
Poly-ε-Caprolactone Implants for Benznidazole Prolonged Release: An Alternative to Chagas Disease Oral Treatment
 ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Drugs and Materials
2.2. Preparation of PCL Implants Containing Benznidazole (BZ–PCL)
2.3. Characterization of PCL Implants Incorporating Benznidazole (BZ–PCL)
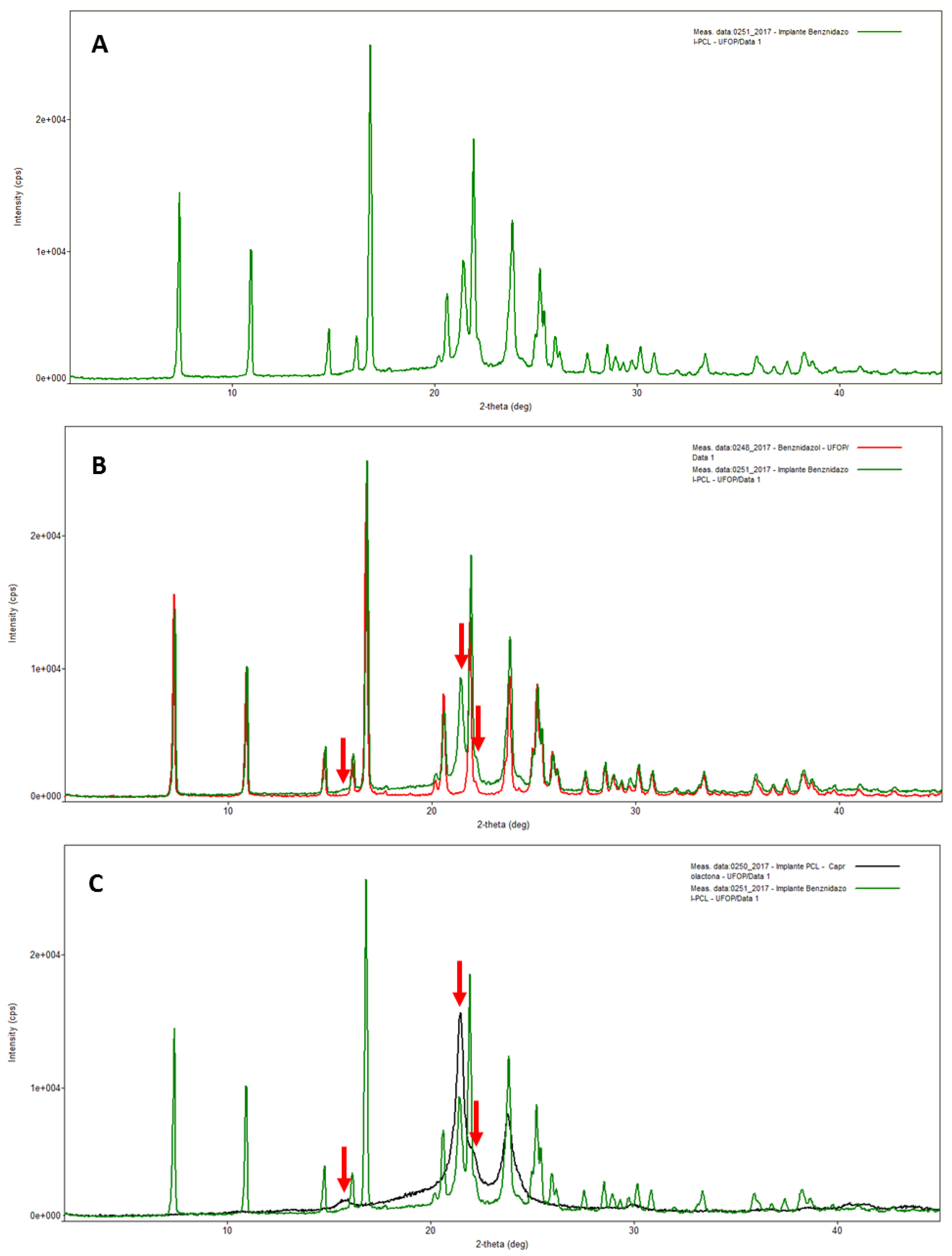
2.3.1. X-ray Diffraction Analysis (XRD)
2.3.2. Scanning Electron Microscopy (SEM)
2.3.3. Differential Scanning Calorimetry (DSC)
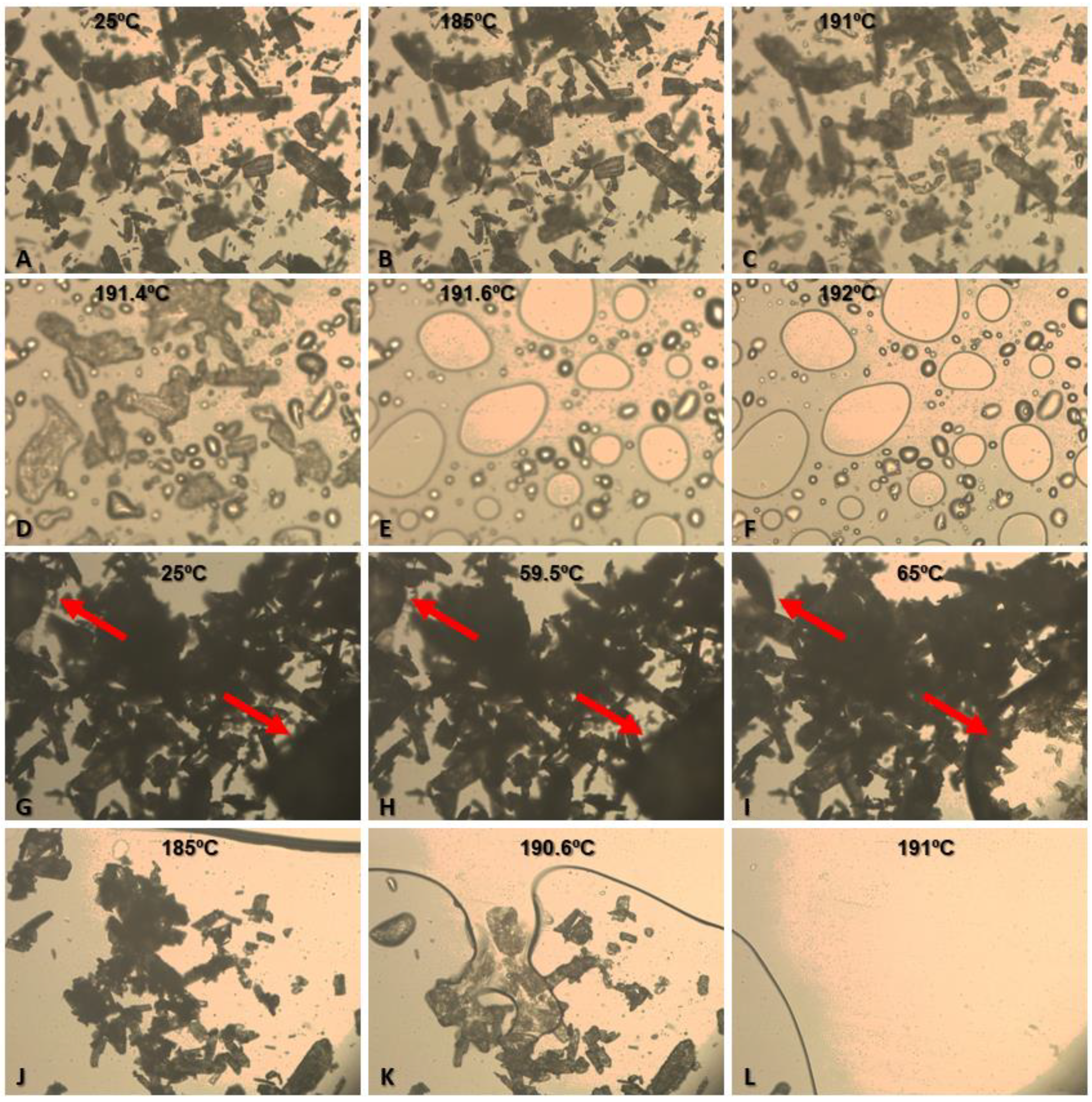
2.3.4. Thermo-Optic Analysis (TOA)
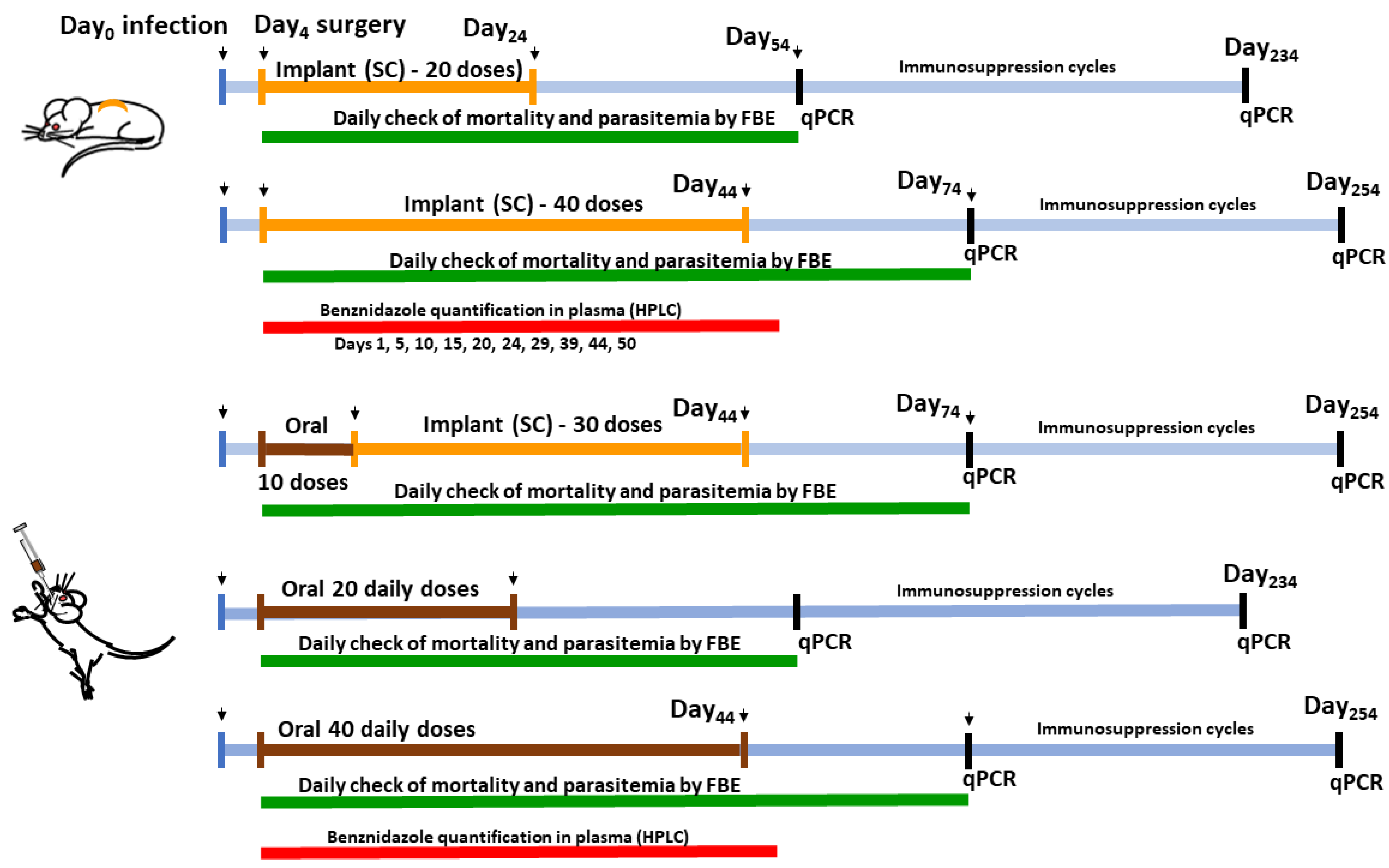
2.4. Animals and Ethical Concerns
2.5. Anti-T. cruzi Efficacy Experiments
2.6. Cure Control Tests
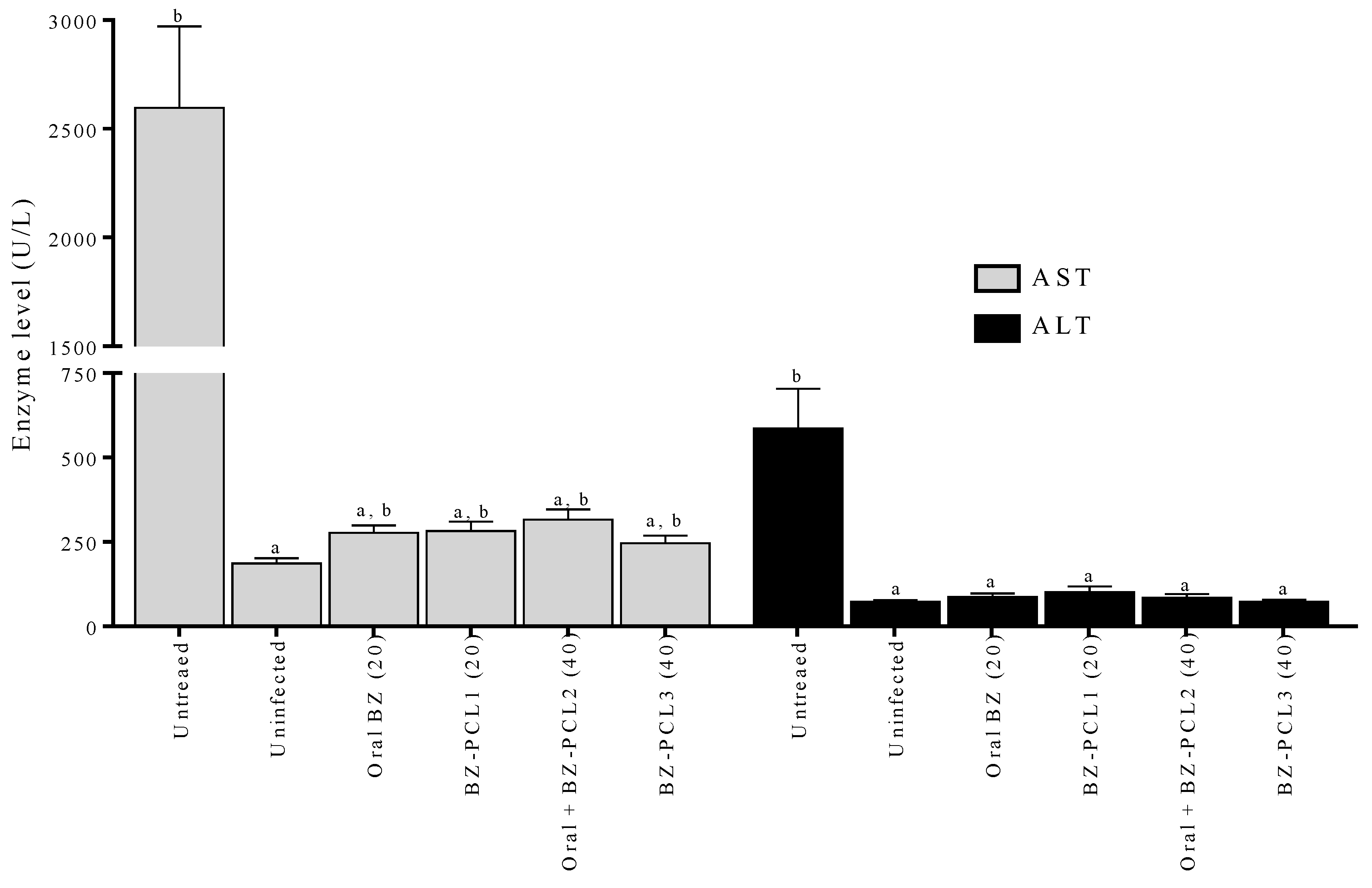
2.7. Hepatic Toxicity Evaluation
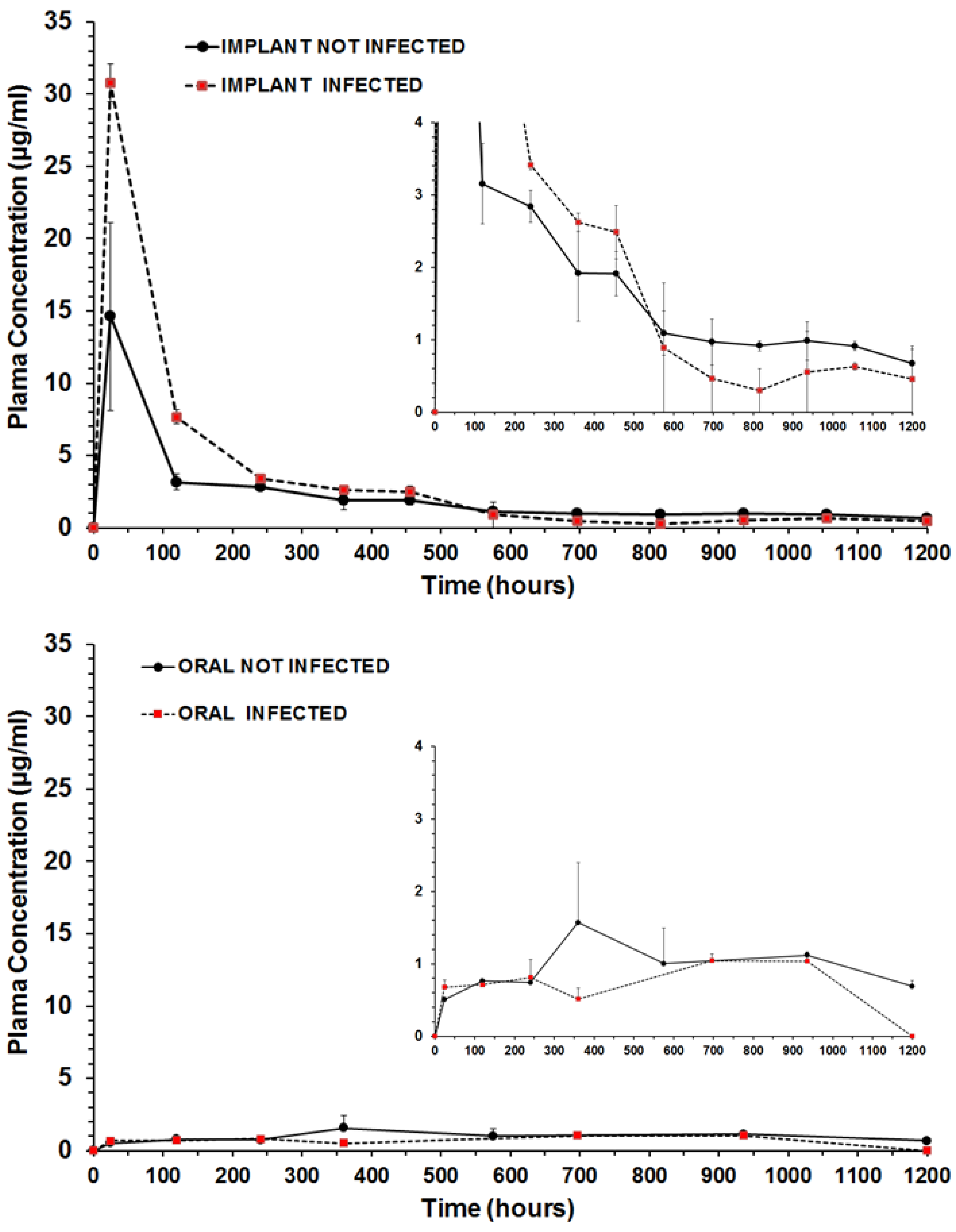
2.8. Benznidazole Plasmatic Concentration following Time
2.9. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Chagas Disease (Also Known as American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 2 September 2022).
- Lee, B.Y.; Bacon, K.M.; Bottazzi, M.E.; Hotez, P.J. Global economic burden of Chagas disease: A computational simulation model. Lancet Infect. Dis. 2013, 13, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.C.P.; Novaes Ramos, A.; Dias Gontijo, E.; Luquetti, A.; Aparecida Shikanai-Yasuda, M.; Rodrigues Coura, J.; Morais Torres, R.; Renan da Cunha Melo, J.; Antonio de Almeida, E.; de Oliveira, W., Jr.; et al. II Consenso Brasileiro em Doença de Chagas, 2015. Epidemiol. E Serviços Saúde 2016, 25, 7–86. [Google Scholar] [CrossRef]
- Pereiro, A.C. Guidelines for the diagnosis and treatment of Chagas disease. Lancet 2019, 393, 1486–1487. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A.; Rassi, A.; Marcondes de Rezende, J. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. North Am. 2012, 26, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Bahia, M.T.; Diniz, L.; Mosqueira, V.C.F. Therapeutical approaches under investigation for treatment of Chagas disease. Expert Opin. Investig. Drugs 2014, 23, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Mazzeti, A.L.; Capelari-Oliveira, P.; Bahia, M.T.; Mosqueira, V.C.F. Review on Experimental Treatment Strategies Against Trypanosoma cruzi. J. Exp. Pharmacol. 2021, 13, 409–432. [Google Scholar] [CrossRef]
