
Pharmacokinetic Boosting of Kinase Inhibitors
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
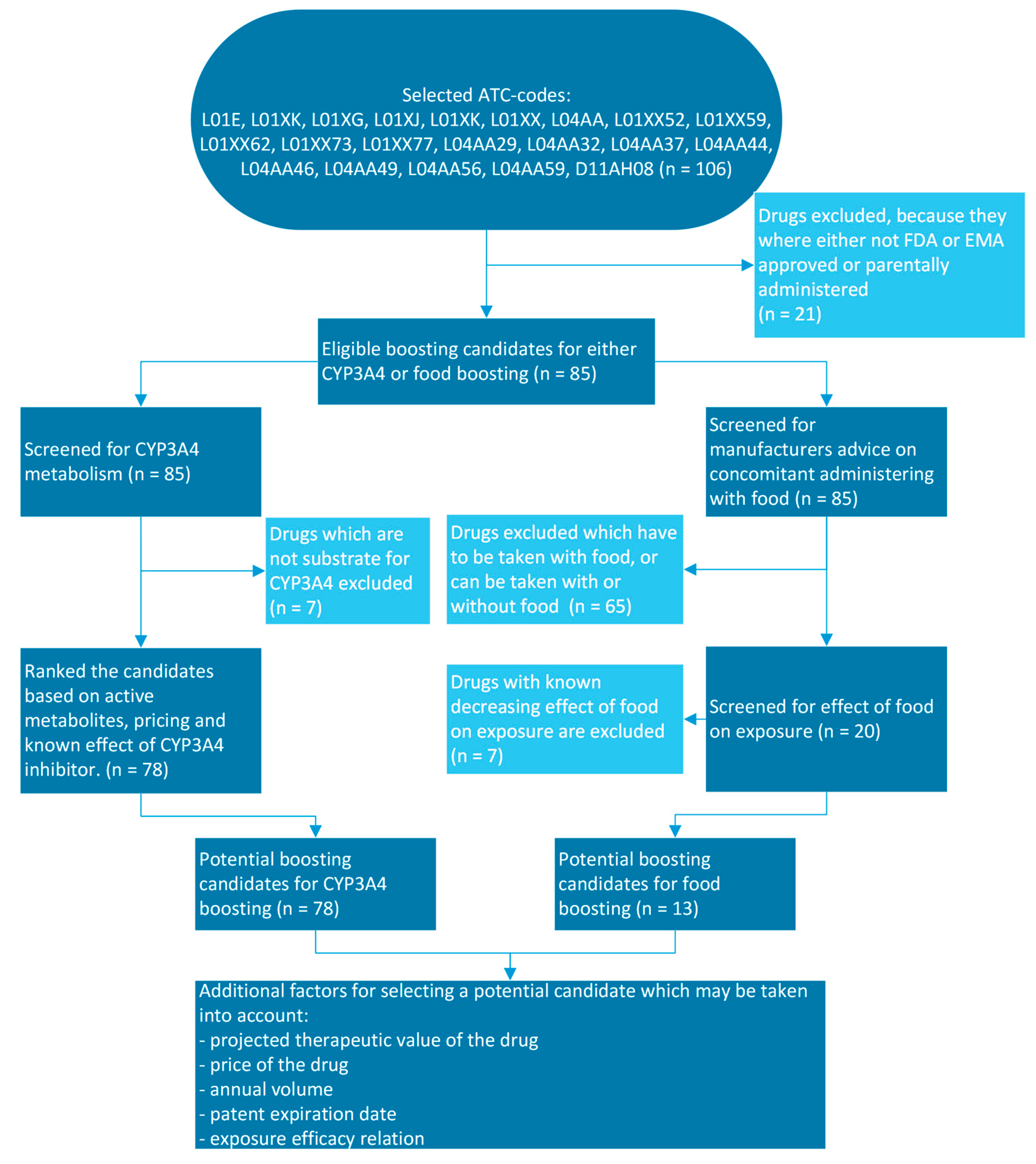
3.1. Pharmacokinetic Boosting Strategies Potentially Useful for Kinase Inhibitors
3.2. Pharmacological Profiling of Candidate Kinase Inhibitors Suitable for Pharmacokinetic CYP3A4 Boosting
3.3. Pharmacological Profiling of Candidate Kinase Inhibitors Suitable for Pharmacokinetic Food Boosting
3.4. Clinical Evidence and Experience with Pharmacokinetic Boosting of Kinase Inhibitors
3.4.1. Axitinib Boosted with Cobicistat
3.4.2. Crizotinib Boosted with Cobicistat
3.4.3. Erlotinib Boosted with Ritonavir
3.4.4. Ibrutinib Boosted with Itraconazole
3.4.5. Imatinib Boosted with Grapefruit Juice
3.4.6. Lapatinib Boosted with Ketoconazole
3.4.7. Nilotinib Boosted with Food
3.4.8. Osimertinib Boosted with Cobicistat
3.4.9. Pazopanib Boosted with Continental Breakfast
3.4.10. Tofacitinib Boosted with Cobicistat
3.4.11. Venetoclax and Ibrutinib Boosted with Itraconazole
3.4.12. Venetoclax Boosted with Posaconazole
3.4.13. Venetoclax Boosted with Grapefruit Juice
3.5. Clinical Trials Currently in Progress
4. Discussion
4.1. Benefits of Pharmacokinetic Boosting of Kinase Inhibitors
4.2. Risks and Disadvantages of Pharmacokinetic Boosting of Kinase Inhibitors
4.3. Factors for Selecting a Pharmacokinetic Boosting Candidate
4.4. Clinical Trial Design of Studies Validating Pharmacokinetic Boosting
4.5. Role of Therapeutic Drug Monitoring in Pharmacokinetic Boosting of Kinase Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krauss, J.; Bracher, F. Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond. Sci. Pharm. 2018, 86, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, K.H.; Woodward, R.; Peters, L.; Verwey, W.F.; Mattis, P.A. The Prolongation of Penicillin Retention in the Body by Means of Para-Aminohippuric Acid. Science 1944, 100, 107–108. [Google Scholar] [CrossRef] [PubMed]
- First, M.R.; Schroeder, T.J.; Weiskittel, P.; Myre, S.A.; Alexander, J.W.; Pesce, A. Concomitant administration of cyclosporin and ketoconazole in renal transplant recipients. Lancet 1989, 334, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Ghosn, J.; Taiwo, B.; Seedat, S.; Autran, B.; Katlama, C. HIV. Lancet 2018, 392, 685–697. [Google Scholar] [CrossRef]
- Kempf, D.J.; Marsh, K.C.; Kumar, G.; Rodrigues, A.D.; Denissen, J.F.; McDonald, E.; Kukulka, M.J.; Hsu, A.; Granneman, G.R.; Baroldi, P.A.; et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Vincent Rajkumar, S. The high cost of prescription drugs: Causes and solutions. Blood Cancer J. 2020, 10, 71. [Google Scholar] [CrossRef]
- Glode, A.E.; May, M.B. Rising Cost of Cancer Pharmaceuticals: Cost Issues and Interventions to Control Costs. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 85–93. [Google Scholar] [CrossRef]
- Stuurman, F.E.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H. Oral Anticancer Drugs: Mechanisms of Low Bioavailability and Strategies for Improvement. Clin. Pharmacokinet. 2013, 52, 399–414. [Google Scholar] [CrossRef]
- Eisenmann, E.D.; Talebi, Z.; Sparreboom, A.; Baker, S.D. Boosting the oral bioavailability of anticancer drugs through intentional drug–drug interactions. Basic Clin. Pharmacol. Toxicol. 2022, 130 (Suppl. S1), 23–35. [Google Scholar] [CrossRef]
- Tucker, G.T. Pharmacokinetic considerations and challenges in oral anticancer drug therapy. Pharm. J. 2019. [Google Scholar] [CrossRef]
- Patel, A.A.; Cahill, K.; Saygin, C.; Odenike, O. Cedazuridine/decitabine: From preclinical to clinical development in myeloid malignancies. Blood Adv. 2021, 5, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef] [PubMed]
- WHO ATC/DDD Index. Available online: https://www.whocc.no/atc_ddd_index/ (accessed on 16 January 2023).
