia, Severine Loisel, Pierre Tuffery, and Angelita Rebollo. 2023. "In Silico and In Vivo Studies of a Tumor-Penetrating and Interfering Peptide with Antitumoral Effect on Xenograft Models of Breast Cancer" Pharmaceutics 15, no. 4: 1180. https://doi.org/10.3390/pharmaceutics15041180