- Soeiro, M.D.N.C. Perspectives for a new drug candidate for Chagas disease therapy. Mem. Inst. Oswaldo Cruz 2022, 117, e220004. [Google Scholar] [CrossRef]
- Bustamante, J.M.; Craft, J.M.; Crowe, B.D.; Ketchie, S.A.; Tarleton, R.L. New, Combined, and Reduced Dosing Treatment Protocols Cure Trypanosoma cruzi Infection in Mice. J. Infect. Dis. 2014, 209, 150–162. [Google Scholar] [CrossRef]
- Diniz, L.D.F.; Mazzeti, A.L.; Caldas, I.S.; Ribeiro, I.; Bahia, M.T. Outcome of E1224-Benznidazole Combination Treatment for Infection with a Multidrug-Resistant Trypanosoma cruzi Strain in Mice. Antimicrob. Agents Chemother. 2018, 62, e00401-18. [Google Scholar] [CrossRef] [Green Version]
- Rial, M.S.; Scalise, M.L.; López Alarcón, M.; Esteva, M.I.; Búa, J.; Benatar, A.F.; Prado, N.G.; Riarte, A.R.; Fichera, L.E. Experimental combination therapy using low doses of benznidazole and allopurinol in mouse models of Trypanosoma cruzi chronic infection. Parasitology 2019, 146, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Mazzeti, A.L.; Diniz, L.D.F.; Gonçalves, K.R.; WonDollinger, R.S.; Assíria, T.; Ribeiro, I.; Bahia, M.T. Synergic Effect of Allopurinol in Combination with Nitroheterocyclic Compounds against Trypanosoma cruzi. Antimicrob. Agents Chemother. 2019, 63, e02264-18. [Google Scholar] [CrossRef] [Green Version]
- Molina-Morant, D.; Fernández, M.L.; Bosch-Nicolau, P.; Sulleiro, E.; Bangher, M.; Salvador, F.; Sanchez-Montalva, A.; Ribeiro, A.L.P.; de Paula, A.M.B.; Eloi, S.; et al. Efficacy and safety assessment of different dosage of benznidazol for the treatment of Chagas disease in chronic phase in adults (MULTIBENZ study): Study protocol for a multicenter randomized Phase II non-inferiority clinical trial. Trials 2020, 21, 328. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.L.; Marson, M.E.; Ramirez, J.C.; Mastrantonio, G.; Schijman, A.G.; Altcheh, J.; Riarte, A.R.; Bournissen, F.G. Pharmacokinetic and pharmacodynamic responses in adult patients with Chagas disease treated with a new formulation of benznidazole. Mem. Inst. Oswaldo Cruz 2016, 111, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Maximiano, F.P.; de Paula, L.M.; Figueiredo, V.P.; de Andrade, I.M.; Talvani, A.; Sá-Barreto, L.C.; Bahia, M.T.; Cunha-Filho, M.S.S. Benznidazole microcrystal preparation by solvent change precipitation and in vivo evaluation in the treatment of Chagas disease. Eur. J. Pharm. Biopharm. 2011, 78, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Berzal, C.; Palmeiro-Roldán, R.; Escario, J.A.; Torrado, S.; Arán, V.J.; Torrado-Santiago, S.; Gómez-Barrio, A. Novel solid dispersions of benznidazole: Preparation, dissolution profile and biological evaluation as alternative antichagasic drug delivery system. Exp. Parasitol. 2015, 149, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares-Sobrinho, J.L.; Santos, F.L.A.; Lyra, M.A.M.; Alves, L.D.S.; Rolim, L.A.; Lima, A.A.N.; Nunes, L.C.C.; Soares, M.F.R.; Rolim-Neto, P.J.; Torres-Labandeira, J.J. Benznidazole drug delivery by binary and multicomponent inclusion complexes using cyclodextrins and polymers. Carbohydr. Polym. 2012, 89, 323–330. [Google Scholar] [CrossRef] [Green Version]
- García, M.C.; Martinelli, M.; Ponce, N.E.; Sanmarco, L.M.; Aoki, M.P.; Manzo, R.H.; Jimenez-Kairuz, A.F. Multi-kinetic release of benznidazole-loaded multiparticulate drug delivery systems based on polymethacrylate interpolyelectrolyte complexes. Eur. J. Pharm. Sci. 2018, 120, 107–122. [Google Scholar] [CrossRef]
- Spósito, P.Á.F.; Mazzeti Silva, A.L.; de Oliveira Faria, C.; Urbina, J.A.; Pound-Lana, G.; Bahia, M.T.; Mosqueira, V.C.F. Ravuconazole self-emulsifying delivery system: In vitro activity against Trypanosoma cruzi amastigotes and in vivo toxicity. Int. J. Nanomed. 2017, 12, 3785–3799. [Google Scholar] [CrossRef] [Green Version]