- European Public Assessment Reports (EPAR)—EMA. Available online: https://www.ema.europa.eu/en/medicines (accessed on 16 January 2023).
- UpToDate. Available online: https://www.uptodate.com/contents/search (accessed on 16 January 2023).
- UpToDate Drug Interactions. Available online: https://www.uptodate.com/drug-interactions (accessed on 16 January 2023).
- Clinical Trials.gov. Available online: https://clinicaltrials.gov/ (accessed on 16 February 2023).
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Loos, N.H.C.; Beijnen, J.H.; Schinkel, A.H. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 2022, 23, 9866. [Google Scholar] [CrossRef]
- Lolodi, O.; Wang, Y.-M.; Wright, W.C.; Chen, T. Differential Regulation of CYP3A4 and CYP3A5 and its Implication in Drug Discovery. Curr. Drug Metab. 2018, 18, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Storelli, F.; Samer, C.; Reny, J.-L.; Desmeules, J.; Daali, Y. Complex Drug–Drug–Gene–Disease Interactions Involving Cytochromes P450: Systematic Review of Published Case Reports and Clinical Perspectives. Clin. Pharmacokinet. 2018, 57, 1267–1293. [Google Scholar] [CrossRef] [Green Version]
- Waring, R.H. Cytochrome P450: Genotype to phenotype. Xenobiotica 2020, 50, 9–18. [Google Scholar] [CrossRef]
- Roy, J.-N.; Lajoie, J.; Zijenah, L.S.; Barama, A.; Poirier, C.; Ward, B.J.; Roger, M. CYP3A5 genetic polymorphisms in different ethnic populations. Drug Metab. Dispos. 2005, 33, 884–887. [Google Scholar] [CrossRef]
- FDA. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table1 (accessed on 8 February 2023).
- Singh, R.S.P.; Toussi, S.S.; Hackman, F.; Chan, P.L.; Rao, R.; Allen, R.; Van Eyck, L.; Pawlak, S.; Kadar, E.P.; Clark, F.; et al. Innovative Randomized Phase I Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID-19 Trial of Nirmatrelvir. Clin. Pharmacol. Ther. 2022, 112, 101–111. [Google Scholar] [CrossRef]
- Larson, K.B.; Wang, K.; Delille, C.; Otofokun, I.; Acosta, E.P. Pharmacokinetic Enhancers in HIV Therapeutics. Clin. Pharmacokinet. 2014, 53, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Eisenmann, E.D.; Garrison, D.A.; Talebi, Z.; Jin, Y.; Silvaroli, J.A.; Kim, J.-G.; Sparreboom, A.; Savona, M.R.; Mims, A.S.; Baker, S.D. Interaction of Antifungal Drugs with CYP3A- and OATP1B-Mediated Venetoclax Elimination. Pharmaceutics 2022, 14, 694. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.; Hughes, C.A.; Wu, J.; Seet, J.; Phillips, E.J. Cobicistat Versus Ritonavir: Similar Pharmacokinetic Enhancers But Some Important Differences. Ann. Pharmacother. 2017, 51, 1008–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, S.; Mathias, A.A.; German, P.; Kearney, B.P. Clinical Pharmacokinetic and Pharmacodynamic Profile of the HIV Integrase Inhibitor Elvitegravir. Clin. Pharmacokinet. 2011, 50, 229–244. [Google Scholar] [CrossRef] [PubMed]
- German, P.; Liu, H.C.; Szwarcberg, J.; Hepner, M.; Andrews, J.; Kearney, B.P.; Mathias, A. Effect of Cobicistat on Glomerular Filtration Rate in Subjects with Normal and Impaired Renal Function. J. Acquir. Immune. Defic. Syndr. 2012, 61, 32–40. [Google Scholar] [CrossRef]
- Koziolek, M.; Alcaro, S.; Augustijns, P.; Basit, A.W.; Grimm, M.; Hens, B.; Hoad, C.L.; Jedamzik, P.; Madla, C.M.; Maliepaard, M.; et al. The mechanisms of pharmacokinetic food-drug interactions—A perspective from the UNGAP group. Eur. J. Pharm. Sci. 2019, 134, 31–59. [Google Scholar] [CrossRef]
- Veerman, G.D.M.; Hussaarts, K.; Jansman, F.G.A.; Koolen, S.W.L.; van Leeuwen, R.W.F.; Mathijssen, R.H.J. Clinical implications of food–drug interactions with small-molecule kinase inhibitors. Lancet Oncol. 2020, 21, e265–e279. [Google Scholar] [CrossRef]
- Van Leeuwen, R.W.; Peric, R.; Hussaarts, K.G.; Kienhuis, E.; Ijzerman, N.; De Bruijn, P.; Van Der Leest, C.; Codrington, H.; Kloover, J.S.; Van Der Holt, B.; et al. Influence of the Acidic Beverage Cola on the Absorption of Erlotinib in Patients with Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Lubberman, F.J.E.; Van Erp, N.P.; Ter Heine, R.; Van Herpen, C.M.L. Boosting axitinib exposure with a CYP3A4 inhibitor, making axitinib treatment personal. Acta Oncol. 2017, 56, 1238–1240. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Igarashi, R.; Suzuki-Honma, N.; Fujiyama, N.; Narita, S.; Inoue, T.; Saito, M.; Akihama, S.; Tsuruta, H.; Miura, M.; et al. Association of pharmacokinetics of axitinib with treatment outcome and adverse events in advanced renal cell carcinoma patients. J. Clin. Oncol. 2015, 33, 506. [Google Scholar] [CrossRef]
- Hohmann, N.; Bozorgmehr, F.; Christopoulos, P.; Mikus, G.; Blank, A.; Burhenne, J.; Thomas, M.; Haefeli, W.E. Pharmacoenhancement of Low Crizotinib Plasma Concentrations in Patients with Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer using the CYP3A Inhibitor Cobicistat. Clin. Transl. Sci. 2021, 14, 487–491. [Google Scholar] [CrossRef]
- FDA—Clinical Pharmacology and Biopharmaceutics Reviews(s)—Crizotinib. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000ClinPharmR.pdf (accessed on 22 February 2023).
