
Dendrimers and Derivatives as Multifunctional Nanotherapeutics for Alzheimer’s Disease

Abstract
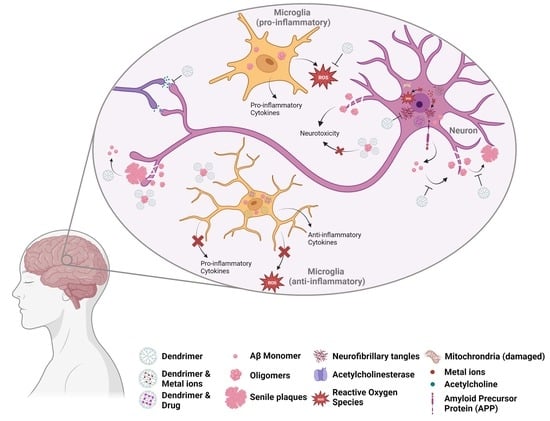
:
1. Introduction
2. Alzheimer’s Disease—Pathogenesis and Therapeutical Routes
3. Dendrimers—A Multivalent and Multifunctional Nanocarrier
4. Dendrimers as Anti-Amyloidogenic Agents
4.1. Dendrimer/Peptide Ratio
4.2. Functional Surface Groups
4.3. Generation
4.4. Topology
5. Dendrimers as Anti-Tau Agents and Inhibitors of Acetylcholinesterase Activity
6. Dendritic Structures as Anti-Inflammatory Agents
6.1. Functional Surface Groups
6.2. Generation/Size and Multivalency
6.3. Internal Structure
7. Dendrimers as Antioxidants and Chelators
8. Dendrimers as Nanocarriers
9. The Other Side of Dendrimers—Caveats and Challenges
10. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- National Insititute on Aging. What Is Dementia? Symptoms, Types, and Diagnosis. Available online: https://www.nia.nih.gov/health/what-dementia-symptoms-types-and-diagnosis (accessed on 16 April 2021).
- World Health Organization. Global Status Report on the Public Health Response to Dementia; WHO: Geneva, Switzerland, 2021; ISBN 9789240033245.
- World Health Organization. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 13 April 2021).
- National Insititute on Aging. What Is Alzheimer’s Disease? Available online: https://www.nia.nih.gov/health/what-alzheimers-disease (accessed on 17 April 2021).
- Patterson, C. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- U.S. Food & Drug Administration. FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 19 June 2021).
- U.S. Food & Drug Administration. FDA Grants Accelerated Approval for Alzheimer’s Drug|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 11 January 2023).
- Alexander, G.C.; Emerson, S.; Kesselheim, A.S. Evaluation of Aducanumab for Alzheimer Disease: Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA—J. Am. Med. Assoc. 2021, 325, 1717–1718. [Google Scholar] [CrossRef]
- Cummings, J. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin. Transl. Sci. 2018, 11, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s Disease Drug Development Pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Gu, X.; Gao, X. Nanotherapeutic Strategies for the Treatment of Neurodegenerative Diseases; Elsevier Ltd.: Amsterdam, The Netherlands, 2019; ISBN 9780128140017. [Google Scholar]
- Fonseca-Santos, B.; Gremião, M.P.D.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for the Treatment of Alzheimer’s Disease. Int. J. Nanomed. 2015, 10, 4981–5003. [Google Scholar] [CrossRef] [Green Version]
- Leiro, V.; Duque Santos, S.; Lopes, C.D.F.; Paula Pêgo, A. Dendrimers as Powerful Building Blocks in Central Nervous System Disease: Headed for Successful Nanomedicine. Adv. Funct. Mater. 2018, 28, 1700313. [Google Scholar] [CrossRef] [Green Version]
- Janaszewska, A.; Lazniewska, J.; Trzepiński, P.; Marcinkowska, M.; Klajnert-Maculewicz, B. Cytotoxicity of Dendrimers. Biomolecules 2019, 9, 330. [Google Scholar] [CrossRef] [Green Version]
- Leiro, V.; Garcia, J.P.; Tomás, H.; Pêgo, A.P. The Present and the Future of Degradable Dendrimers and Derivatives in Theranostics. Bioconjug. Chem. 2015, 26, 1185–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignani, S.; Bryszewska, M.; Zablocka, M.; Klajnert-Maculewicz, B.; Cladera, J.; Shcharbin, D.; Majoral, J.P. Can Dendrimer Based Nanoparticles Fight Neurodegenerative Diseases? Current Situation versus Other Established Approaches. Prog. Polym. Sci. 2017, 64, 23–51. [Google Scholar] [CrossRef]
- Gajbhiye, V.; Palanirajan, V.K.; Tekade, R.K.; Jain, N.K. Dendrimers as Therapeutic Agents: A Systematic Review. J. Pharm. Pharmacol. 2009, 61, 989–1003. [Google Scholar] [CrossRef]
- Klajnert, B.; Cortijo-Arellano, M.; Cladera, J.; Bryszewska, M. Influence of Dendrimer’s Structure on Its Activity against Amyloid Fibril Formation. Biochem. Biophys. Res. Commun. 2006, 345, 21–28. [Google Scholar] [CrossRef]