- Mazzeti, A.L.; Oliveira, L.T.; Gonçalves, K.R.; Schaun, G.C.; Mosqueira, V.C.F.; Bahia, M.T. Benznidazole self-emulsifying delivery system: A novel alternative dosage form for Chagas disease treatment. Eur. J. Pharm. Sci. 2020, 145, 105234. [Google Scholar] [CrossRef]
- Stewart, S.; Domínguez-Robles, J.; Donnelly, R.; Larrañeta, E. Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers 2018, 10, 1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohtashami, Z.; Esmaili, Z.; Vakilinezhad, M.A.; Seyedjafari, E.; Akbari Javar, H. Pharmaceutical implants: Classification, limitations and therapeutic applications. Pharm. Dev. Technol. 2020, 25, 116–132. [Google Scholar] [CrossRef]
- Carcaboso, Á.M.; Chiappetta, D.A.; Höcht, C.; Blake, M.G.; Boccia, M.M.; Baratti, C.M.; Sosnik, A. In vitro/in vivo characterization of melt-molded gabapentin-loaded poly(epsilon-caprolactone) implants for sustained release in animal studies. Eur. J. Pharm. Biopharm. 2008, 70, 666–673. [Google Scholar] [CrossRef]
- Cheng, L.; Guo, S.; Wu, W. Characterization and in vitro release of praziquantel from poly(ɛ-caprolactone) implants. Int. J. Pharm. 2009, 377, 112–119. [Google Scholar] [CrossRef]
- Lao, L.L.; Venkatraman, S.S.; Peppas, N.A. Modeling of drug release from biodegradable polymer blends. Eur. J. Pharm. Biopharm. 2008, 70, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Lemmouchi, Y.; Schacht, E.; Kageruka, P.; De Deken, R.; Diarra, B.; Diall, O.; Geerts, S. Biodegradable polyesters for controlled release of trypanocidal drugs: In vitro and in vivo studies. Biomaterials 1998, 19, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.D.F.; Pereira, L.G.R.; Barbosa, L.A.D.O.; Fialho, S.L.; Pereira, B.G.; Patricio, P.S.D.O.; Pinto, F.C.H.; Da Silva, G.R. Efficacy of methotrexate-loaded poly( ε -caprolactone) implants in Ehrlich solid tumor-bearing mice. Drug Deliv. 2013, 20, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.; Min, W.; Cruz, T.; Cindric, S.; Arsenault, L.; Von Hoff, D.; Degan, D.; Hunter, W.; Burt, H. A polymer-based drug delivery system for the antineoplastic agent bis(maltolato)oxovanadium in mice. Br. J. Cancer 1997, 75, 1014–1020. [Google Scholar] [CrossRef] [Green Version]