- Boosman, R.J.; de Gooijer, C.J.; Groenland, S.L.; Burgers, J.A.; Baas, P.; van der Noort, V.; Beijnen, J.H.; Huitema, A.D.R.; Steeghs, N. Ritonavir-Boosted Exposure of Kinase Inhibitors: An Open Label, Cross-over Pharmacokinetic Proof-of-Concept Trial with Erlotinib. Pharm. Res. 2022, 39, 669–676. [Google Scholar] [CrossRef]
- Tapaninen, T.; Olkkola, A.M.; Tornio, A.; Neuvonen, M.; Elonen, E.; Neuvonen, P.J.; Niemi, M.; Backman, J.T. Itraconazole Increases Ibrutinib Exposure 10-Fold and Reduces Interindividual Variation—A Potentially Beneficial Drug-Drug Interaction. Clin. Transl. Sci. 2020, 13, 345–351. [Google Scholar] [CrossRef]
- Kimura, S.-I.; Kako, S.; Wada, H.; Sakamoto, K.; Ashizawa, M.; Sato, M.; Terasako, K.; Kikuchi, M.; Nakasone, H.; Okuda, S.; et al. Can grapefruit juice decrease the cost of imatinib for the treatment of chronic myelogenous leukemia? Leuk. Res. 2011, 35, e11–e12. [Google Scholar] [CrossRef]
- Chien, A.J.; Munster, P.N.; Melisko, M.E.; Rugo, H.S.; Park, J.W.; Goga, A.; Auerback, G.; Khanafshar, E.; Ordovas, K.; Koch, K.M.; et al. Phase I Dose-Escalation Study of 5-Day Intermittent Oral Lapatinib Therapy in Patients with Human Epidermal Growth Factor Receptor 2–Overexpressing Breast Cancer. J. Clin. Oncol. 2014, 32, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Boons, C.; den Hartog, Y.M.; Janssen, J.; Zandvliet, A.S.; Vos, R.M.; Swart, E.L.; Hendrikse, N.H.; Hugtenburg, J.G. Food-effect study of nilotinib in chronic myeloid leukaemia (NiFo study): Enabling dose reduction and relief of treatment burden. Eur. J. Haematol. 2020, 105, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Akamine, Y.; Abumiya, M.; Takahashi, S.; Yoshioka, T.; Kameoka, Y.; Takahashi, N.; Miura, M. Relationship between achievement of major molecular response or deep molecular response and nilotinib plasma concentration in patients with chronic myeloid leukemia receiving first-line nilotinib therapy. Cancer Chemother. Pharmacol. 2022, 89, 609–616. [Google Scholar] [CrossRef]
- van Veelen, A.; Gulikers, J.; Hendriks, L.E.L.; Dursun, S.; Ippel, J.; Smit, E.F.; Dingemans, A.C.; van Geel, R.; Croes, S. Pharmacokinetic boosting of osimertinib with cobicistat in patients with non-small cell lung cancer: The OSIBOOST trial. Lung Cancer 2022, 171, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Lubberman, F.J.E.; Gelderblom, H.; Hamberg, P.; Vervenne, W.L.; Mulder, S.F.; Jansman, F.G.A.; Colbers, A.; Van Der Graaf, W.T.A.; Burger, D.M.; Luelmo, S.; et al. The Effect of Using Pazopanib With Food vs. Fasted on Pharmacokinetics, Patient Safety, and Preference (DIET Study). Clin. Pharmacol. Ther. 2019, 106, 1076–1082. [Google Scholar] [CrossRef]
- van der Togt, C.J.T.; Verhoef, L.M.; van den Bemt, B.J.F.; den Broeder, N.; ter Heine, R.; den Broeder, A.A. Pharmacokinetic boosting to enable a once-daily reduced dose of tofacitinib in patients with rheumatoid arthritis and psoriatic arthritis (the PRACTICAL study). Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X221142277. [Google Scholar] [CrossRef]
- De la Garza-Salazar, F.; Colunga-Pedraza, P.R.; Gómez-Almaguer, D. Cytochrome P450 inhibition to decrease dosage and costs of venetoclax and ibrutinib: A proof-of-concept case study. Br. J. Clin. Pharmacol. 2023, 89, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; DiNardo, C.D.; Potluri, J.; Dunbar, M.; Kantarjian, H.M.; Humerickhouse, R.A.; Wong, S.L.; Menon, R.M.; Konopleva, M.Y.; Salem, A.H. Management of Venetoclax-Posaconazole Interaction in Acute Myeloid Leukemia Patients: Evaluation of Dose Adjustments. Clin. Ther. 2017, 39, 359–367. [Google Scholar] [CrossRef]
- Long, Z.; Ruan, M.; Wu, W.; Zeng, Q.; Li, Q.; Huang, Z. The successful combination of grapefruit juice and venetoclax in an unfit acute myeloid leukemia patient with adverse risk: A case report. Front. Oncol. 2022, 12, 912696. [Google Scholar] [CrossRef]
- Hellriegel, E.T.; Bjornsson, T.D.; Hauck, W.W. Interpatient variability in bioavailability is related to the extent of absorption: Implications for bioavailability and bioequivalence studies. Clin. Pharmacol. Ther. 1996, 60, 601–607. [Google Scholar] [CrossRef]
- Chitikela, S.; Kumar, L.; Sahoo, R.K. Azacitidine and Venetoclax in AML. New Engl. J. Med. 2020, 383, 2087–2089. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the Investigation of Bioequivalence (rev1). Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (accessed on 6 February 2023).
- Glaeser, H. Importance of P-glycoprotein for Drug–Drug Interactions. Handb. Exp. Pharmacol. 2011, 285–297. [Google Scholar] [CrossRef]
- van Erp, N.P.; Gelderblom, H.; Guchelaar, H.-J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat. Rev. 2009, 35, 692–706. [Google Scholar] [CrossRef]
- Truong, J.; Chan, K.K.W.; Mai, H.; Chambers, A.; Sabharwal, M.; Trudeau, M.E.; Cheung, M.C. The impact of pricing strategy on the costs of oral anti-cancer drugs. Cancer Med. 2019, 8, 3770–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.S.; Bose, P.; Cruz, N.D.; Jiang, Y.; Wu, Q.; Thompson, P.A.; Feng, S.; Kroll, M.H.; Qiao, W.; Huang, X.; et al. A pilot study of lower doses of ibrutinib in patients with chronic lymphocytic leukemia. Blood 2018, 132, 2249–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.Y. After Outcry, drugmakers Decide Not to Triple the Price of a Cancer Pill—The Washington Post. Available online: https://www.washingtonpost.com/news/wonk/wp/2018/05/15/after-outcry-drugmakers-decide-not-to-triple-the-price-of-a-cancer-pill/ (accessed on 15 March 2023).