- Klementieva, O.; Benseny-Cases, N.; Gella, A.; Appelhans, D.; Voit, B.; Cladera, J. Dense Shell Glycodendrimers as Potential Nontoxic Anti-Amyloidogenic Agents in Alzheimer’s Disease. Amyloid-Dendrimer Aggregates Morphology and Cell Toxicity. Biomacromolecules 2011, 12, 3903–3909. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, T.; Ionov, M.; Nieznanski, K.; Nieznanska, H.; Klementieva, O.; Granell, M.; Cladera, J.; Majoral, J.P.; Caminade, A.M.; Klajnert, B. Phosphorus Dendrimers Affect Alzheimer’s (Aβ1–28) Peptide and MAP-Tau Protein Aggregation. Mol. Pharm. 2012, 9, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Klajnert, B.; Cladera, J.; Bryszewska, M. Molecular Interactions of Dendrimers with Amyloid Peptides: PH Dependence. Biomacromolecules 2006, 7, 2186–2191. [Google Scholar] [CrossRef]
- Shcharbin, D.; Jokiel, M.; Klajnert, B.; Bryszewska, M. Effect of Dendrimers on Pure Acetylcholinesterase Activity and Structure. Bioelectrochemistry 2006, 68, 56–59. [Google Scholar] [CrossRef]
- Wasiak, T.; Marcinkowska, M.; Pieszynski, I.; Zablocka, M.; Caminade, A.M.; Majoral, J.P.; Klajnert-Maculewicz, B. Cationic Phosphorus Dendrimers and Therapy for Alzheimer’s Disease. New J. Chem. 2015, 39, 4852–4859. [Google Scholar] [CrossRef]
- Klajnert, B.; Sadowska, M.; Bryszewska, M. The Effect of Polyamidoamine Dendrimers on Human Erythrocyte Membrane Acetylcholinesterase Activity. Bioelectrochemistry 2004, 65, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Martinsson, I.; Appelhans, D.; Effenberg, C.; Benseny-Cases, N.; Cladera, J.; Gouras, G.; Ferrer, I.; Klementieva, O. Poly(Propylene Imine) Dendrimers with Histidine-Maltose Shell as Novel Type of Nanoparticles for Synapse and Memory Protection. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 198–209. [Google Scholar] [CrossRef]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s Disease: Strategies for Disease Modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Parihar, M.S.; Hemnani, T. Alzheimer’s Disease Pathogenesis and Therapeutic Interventions. J. Clin. Neurosci. 2004, 11, 456–467. [Google Scholar] [CrossRef]
- Wen, M.M.; El-Salamouni, N.S.; El-Refaie, W.M.; Hazzah, H.A.; Ali, M.M.; Tosi, G.; Farid, R.M.; Blanco-Prieto, M.J.; Billa, N.; Hanafy, A.S. Nanotechnology-Based Drug Delivery Systems for Alzheimer’s Disease Management: Technical, Industrial, and Clinical Challenges. J. Control. Release 2017, 245, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Gupta, S.; Knight, A.G.; Beckett, T.L.; McMullen, J.M.; Davis, P.R.; Murphy, M.P.; Van Eldik, L.J.; St Clair, D.; Keller, J.N. Cognitive Impairment in Humanized APP × PS1 Mice Is Linked to Aβ1–42 and NOX Activation. Neurobiol. Dis. 2011, 44, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Barage, S.H.; Sonawane, K.D. Amyloid Cascade Hypothesis: Pathogenesis and Therapeutic Strategies in Alzheimer’s Disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Almeida, Z.L.; Brito, R.M.M. Structure and Aggregation Mechanisms in Amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef] [Green Version]
- Kulikova, A.A.; Makarov, A.A.; Kozin, S.A. Roles of Zinc Ions and Structural Polymorphism of β-Amyloid in the Development of Alzheimer’s Disease. Mol. Biol. 2015, 49, 217–230. [Google Scholar] [CrossRef]
- Capetillo-Zarate, E.; Gracia, L.; Yu, F.; Banfelder, J.R.; Lin, M.T.; Tampellini, D.; Gouras, G.K. High-Resolution 3D Reconstruction Reveals Intra-Synaptic Amyloid Fibrils. Am. J. Pathol. 2011, 179, 2551–2558. [Google Scholar] [CrossRef]