- Benhabbour, S.R.; Kovarova, M.; Jones, C.; Copeland, D.J.; Shrivastava, R.; Swanson, M.D.; Sykes, C.; Ho, P.T.; Cottrell, M.L.; Sridharan, A.; et al. Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery. Nat. Commun. 2019, 10, 4324. [Google Scholar] [CrossRef] [Green Version]
- Golenser, J.; Domb, A.; Teomim, D.; Tsafack, A.; Nisim, O.; Ponka, P.; Eling, W.; Cabantchik, Z.I. The treatment of animal models of malaria with iron chelators by use of a novel polymeric device for slow drug release. J. Pharmacol. Exp. Ther. 1997, 281, 1127–1135. [Google Scholar]
- Chen, B.Z.; Yang, Y.; Wang, B.B.; Ashfaq, M.; Guo, X.D. Self-implanted tiny needles as alternative to traditional parenteral administrations for controlled transdermal drug delivery. Int. J. Pharm. 2019, 556, 338–348. [Google Scholar] [CrossRef]
- Filardi, L.S.; Brener, Z. Susceptibility and natural resistance of Trypanosoma cruzi strains to drugs used clinically in Chagas disease. Trans. R. Soc. Trop. Med. Hyg. 1987, 81, 755–759. [Google Scholar] [CrossRef]
- Caldas, S.; Santos, F.M.; Lana, M.D.; Diniz, L.F.; Machado-Coelho, G.L.L.; Veloso, V.M.; Bahia, M.T. Trypanosoma cruzi: Acute and long-term infection in the vertebrate host can modify the response to benznidazole. Exp. Parasitol. 2008, 118, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Brener, Z. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev. Inst. Med. Trop. Sao Paulo 1962, 4, 389–396. [Google Scholar]
- Cummings, K.L.; Tarleton, R.L. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Mol. Biochem. Parasitol. 2003, 129, 53–59. [Google Scholar] [CrossRef]
- Perin, L.; Moreira da Silva, R.; Fonseca, K.D.S.; Cardoso, J.M.D.O.; Mathias, F.A.S.; Reis, L.E.S.; Molina, I.; Correa-Oliveira, R.; Vieira, P.M.D.A.; Carneiro, C.M. Pharmacokinetics and Tissue Distribution of Benznidazole after Oral Administration in Mice. Antimicrob. Agents Chemother. 2017, 61, e02410-16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Spósito, P.Á.; Mazzeti, A.L.; de Castro, K.C.M.P.; Mendes, P.F.; Urbina, J.A.; Bahia, M.T.; Mosqueira, V.C.F. Higher oral efficacy of ravuconazole in self-nanoemulsifying systems in shorter treatment in experimental chagas disease. Exp. Parasitol. 2021, 228, 108142. [Google Scholar] [CrossRef]