- Navid, F.; Christensen, R.; Inaba, H.; Li, L.; Chen, Z.; Cai, X.; Regel, J.; Baker, S.D. Alternative formulations of sorafenib for use in children. Pediatr. Blood Cancer 2013, 60, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, J.K.; ter Heine, R.T.; Verheul, H.M.W.; Chatelut, E.; Rudek, M.A.; Gurney, H.; Plummer, R.; Gilbert, D.C.; Buclin, T.; Burger, D.M.; et al. Off-label, but on target: The evidence needed to implement alternative dosing regimens of anticancer drugs. ESMO Open 2023, 8, 100749. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.; Sparreboom, A.; Verweij, J. Determining the optimal dose in the development of anticancer agents. Nat. Rev. Clin. Oncol. 2014, 11, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Groenland, S.L.; van Eerden, R.A.G.; Westerdijk, K.; Meertens, M.; Koolen, S.L.W.; Moes, D.; de Vries, N.; Rosing, H.; Otten, H.; Vulink, A.J.E.; et al. Therapeutic drug monitoring-based precision dosing of oral targeted therapies in oncology: A prospective multicenter study. Ann. Oncol. 2022, 33, 1071–1082. [Google Scholar] [CrossRef]
- Mueller-Schoell, A.; Groenland, S.L.; Scherf-Clavel, O.; van Dyk, M.; Huisinga, W.; Michelet, R.; Jaehde, U.; Steeghs, N.; Huitema, A.D.R.; Kloft, C. Therapeutic drug monitoring of oral targeted antineoplastic drugs. Eur. J. Clin. Pharmacol. 2021, 77, 441–464. [Google Scholar] [CrossRef] [PubMed]
- van Gelder, T. The Appropriately Designed TDM Clinical Trial: Endpoints, Pitfalls, and Perspectives. Ther. Drug Monit. 2023, 45, 6–10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Active Metabolites | Known Effect of CYP3A4 Inhibitor on AUC | Pricing of Different Drug Strengths |
|---|---|---|
| 0—no active metabolites or unknown | 0—>200% increase in AUC | 0—mg-based pricing or one strength available |
| 1—minor active metabolites (<10% responsible for efficacy) | 1—100–200% increase in AUC | 1—flat-based pricing for all available strengths |
| 2—major active metabolites (>10% responsible for efficacy) | 2—50–100% increase in AUC | |
| 5—<50% or unknown increase in AUC |
| Ritonavir | Cobicistat | |
|---|---|---|
| Antiviral activity | Yes, HIV protease inhibitor; inhibits HIV-1 and HIV-2 | No |
| Dosage as pharmacokinetic enhancer | 100 mg or 200 mg; QD or BID | 150 mg QD |
| Protein binding | 99% | 98% |
| Half-life | 3–5 h | 3–4 h |
| Distribution volume | 20–40 L | Not known |
| Metabolized by | CYP3A4 and to lesser extent CYP2D6 | CYP3A4 and to lesser extent CYP2D6 |
| Inhibitor of | CYP3A4 (strong), CYP2D6 (minor), P-gp and OATP1B1 | CYP3A4 (strong) CYP2D6 (minor), P-gp, BCRP, MATE1, OATP1B1, OATP1B3 |
| Inducer of | CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, UGT | - |
| In vitro CYP3A4 inhibition duration | Irreversible | Irreversible |
| Drug | Pricing of Different Strengths | Metabolism | Substrate of Transporters | Bioavailability | CYP3A4 Inhibitory Drug | Effect on AUC (Fold Change) | Effect on Cmax (Fold Change) | Ranking Score |
|---|---|---|---|---|---|---|---|---|
| Adagrasib | One strength available | CYP3A4, however at steady state, adagrasib inhibits its own CYP3A4 metabolism, which allows CYP2C8, CYP1A2, CYP2B6, CYP2C9, and CYP2D6 to contribute to metabolism. | BCRP/ABCG2 | - | Itraconazole | 4 | 2.4 | 0 |
| Bosutinib | Strength-based pricing | CYP3A4 to primarily inactive metabolites M2, M5 and M6 | - | 34% when administered with food | Ketoconazole | 8.6 | 5.2 | 0 |
| Cobimetinib | One strength available | CYP3A4 | P-gp/ABCB1 | 46% | Itraconazole | 6.7 | 3.2 | 0 |
| Duvelisib | Strength-based pricing | CYP3A4 | BCRP/ABCG2 | 42% | Ketoconazole | 4 | 1.7 | 0 |
| Encorafenib | One strength available | CYP3A4 and to a lesser extent by CYP2C19 and CYP2D6 | P-gp/ABCB1 | ≥86% of the dose is absorbed | Posaconazole | 3 | 1.7 | 0 |
| Fedratinib | One strength available | CYP3A4, CYP2C19, and flavin-containing monooxygenase 3 (FMO3) | OATP1B1/1B3 (SLCO1B1/1B3) | - | Ketoconazole | 3.1–3.9 | 1.9 | 0 |
| Lapatinib | One strength available | CYP3A4 and CYP3A5 and to a lesser extent by CYP2C19 and CYP2C8 to metabolites | BCRP/ABCG2 and P-gp/ABCB1 | - | Ketoconazole | 3.6 | 2.1 | 0 |
| Larotrectinib | Strength-based pricing | CYP3A4; forms a metabolite (activity unknown) | BCRP/ABCG2 and P-gp/ABCB1 | 34% | Itraconazole | 4.3 | 2.8 | 0 |
| Pralsetinib | One strength available | CYP3A4 and to a lesser extent CYP2D6 and CYP1A2 | BCRP/ABCG2 and P-gp/ABCB1 | - | Itraconazole | 3.5 | 1.8 | 0 |
| Zanubrutinib | One strength available | CYP3A4 | - | - | Ketoconazole | 3.8 | 2.6 | 0 |
| Avacopan | One strength available | CYP3A4 | - | - | Itraconazole | 2.19 | 1.87 | 1 |
| Avapritinib | Flat pricing | CYP3A4 and CYP3A5 and to lesser extent CYP2C9, which forms the metabolite M690 | - | - | Itraconazole | 7 | - | 1 |
| Axitinib | Strength-based pricing | CYP3A4/5 and to a lesser extent CYP1A2, CYP2C19 and UGT1A1 | 58% | Ketoconazole | 2.1 | 1.5 | 1 | |
| Brigatinib | Strength-based pricing | CYP2C8 and CYP3A4 | BCRP/ABCG2 and P-gp/ABCB1 | - | Itraconazole | 2.01 | - | 1 |
| Ceritinib | One strength available | CYP3A4 | P-gp/ABCB1 | - | Ketoconazole | 2.9 | 1.2 | 1 |
| Crizotinib | Flat pricing | CYP3A4/5 | P-gp/ABCB1 | 43% | Ketoconazole | 3.2 | 1.7 | 1 |
| Dasatinib | Strength-based pricing | CYP3A4, flavin-containing mono-oxygenase-3 (FOM-3) and UGT to an active metabolite (minor role in the efficacy) | - | - | Ketoconazole | 4.8 | 3.6 | 1 |
| Glasdegib | Strength-based pricing | CYP3A4 and to a lesser extent CYP2C8 and UGT1A9 | BCRP/ABCG2 | 77% | Ketoconazole | 2.4 | 1.4 | 1 |