- Lorenzo, A.; Yankner, B.A. Amyloid Fibril Toxicity in Alzheimer’s Disease and Diabetes. Ann. N. Y. Acad. Sci. 1996, 777, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. Amyloid Fibrils Induce Dysfunction of Hippocampal Glutamatergic Silent Synapses. Hippocampus 2018, 28, 549–556. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severity of Alzheimer’s Disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and Neuron Numbers, but Not Amyloid Load, Predict Cognitive Status in Alzheimer’s Disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Verma, M.; Vats, A.; Taneja, V. Toxic Species in Amyloid Disorders: Oligomers or Mature Fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138. [Google Scholar]
- Picone, P.; Nuzzo, D.; Giacomazza, D.; Di Carlo, M. β-Amyloid Peptide: The Cell Compartment Multi-Faceted Interaction in Alzheimer’s Disease. Neurotox. Res. 2020, 37, 250–263. [Google Scholar] [CrossRef]
- Vestergaard, M.; Kerman, K. Analytical Tools for Detecting Amyloid Beta Oligomerisation and Assembly. Curr. Pharm. Anal. 2009, 5, 229–245. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The Amyloid β-Peptide Is Imported into Mitochondria via the TOM Import Machinery and Localized to Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M.D. Mitochondrial Dysfunction: Different Routes to Alzheimer’s Disease Therapy. Oxid. Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seiça, R.M.; Moreira, P.I. Alzheimer’s Disease: From Mitochondrial Perturbations to Mitochondrial Medicine. Brain Pathol. 2016, 26, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M.F. The Release of Calcium from the Endoplasmic Reticulum Induced by Amyloid-Beta and Prion Peptides Activates the Mitochondrial Apoptotic Pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.O.; Ferreiro, E.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M.F. ER Stress-Mediated Apoptotic Pathway Induced by Aβ Peptide Requires the Presence of Functional Mitochondria. J. Alzheimer’s Dis. 2010, 20, 625–636. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Camero, S.; Benítez, M.J.; Cuadros, R.; Hernández, F.; Ávila, J.; Jiménez, J.S. Thermodynamics of the Interaction between Alzheimer’s Disease Related Tau Protein and DNA. PLoS ONE 2014, 9, e104690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barucker, C.; Harmeier, A.; Weiske, J.; Fauler, B.; Albring, K.F.; Prokop, S.; Hildebrand, P.; Lurz, R.; Heppner, F.L.; Huber, O.; et al. Nuclear Translocation Uncovers the Amyloid Peptide Aβ42 as a Regulator of Gene Transcription. J. Biol. Chem. 2014, 289, 20182–20191. [Google Scholar] [CrossRef] [Green Version]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A Randomized, Double-Blind, Phase 2b Proof-of-Concept Clinical Trial in Early Alzheimer’s Disease with Lecanemab, an Anti-Aβ Protofibril Antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Haeberlein, S.B.; von Hehn, C.; Tian, Y.; Chalkias, S.; Muralidharan, K.K.; Chen, T.; Wu, S.; Li, J.; Skordos, L.; Nisenbaum, L.; et al. EMERGE and ENGAGE Topline Results: Two Phase 3 Studies to Evaluate Aducanumab in Patients With Early Alzheimer’s Disease. Clin. Trials Alzheimer’s Dis. 2019, 5, 1–60. [Google Scholar]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The Murine Version of BAN2401 (MAb158) Selectively Reduces Amyloid-β Protofibrils in Brain and Cerebrospinal Fluid of Tg-ArcSwe Mice. J. Alzheimer’s Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and Kinetic Basis for the Selectivity of Aducanumab for Aggregated Forms of Amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab Produced a Clinically Meaningful Benefit in Association with Amyloid Lowering. Alzheimer’s Res. Ther. 2021, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Yuan, C.; Li, C.; Liu, H.; Wang, X. Multifunctional Nano-Enabled Delivery Systems in Alzheimer’s Disease Management. Biomater. Sci. 2020, 8, 5538–5554. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent Developments in Clinical Biomarkers and Therapies Targeting Tau Phosphorylation in Alzheimer’s Disease and Other Tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Ferreira, E.; Tran, A.; Turner, E.C.; Belfiore, R.; Branca, C.; Oddo, S. Acute Tau Knockdown in the Hippocampus of Adult Mice Causes Learning and Memory Deficits. Aging Cell 2018, 17, e12775. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a Pivotal Kinase in Alzheimer Disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed] [Green Version]