- Mazzeti, A.L.; Diniz, L.D.F.; Gonçalves, K.R.; Nascimento, A.F.S.; Spósito, P.A.F.; Mosqueira, V.C.F.; Machado-Coelho, G.L.L.; Ribeiro, I.; Bahia, M.T. Time and dose-dependence evaluation of nitroheterocyclic drugs for improving efficacy following Trypanosoma cruzi infection: A pre-clinical study. Biochem. Pharmacol. 2018, 148, 213–221. [Google Scholar] [CrossRef]
- Honorato, S.B.; Mendonça, J.S.; Boechat, N.; Oliveira, A.C.; Mendes Filho, J.; Ellena, J.; Ayala, A.P. Novel polymorphs of the anti-Trypanosoma cruzi drug benznidazole. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2014, 118, 389–394. [Google Scholar] [CrossRef]
- Leonardi, D.; Bombardiere, M.E.; Salomon, C.J. Effects of benznidazole:cyclodextrin complexes on the drug bioavailability upon oral administration to rats. Int. J. Biol. Macromol. 2013, 62, 543–548. [Google Scholar] [CrossRef]
- Soares-Sobrinho, J.L.; de La Roca Soares, M.F.; Lopes, P.Q.; Correia, L.P.; de Souza, F.S.; Macêdo, R.O.; Rolim-Neto, P.J. A Preformulation Study of a New Medicine for Chagas Disease Treatment: Physicochemical Characterization, Thermal Stability, and Compatibility of Benznidazole. AAPS PharmSciTech 2010, 11, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdellah Ali, S.F. Mechanical and thermal properties of promising polymer composites for food packaging applications. IOP Conf. Ser. Mater. Sci. Eng. 2016, 137, 012035. [Google Scholar] [CrossRef] [Green Version]
- van Natta, F.J.; Hill, J.W.; Carothers, W.H. Studies of Polymerization and Ring Formation. XXIII. 1 ε-Caprolactone and its Polymers. J. Am. Chem. Soc. 1934, 56, 455–457. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Molina, I.; Perin, L.; Aviles, A.S.; de Abreu Vieira, P.M.; da Silva Fonseca, K.; Cunha, L.M.; Carneiro, C.M. The effect of benznidazole dose among the efficacy outcome in the murine animal model. A quantitative integration of the literature. Acta Trop. 2020, 201, 105218. [Google Scholar] [CrossRef] [PubMed]