| Ibrutinib | Strength-based pricing | CYP3A and to a lesser extent CYP2D6 to form active metabolite PCI-45227(minor role in the efficacy) | - | 2.9% | Ketoconazole | 24 | 29 | 1 |
| Ivosidenib | One strength available | CYP3A4 and to a lesser extent the N-dealkylation and hydrolytic pathways | P-gp/ABCB1 | - | Itraconazole | 2.7 | No change | 1 |
| Nilotinib | Flat pricing | CYP3A4 to primarily inactive metabolites | P-gp/ABCB1 | 50% | Ketoconazole | 3 | 1.8 | 1 |
| Olaparib | One strength available | CYP3A4 | P-gp/ABCB1 | - | Itraconazole | 2.7 | 1.4 | 1 |
| Ribociclib | One strength available | CYP3A4 to metabolites M13, M4 and M1 (minor role in the efficacy) | - | - | Ritonavir | 3.2 | 1.7 | 1 |
| Selpercatinib | Strength-based pricing | CYP3A4 | BCRP/ABCG2 | 73% | Itraconazole | 2.3 | 1.3 | 1 |
| Venetoclax | Strength-based pricing | CYP3A to form the major metabolite M27 (BCL-2 inhibitory activity 58-fold lower) | BCRP/ABCG2 and P-gp/ABCB1 | - | Ritonavir | 6.1–8.1 | 2.3–2.4 | 1 |
| Acalabrutinib | One strength available | CYP3A4 enzymes and to a lesser extent glutathione conjugation and amide hydrolysis to form active metabolite ACP-5862 | BCRP/ABCG2 and P-gp/ABCB1 | 25% | Itraconazole | 5.1 | 3.7–3.9 | 2 |
| Dabrafenib | Strength-based pricing | CYP2C8 and CYP3A4 to form active metabolite hydroxy-dabrafenib | BCRP/ABCG2 and P-gp/ABCB1 | 95% | Ketoconazole | 1.71 | - | 2 |
| Erlotinib | Strength-based pricing | CYP3A4 and to a lesser extent CYP1A1, CYP1A2, and CYP1C to form metabolites (activity unknown) | - | 60% without food, 100% with food | Ketoconazole | 1.69 | 1.52 | 2 |
| Everolimus | Flat pricing | CYP3A4 and forms six metabolites with minor activity | P-gp/ABCB1 | 30% | Ketoconazole | 15 | 3.9 | 2 |
| Gefitinib | One strength available | CYP3A4 and to a lesser extent CYP2D6. Forms metabolites | BCRP/ABCG2 | 60% | Itraconazole | 1.16–1.78 | 1.32–1.51 | 2 |
| Gilteritinib | One strength available | CYP3A4 to form active metabolites M17, M16 and M10 (minor role in the efficacy) | P-gp/ABCB1 | - | Itraconazole | 2.2 | 1.2 | 2 |
| Infigratinib | Strength-based pricing | CYP3A4 and to a lesser extent FMO3 to form active metabolites BHS697 and CQM157 | BCRP/ABCG2 and P-gp/ABCB1 | - | Itraconazole | 7.22 | 2.64 | 2 |
| Mobocertinib | One strength available | CYP3A to form active metabolites AP32960 and AP32914 | P-gp/ABCB1 | 37% | Itraconazole | 6.3 | 2.9 | 2 |
| Neratinib | One strength available | CYP3A4 and flavin-containing monooxygenase to form active metabolites M3, M6, M7, and M11 | P-gp/ABCB1 | - | Ketoconazole | 4.8 | 3.2 | 2 |
| Pacritinib | One strength available | CYP3A4 and forms the 2 major metabolites M1 and M2 (activity unknown) | - | - | Clarithromycin | 1.8 | 1.3 | 2 |
| Pazopanib | One strength available | CYP3A4 and P-gp/ABCB1 and to a lesser extent by CYP1A2, CYP2C8 and BCRP/ABCG2 | BCRP/ABCG2 and P-gp/ABCB1 | - | Ketoconazole | 1.66 | 1.45 | 2 |
| Pexidartinib | One strength available | CYP3A4 and glucuronidation via UGT1A4 to form an inactive metabolite | - | - | Itraconazole | 1.73 | 1.48 | 2 |
| Sonidegib | One strength available | CYP3A4 | - | <10% | Ketoconazole | 2.2 | 1.5 | 2 |
| Tofacitinib | Flat pricing | CYP3A4 and CYP2C19 to form inactive metabolites | - | 74% | Itraconazole | 2.04 | 1.15 | 2 |
| Upadacitinib | Strength-based pricing | CYP3A4 | - | - | Ketoconazole | 1.75 | 1.7 | 2 |
| Entrectinib | Flat pricing | CYP3A4 to form the active metabolite M5 | - | - | Itraconazole | 6 | 1.7 | 3 |
| Idelalisib | Flat pricing | Aldehyde oxidase and CYP3A, which forms major metabolite GS-563117, to a lesser extent UGT1A4 | BCRP/ABCG2 and P-gp/ABCB1 | - | Ketoconazole | 1.8 | No change | 3 |
| Nintedanib | Flat pricing | Hydrolytic cleavage by esterases to inactive metabolite BIBF 1202, which is further UGT 1A1, UGT 1A7, UGT 1A8, and UGT 1A10 to BIBF 1202 glucuronide, and to a lesser extent CYP3A4 | OCT1 and P-gp/ABCB1 | 5% | Ketoconazole | 1.6 | 1.8 | 3 |
| Palbociclib | Flat pricing | CYP3A4 and SULT2A1 | - | 46% | Itraconazole | 1.87 | 1.34 | 3 |
| Pemigatinib | Flat pricing | CYP3A4 | BCRP/ABCG2 and P-gp/ABCB1 | - | Itraconazole | 1.88 | 1.17 | 3 |
| Ponatinib | Flat pricing | CYP3A4 and to a lesser extent CYP2C8, CYP2D6, and CYP3A5 | BCRP/ABCG2 and P-gp/ABCB1 | - | Ketoconazole | 1.78 | 1.47 | 3 |
| Ripretinib | One strength available | CYP3A4 and to a lesser extent CYP2C8 and CYP2D6 to form active metabolite DP-5439(activity unknown) | BCRP/ABCG2 and P-gp/ABCB1 | - | Itraconazole | 1.99 | 1.36 | 3 |
| Abemaciclib | Flat pricing | CYP3A4 to form active metabolites M2, M20, M18 and M1 | BCRP/ABCG2 and P-gp/ABCB1 | 45% | Clarithromycin | 2.5 | - | 4 |
| Midostaurin | One strength available | CYP3A4 to form active metabolites CGP62221 and CGP52421 | - | - | Ketoconazole | 10.4 | 1.8 | 4 |
| Sunitinib | Strength-based pricing | CYP3A4 to form active metabolite SU12662 | - | - | Ketoconazole | 1.51 | 1.49 | 4 |
| Apremilast | One strength available | CYP3A4 and to a lesser extent CYP1A2 and CYP2A6 | P-gp/ABCB1 | 73% | - | - | - | 5 |
| Baricitinib | Strength-based pricing | CYP3A4 | BCRP/ABCG2 and P-gp/ABCB1 | 80% | - | - | - | 5 |
| Erdafitinib | Strength-based pricing | CYP2C9 and CYP3A4 | P-gp/ABCB1 | Itraconazole | 1.34 | No change | 5 | |
| Lorlatinib | Strength-based pricing | CYP3A4 and UGT1A4 and to a lesser extent CYP2C8, CYP2C19, CYP3A5 and UGT1A3 | - | 81% | Itraconazole | 1.42 | 1.24 | 5 |
| Selumetinib | Strength-based pricing | CYP3A4 and to a lesser extent BCRP/ABCG2, CYP1A2, CYP2A6, CYP2C19, CYP2C9, CYP2E1, CYP3A4, P-glycoprotein/ABCB1, UGT1A1 and UGT1A3 | BCRP/ABCG2 and P-gp/ABCB1 | 62% | Itraconazole | 1.49 | 1.19 | 5 |
| Sotorasib | One strength available | CYP3A4, CYP3A5 and CYP2C8 | - | - | - | - | - | 5 |
| Tucatinib | Strength-based pricing | CYP2C8 and to a lesser extent CYP3A | BCRP/ABCG2 | - | - | - | - | 5 |
| Vemurafenib | One strength available | BCRP/ABCG2 and CYP3A4 and to a lesser extent P-gp/ABCB1 | BCRP/ABCG2 and P-gp/ABCB1 | 64% | Itraconazole | 1.4 | 1.4 | 5 |
| Ruxolitinib | Flat pricing | CYP3A4 and to lesser extent CYP2C9 to form active metabolites | - | - | Ketoconazole | 1.91 | 1.33 | 5 |
| Alpelisib | Flat pricing | Chemical and enzymatic hydrolysis to form its metabolite and to a lesser extent CYP3A4 | BCRP/ABCG2 | - | - | - | - | 6 |
| Asciminib | Flat pricing | CYP3A4, UGT2B7 and UGT2B17 | BCRP/ABCG2 and P-gp/ABCB1 | - | Clarithromycin | 1.36 | 1.19 | 6 |
| Cabozantinib | Flat pricing | CYP3A4 | - | - | Ketoconazole | 1.36 | - | 6 |
| Capmatinib | Flat pricing | CYP3A4 and aldehyde oxidase | P-gp/ABCB1 | >70% | Itraconazole | 1.42 | No change | 6 |
| Enasidenib | Flat pricing | CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A1, UGT1A3, UGT1A4, UGT1A9, UGT2B7 and UGT2B15 | - | 57% | - | - | - | 6 |
| Futibatinib | unknown | CYP3A, and to a lesser extent CYP2C9 and CYP2D6 | BCRP/ABCG2 and P-gp/ABCB1 | - | Itraconazole | 1.41 | 1.51 | 6 |
| Ixazomib | Flat pricing | CYP3A4, CYP1A2, CYP2B6, CYP2C8, CYP2D6, CYP2C19 and CYP2C9 | P-gp/ABCB1 | 58% | - | - | - | 6 |
| Lenvatinib | Flat pricing | CYP3A4 and aldehyde oxidase | BCRP/ABCG2 and P-gp/ABCB1 | - | - | - | - | 6 |
| Rucaparib | Flat pricing | CYP2D6 and to a lesser extent CYP1A2 and CYP3A4 | BCRP/ABCG2 and P-gp/ABCB1 | 36% | - | - | - | 6 |
| Tepotinib | One strength available | CYP3A4 and CYP2C8 to form an active metabolite | P-gp/ABCB1 | 71.6% | - | - | - | 6 |
| Tivozanib | Flat pricing | CYP3A4 | - | - | Ketoconazole | 1.12 | - | 6 |
| Alectinib | One strength available | CYP3A4 to form active metabolite M4 | - | 37% | - | - | - | 7 |
| Imatinib | Strength-based pricing | CYP3A4 and to a lesser extent CYP1A2, CYP2D6, CYP2C9 and CYP2C19 to form active metabolite CGP74588 | OCT1 and P-gp/ABCB1 | 98% | Ketoconazole | 1.4 | 1.26 | 7 |
| Regorafenib | One strength available | CYP3A4 and UGT1A9 to form active metabolites M2 and M5 | - | - | Ketoconazole | 1.33 | - | 7 |
| Sorafenib | One strength available | CYP3A4 and UGT1A9 to form an active metabolite | - | 38–49% | Itraconazole | No change | No change | 7 |
| Vandetanib | Strength-based pricing | CYP3A4 to form active metabolites N-desmethyl vandetanib and vandetanib-N-oxide | - | - | Itraconazole | 1.09 | No change | 7 |
| Abrocitinib | Flat pricing | CYP2C19 and to a lesser extent CYP2C9, CYP3A4 and CYP2B6 to form active metabolites 3-hydroxypropyl abrocitinib and 2-hydroxypropyl abrocitinib | OAT1/3 | 60% | - | - | - | 8 |
| Dacomitinib | Flat pricing | Oxidation and glutathione conjugation and by CYP2D6 and CYP3A4 to form active metabolite O-desmethyl dacomitinib | BCRP/ABCG2 | 80% | - | - | - | 8 |
| Osimertinib | Flat pricing | CYP3A4 to form active metabolites Z7550 and AZ5104 | BCRP/ABCG2 and P-gp/ABCB1 | Itraconazole | 1.24 | .8 | 8 |
| Drug | Pricing of Different Strengths | Bioavailability | Food Effect |
|---|---|---|---|
| Avapritinib | Flat pricing | - | AUC and Cmax increased 1.29 and 1.59-fold, respectively, when administered with a high-fat, high-calorie meal |
| Cabozantinib | Flat pricing | - | AUC and Cmax increased 1.57 and 1.41-fold, respectively, when administered with a high-fat meal |
| Erlotinib | Strength-based pricing | 60% without food | Absorption 60% in fasted state, food increases absorption to 100% |
| Ibrutinib | Strength-based pricing | 2.9% | AUC and Cmax increased two-fold and two- to four-fold, respectively, when administered with a high-fat, high-calorie meal |
| Infigratinib | Strength-based pricing | - | AUC and Cmax increased 1.8- to 2.2-fold and 1.6- to 1.8-fold, respectively, when administered with a high-fat, high-calorie meal |
| Ivosidenib | One strength available | - | AUC and Cmax increased 1.24 and 1.98-fold, respectively, when administered with a high-fat meal |
| Lapatinib | One strength available | - | AUC increased three- to four-fold when administered with food |
| Nilotinib | Flat pricing | 50% | Bioavailability increased 1.82-fold when administered 30 min after a high-fat meal |
| Pazopanib | One strength available | - | AUC increased two-fold when administered with a high-fat or low-fat meal |
| Pexidartinib | One strength available | - | AUC and Cmax increased two-fold when administered with a high-fat meal |
| Pralsetinib | One strength available | - | AUC and Cmax increased 2.22 and 2.04-fold, respectively, when administered with a high-fat meal |
| Sonidegib | One strength available | <10% | AUC increased seven- to eight-fold when administered with a high-fat meal |
| Sotorasib | One strength available | - | AUC increased 1.25-fold when administered with a high-fat meal |
| Target Drug | Boosting Agent | Study Aim | Study Design | Outcomes | Results | Conclusion | Reference |
|---|---|---|---|---|---|---|---|
| Axitinib | Cobicistat 150 mg QID | Boost axitinib exposure with cobicistat | Case report, one patient | Cmin | Axitinib 10 mg QID + cobicistat 150 mg QID resulted in a 15-month stable response | Boosting axitinib with cobicistat can be a promising strategy to boost patients with sub-optimal axitinib exposure. | [34] |