- Novaes, R.D.; Santos, E.C.; Cupertino, M.C.; Bastos, D.S.S.; Oliveira, J.M.; Carvalho, T.V.; Neves, M.M.; Oliveira, L.L.; Talvani, A. Trypanosoma cruzi infection and benznidazole therapy independently stimulate oxidative status and structural pathological remodeling of the liver tissue in mice. Parasitol. Res. 2015, 114, 2873–2881. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
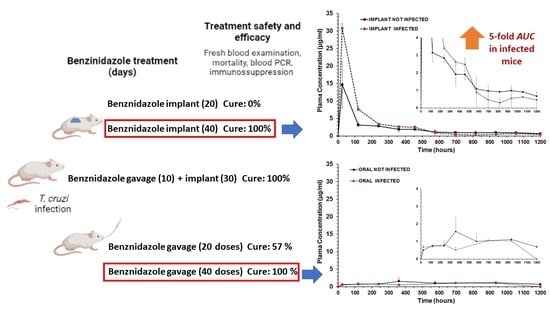
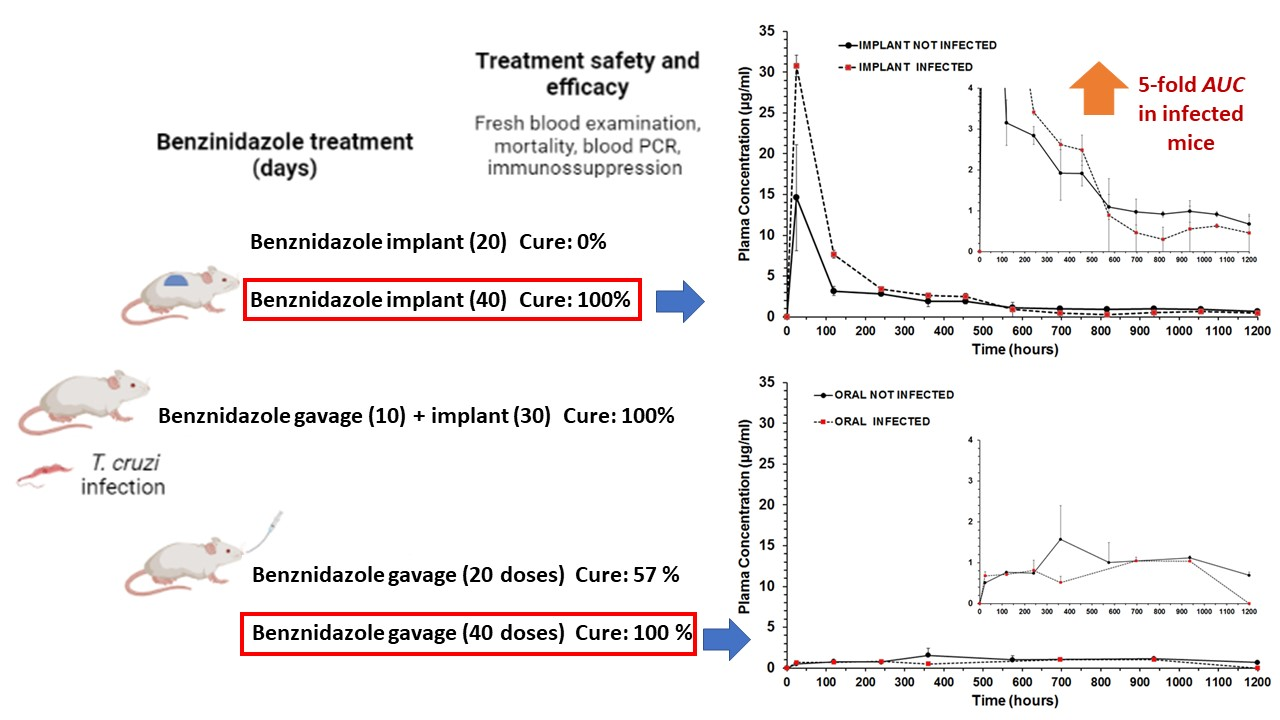
| Treatment Schedule | Benznidazole Oral Dose (Days) | Benznidazole Implant Dose (Days) | Implant PCL:BZ Ratio | Benznidazole Total Dose/Per Mouse 1 |
|---|---|---|---|---|
| blank-PCL implant | 0 mg/kg | 0 mg/kg | 1:0 | 0 mg |
| BZ–PCL 1 | 0 mg/kg | 100 mg/kg (20) | 1:0.66 | 50 mg |
| BZ–PCL 2 $ | 100 mg/kg (10) | 100 mg/kg (30) | 1:1 | 75 mg |
| BZ–PCL 3 | 0 mg/kg | 100 mg/kg (40) | 1:1.33 | 100 mg |
| Oral BZ & | 100 mg/kg (20) | 0 mg/kg | - | 50 mg |
| Oral BZ | 100 mg/kg (40) | 0 mg/kg | - | 100 mg |
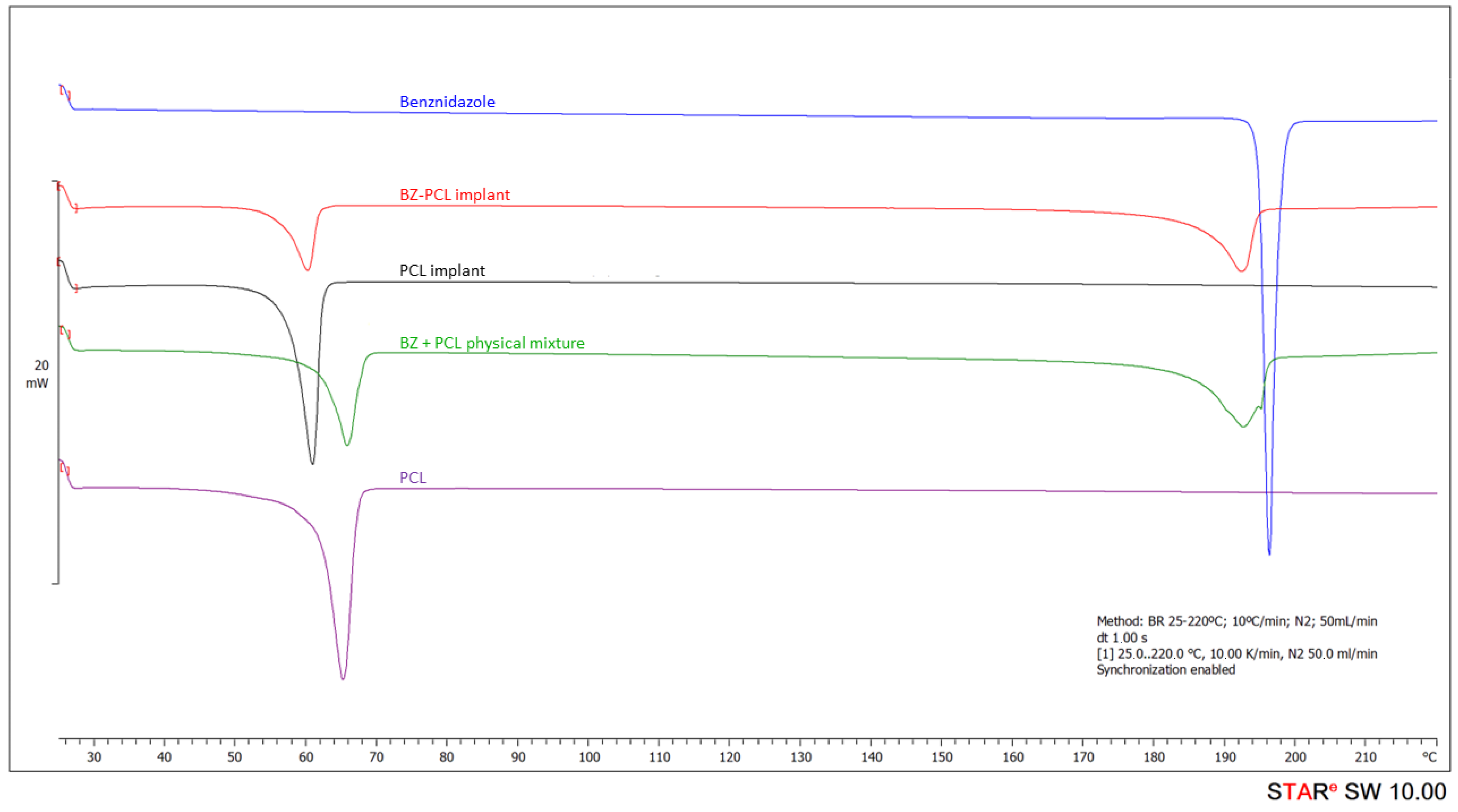
| Sample | DSC—PCL | DSC—Benznidazol | ||||||
|---|---|---|---|---|---|---|---|---|
| Tonset (°C) | Tpeak (°C) | Tendset (°C) | Enthalpy (J/g) | Tonset (°C) | Tpeak (°C) | Tendset (°C) | Enthalpy (J/g) | |
| Benznidazole | -- | -- | -- | -- | 194.54 | 195.19 | 197.62 | −125.62 |
| PCL polymer | 61.59 | 64.83 | 67.18 | −88.30 | -- | -- | -- | -- |
| PCL implant | 57.35 | 60.68 | 62.31 | −92.67 | -- | -- | -- | -- |
| BZ–PCL implant | 56.35 | 60.19 | 61.80 | −36.09 | 186.69 | 192.27 | 194.61 | −59.12 |
| Physical mixture BZ + PCL | 62.11 | 65.65 | 68.08 | −41.77 | 185.34 | 192.42 | 196.09 | −59.17 |
| Treatment Schedule/Groups | Nº of Daily Doses or Doses in Implants (mg/kg) | Parasitemia Suppression by FBE (Days) | Parasitemia Reactivation (Days) by FBE 2 | Negative Total Tests 3 |
|---|---|---|---|---|
| Uninfected | 0 | -/7 | - | -/7 (100%) |
| Infected and untreated | 0 | 0/7 | - | 0/7 (0%) |
| Implant BZ–PCL (20) | 20 × 100 mg (implant) | 5/5 (1.20 ± 0.45) | 5/5 (22.8 ± 7.53) | 0/5 (0%) |
| Oral + Implant BZ–PCL | 10 × 100 mg (oral) + 30 × 100 (implant) | 6/6 (1.17± 0.41) | 0/6 (ND) | 6/6 (100%) |
| Implant BZ–PCL3 (40) | 40 × 100 mg (implant) | 8/8 (1 ± 0) | 0/8 (ND) | 8/8 (100%) |
| Oral 100 mg/kg (20) 1,@ | 20 × 100 mg (oral) | 7/7 (1.47 ± 0.77) | 1/7 (27 ± 0) | 4/7 (57.1%) |
| Oral 100 mg/kg (40) 1,#,@ | 40 × 100 mg (oral) | 7/7 (1.47 ± 0.77) | 0/7 (ND) | 7/7 (100%) |
| Parameter | Unit | Treatments and Formulations | |||
|---|---|---|---|---|---|
| Oral Not Infected | Oral Infected | Subcutaneous Implant Not Infected | Subcutaneous Implant Infected | ||
| Cmax | µg/mL | 1.5718 | 1.0444 | 14.6115 | 30.7782 |
| Clast obs | µg/mL | 0.6886 | 1.0374 | 0.6712 | 0.4558 |
| Clast pred | µg/mL | 0.7481 | nd | 0.6169 | 0.3134 |
| Tmax | h | 360 | 696 | 24 | 24 |
| AUC0-t | µg/mL.h | 1185.022 | 755.789 | 2471.565 | 3737.566 |
| MRT0-t | h | 616.99 | 531.76 | 349.74 | 220.69 |
| Cl/F | L/h | 0.0484 | nd | 0.0339 | 0.0255 |
| ke | 1/h | 0.000783 | nd | 0.001405 | 0.002492 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzeti, A.L.; Gonçalves, K.R.; Boasquívis, P.F.; Barbosa, J.; Pereira, B.G.; Soeiro, M.d.N.C.; Mosqueira, V.C.F.; Bahia, M.T. Poly-ε-Caprolactone Implants for Benznidazole Prolonged Release: An Alternative to Chagas Disease Oral Treatment. Pharmaceutics 2023, 15, 1126. https://doi.org/10.3390/pharmaceutics15041126
Mazzeti AL, Gonçalves KR, Boasquívis PF, Barbosa J, Pereira BG, Soeiro MdNC, Mosqueira VCF, Bahia MT. Poly-ε-Caprolactone Implants for Benznidazole Prolonged Release: An Alternative to Chagas Disease Oral Treatment. Pharmaceutics. 2023; 15(4):1126. https://doi.org/10.3390/pharmaceutics15041126
Chicago/Turabian StyleMazzeti, Ana Lia, Karolina R. Gonçalves, Patrícia Ferreira Boasquívis, Jamile Barbosa, Bruno G. Pereira, Maria de Nazaré Correia Soeiro, Vanessa Carla Furtado Mosqueira, and Maria Terezinha Bahia. 2023. "Poly-ε-Caprolactone Implants for Benznidazole Prolonged Release: An Alternative to Chagas Disease Oral Treatment" Pharmaceutics 15, no. 4: 1126. https://doi.org/10.3390/pharmaceutics15041126