| Crizotinib | Cobicistat 150 mg QD | Patients with low crizotinib exposure (Cmin,ss ≤ 310 ng/mL) were boosted with cobicistat | Open-label, non-randomized, within group crossover study, one patient | Change in AUC0–24,ss and Cmin0–24,ss | The AUC and Cmin,ss increased by 78% and 164% respectively when crizotinib was boosted by cobicistat. | Cobicistat enhanced the exposure of crizotinib. Only one patient was enrolled because the next-generation ALK inhibitor alectinib was approved for the treatment of the same population with better outcomes. | [36] |
| Erlotinib | Ritonavir 200 mg QD | Bioequivalence of erlotinib 150 mg QD compared to erlotinib 75 mg QD + ritonavir 200 mg QD to save treatment costs | Open-label, non-randomized, within group crossover study, nine patients | GMR of AUC0–24h, Cmax and Cmin | GMR of erlotinib 150 mg QD vs. erlotinib 75 mg + ritonavir 200 mg QD for AUC0–24h, Cmax and Cmin were 0.99 (CI 95% 0.58–1.69, p = 0.545), 0.91 (CI 95% 0.55–1.49, p= 0.500) and 1.06 (CI 95% 0.59–1.93, p = 0.150), respectively. | Erlotinib 150 mg QD compared to erlotinib 75 mg + ritonavir 200 mg is bioequivalent and can be a strategy to reduce the erlotinib dosage by 50% and thus save treatment costs. | [38] |
| Ibrutinib | Itraconazole 200 mg BID | Evaluate exposure of Ibrutinib 15 mg + itraconazole compared to ibrutinib 140 mg + placebo | Randomized placebo-controlled crossover study with 11 healthy volunteers | GMR of AUC0-∞ and Cmax | GMR of ibrutinib 15 mg + itraconazole vs. ibrutinib 140 mg + placebo AUC0-∞ and Cmax were 1.07 (CI 90% 0.77–1.49; p = 0.719) and 0.94 (CI 90% 0.68–1.30, p = 0.727), respectively, the GMR CVs of AUC0-∞ and Cmax for ibrutinib 15 mg boosted + itraconazole were 0.55 and 0.53, respectively, and for ibrutinib 140 mg + placebo 1.04 and 0.99, respectively. | The interindividual variability of exposure of ibrutinib is high; boosting with itraconazole and a reduced dose of ibrutinib could lower the interindividual variability. Boosting with itraconazole is cost-effective and can potentially reduce the treatment costs associate with ibrutinib by 90%. Cost savings in the United States are projected to be more than $10,000 annually per patient. | [39] |
| Imatinib | Grapefruit juice | Evaluate whether reduction of imatinib is feasible to reduce treatment costs with grapefruit juice | Open-label, non-randomized, within group crossover study, four patients | Cmin and Cmax | The median Cmin was 1080 ng/mL (range: 1060–1360 ng/mL) and 1102 ng/mL (range: 772–1450 ng/mL) for imatinib 400 mg and imatinib 400 mg in combination with grapefruit juice, respectively. The median Cmax was 2495 ng/mL (range: 2380–2680 ng/mL) and 2455 ng/mL (range: 1870–2750 ng/mL) for imatinib 400mg and imatinib 400mg in combination with grapefruit, juice respectively. | Pharmacokinetic of imatinib 400 mg QD compared to imatinib 400 mg QD boosted with grapefruit juice was not significantly different. The study was prematurely terminated because no significant effect of grapefruit juice on imatinib pharmacokinetics was observed. | [40] |
| Lapatinib | Ketoconazole 200 mg BID | Evaluate dose-escalating strategies for lapatinib | Phase I dose escalation study, 12 patients in the cohorts boosted with ketoconazole | Lapatinib concentration | Concomitant administration of lapatinib + ketoconazole increased lapatinib exposure 2.7 fold. | Lapatinib exposure can be enhanced by ketoconazole. | [41] |
| Nilotinib | Food; low-fat, medium-fat and high-fat meals | Evaluate whether nilotinib exposure can be enhanced with food | Open-label, non-randomized, within-group crossover study, 15 patients | GMR of AUC0–12h, Cmax and Cmin | The GMR of the morning dose of AUC0–12h, Cmax and Cmin was 0.89 (CI 90% 0.81–0.98), 0.90 (CI 90% 0.8–1.02) and 0.88 (CI 90% 0.84–0.92), respectively, and were within acceptance limits for bioequivalence. The GMR of the evening dose of AUC0–12h, Cmax and Cmin was 0.84 (CI 90% 0.73–0.97), 0.8 (CI 90% 0.68–0.93) and 1.06 (CI 90% 0.92–1.22), respectively. | Bioequivalence for Cmin was reached; AUC0–12h and Cmax were not bioequivalent. Nilotinib efficacy is associated with Cmin, meaning that nilotinib 200 mg BID with food can still be a viable option. | [42] |
| Osimertinib | Cobicistat 150 mg QD | Patients with low osimertinib exposure (Cmin,ss ≤ 195 ng/mL) were boosted with cobicistat | Open-label, non-randomized, within-group crossover study, 11 patients | Change in AUC0–24,ss (primary) and Cmin | The mean AUC0–24,ss increase with cobicistat was 60% | Concurrent use of cobicistat and osimertinib increased the exposure of osimertinib and its metabolite AZ5104 in all patients and can be an option to reduce the osimertinib dose. | [44] |
| Pazopanib | Food; continental breakfast | Evaluate whether pazopanib exposure can be enhanced with food | Open-label, randomized, within-group crossover study, 19 patients in part 1, 78 patients in part 2 | GMR of AUC0–24h, Cmax, Cmin, gastrointestinal toxicities and patient preference | The GMR of steady state AUC0–24h, Cmax and Cmin was 1.09 (CI 90% 1.02–1.17), 1.12 (CI 90% 1.04–1.20) and 1.10 (CI 90% 1.02–1.18), respectively. | Pazopanib 800 mg QD compared to pazopanib 600 mg QD + continental breakfast is bioequivalent, gastrointestinal AEs were comparable in both groups. Pazopanib + continental breakfast can achieve savings of approximately $8500 per patient for metastatic renal cell carcinoma and approximately $3800 per patient for soft tissue sarcoma in the Netherlands. | [45] |
| Tofacitinib | Cobicistat 150 mg QD | Bioequivalence of tofacitinib 5 mg BID compared to tofacitinib 5 mg QD + cobicistat 150 mg QD | Open-label, non-randomized, within-group crossover study, 25 patients | GMR of Cavg,ss | GMR of tofacitinib Cavg,ss for tofacitinib 5 mg BID vs. tofacitinib 5 mg QD + cobicistat 150 mg QD was 85% (CI 75–96%) | Tofacitinib 5 mg BID compared to tofacitinib 5 mg QD + cobicistat 150 mg are not pharmacokinetically bioequivalent. Disease activity remained stable, indicating similar efficacy. The tofacitinib 5 mg QD + cobicistat can potentially achieve annual cost savings of approximately €6500 per patient in the European Union and approximately €21,500 in the United States until the patent expiry date of 2028. | [46] |
| Venetoclax and ibrutinib | Itraconazole 100 mg BID | Evaluate whether a 75% dose reduction of venetoclax and ibrutinib is feasible when co-administered with itraconazole | Case report, one patient | Efficacy | A 22-year-old man was successfully treated with a 75% reduced dose of venetoclax 100 mg QD + itraconazole 100 mg BID and ibrutinib 75% reduced dose of 140 mg QD + itraconazole 100 mg BID. | CYP3A4 boosting with itraconazole to reduce the treatment costs of venetoclax and ibrutinib can be a promising strategy. A total of approximately $10,900 in cost savings was achieved in this patient by boosting venetoclax and ibrutinib. More research to validate this hypothesis is warranted; especially prospective studies are required. | [47] |
| Venetoclax | Posaconazole 300 mg QD | Evaluate which dose adjustment is necessary when venetoclax is concurrently administered with posaconazole | Drug–drug interaction study, 12 patients | AUC0–24h and Cmax | Venetoclax 50 mg QD + posaconazole increased the mean AUC0–24h and Cmax by 76% and 53%, respectively, vs. venetoclax 400 mg QD alone. Venetoclax 100 mg QD + posaconazole increased the mean AUC0–24h and Cmax by 155% and 93% vs. venetoclax 400 mg QD alone. | The venetoclax dose should be reduced by at least 75% when co-administered with posaconazole. | [48] |
| Venetoclax | Grapefruit juice | Evaluate whether venetoclax 100 mg QD could be boosted by grapefruit juice so that the therapy becomes more affordable | Case-report, one patient | Cmax and efficacy | The venetoclax Cmax was 1440 ng/mL and 1920 ng/mL on day 7 and day 14 after receiving the combination venetoclax 100 mg QD and grapefruit juice 200 mL TID. The venetoclax Cmax was inside the efficacy boundary of 1000 ng–3000 ng/mL. The patient was in remission for at least five cycles of 28 days; no serious AEs were observed. | Boosting venetoclax to make the treatment more affordable with grapefruit juice was safe and effective for this patient. The venetoclax-associated monthly costs were reduced from 38,880 RMD yuan (approx. €5281) to 9720 RMD Yuan (approx. €1319). | [49] |
| Study Name | Target Drug | Boosting Agent | Study Aim | Study Design | Outcomes | NCT Number |
|---|---|---|---|---|---|---|
| Cytochrome P450 Inhibition to Decrease Dosage of Dasatinib for Chronic Myelogenous Leukemia | Dasatinib | Ketoconazole | Investigate whether a 75% dasatinib dose reduction when boosted with ketoconazole is feasible to reduce treatment costs | Phase II open-label single-arm study with 15 participants | Primary: Cytogenetic and molecular response rates and AEs | NCT05638763 |
| Efficacy and Safety of Low-dose Ibrutinib and Itraconazole in Chronic Graft Versus Host Disease | Ibrutinib | Itraconazole | Investigate whether a 75% ibrutinib dose reduction when boosted with Itraconazole is feasible to reduce treatment costs | Phase II open-label single-arm study with 13 participants | Primary: Overall response rate and AEs | NCT05348096 |
| Pharmacokinetic Boosting of Olaparib to Improve Exposure, Tolerance and Cost-effectiveness (PROACTIVE) | Olaparib | Cobicistat | Ascertain bioequivalence of olaparib 300 mg BID vs. olaparib 100 mg BID + cobicistat 150 mg BID to reduce treatment costs | Part 1: bioequivalence in a cross-over olaparib vs. boosted olaparib. Part 2: non-inferiority of olaparib vs. boosted Olaparib, 160 participants | Primary: AUC0–12h, progression-free survival, number of dose reductions as a measure of toxicity | NCT05078671 |
| A Study of Extending Relugolix Dosing Intervals Through Addition of Itraconazole or Ritonavir in Prostate Cancer Patients | Relugolix | Itraconazole or ritonavir | Investigate safety and efficacy of relugolix when combined with itraconazole or ritonavir to extend dosing interval of relugolix to reduce treatment costs | Phase Ib in 100 participants | Primary: testosterone suppression | NCT05679388 |
| Low-dose Venetoclax and Azacitidine as Front-line Therapy in Newly Diagnosed AML | Venetoclax | Itraconazole | Investigate whether a 75% venetoclax dose reduction when boosted with Itraconazole is feasible to reduce treatment costs | Phase II open-label single-arm study with 15 participants | Primary: number of patients who are hospitalized, number of deceased patients in predefined time frames | NCT05048615 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westra, N.; Touw, D.; Lub-de Hooge, M.; Kosterink, J.; Oude Munnink, T. Pharmacokinetic Boosting of Kinase Inhibitors. Pharmaceutics 2023, 15, 1149. https://doi.org/10.3390/pharmaceutics15041149
Westra N, Touw D, Lub-de Hooge M, Kosterink J, Oude Munnink T. Pharmacokinetic Boosting of Kinase Inhibitors. Pharmaceutics. 2023; 15(4):1149. https://doi.org/10.3390/pharmaceutics15041149
Chicago/Turabian StyleWestra, Niels, Daan Touw, Marjolijn Lub-de Hooge, Jos Kosterink, and Thijs Oude Munnink. 2023. "Pharmacokinetic Boosting of Kinase Inhibitors" Pharmaceutics 15, no. 4: 1149. https://doi.org/10.3390/pharmaceutics15041149