- Kimura, T.; Ishiguro, K.; Hisanaga, S.I. Physiological and Pathological Phosphorylation of Tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, K.; Omori, A.; Ohtake, A.; Sato, K.; Takahashi, M. Tau-Tubulin Kinase Phosphorylates Tau at Ser-208 and Ser-210, Sites Found in Paired Helical Filament-Tau. FEBS Lett. 2001, 492, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Matenia, D.; Mandelkow, E.M. The Tau of MARK: A Polarized View of the Cytoskeleton. Trends Biochem. Sci. 2009, 34, 332–342. [Google Scholar] [CrossRef]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of Tau by Fyn: Implications for Alzheimer’s Disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer’s Disease Abnormally Phosphorylated Tau by Protein Phosphatase-2A. Neuroscience 1994, 61, 765–772. [Google Scholar] [CrossRef]
- Abbott, A.; Dolgin, E. Failed Alzheimer’s Trial Does Not Kill Leading Theory of Disease. Nature 2016, 540, 15–16. [Google Scholar] [CrossRef] [Green Version]
- Vögtle, F.; Buhleier, E.W.; Wehner, W. Cascade and Nonskid-Chain-Like Syntheses of Molecular Cavity Topologies. Synthesis 1978, 2, 155–158. [Google Scholar]
- Heegaard, P.M.H.; Boas, U.; Otzen, D.E. Dendrimer Effects on Peptide and Protein Fibrillation. Macromol. Biosci. 2007, 7, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Hayder, M.; Fruchon, S.; Fournié, J.J.; Poupot, M.; Poupot, R. Anti-Inflammatory Properties of Dendrimers per Se. Sci. World J. 2011, 11, 1367–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shcharbin, D.; Shcharbina, N.; Dzmitruk, V.; Pedziwiatr-Werbicka, E.; Ionov, M.; Mignani, S.; de la Mata, F.J.; Gómez, R.; Muñoz-Fernández, M.A.; Majoral, J.P.; et al. Dendrimer-Protein Interactions versus Dendrimer-Based Nanomedicine. Colloids Surf. B Biointerfaces 2017, 152, 414–422. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Li, T.; Tian, W.; Zhang, Q.; Cheng, Y. Generation 9 Polyamidoamine Dendrimer Encapsulated Platinum Nanoparticle Mimics Catalase Size, Shape, and Catalytic Activity. Langmuir 2013, 29, 5262–5270. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Hall, M.; Kallos, G.; Martin, S.; Ryder, J.; Smith, P. Dendritic Macromolecules: Synthesis of Starburst Dendrimers. Macromolecules 1986, 19, 2466–2468. [Google Scholar] [CrossRef]
- Starpharma Ltd. SPL7013 Products. Available online: https://starpharma.com/spl7013 (accessed on 15 February 2023).
- Mendling, W.; Holzgreve, W. Astodrimer Sodium and Bacterial Vaginosis: A Mini Review. Arch. Gynecol. Obstet. 2022, 306, 101–108. [Google Scholar] [CrossRef]
- Caminade, A.M. Dendrimers, an Emerging Opportunity in Personalized Medicine? J. Pers. Med. 2022, 12, 1334. [Google Scholar] [CrossRef]
- Starpharma Ltd. VIRALEZE—How it works. Available online: https://viraleze.co/how-it-works/ (accessed on 15 February 2023).
- Paull, J.R.A.; Heery, G.P.; Bobardt, M.D.; Castellarnau, A.; Luscombe, C.A.; Fairley, J.K.; Gallay, P.A. Virucidal and Antiviral Activity of Astodrimer Sodium against SARS-CoV-2 in Vitro. Antivir. Res. 2021, 191, 105089. [Google Scholar] [CrossRef]
- Paull, J.R.A.; Luscombe, C.A.; Castellarnau, A.; Heery, G.P.; Bobardt, M.D.; Gallay, P.A. Protective Effects of Astodrimer Sodium 1% Nasal Spray Formulation against Sars-Cov-2 Nasal Challenge in K18-Hace2 Mice. Viruses 2021, 13, 1656. [Google Scholar] [CrossRef]
- Castellarnau, A.; Heery, G.P.; Seta, A.; Luscombe, C.A.; Kinghorn, G.R.; Button, P.; McCloud, P.; Paull, J.R.A. Astodrimer Sodium Antiviral Nasal Spray for Reducing Respiratory Infections Is Safe and Well Tolerated in a Randomized Controlled Trial. Sci. Rep. 2022, 12, 10210. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Navath, R.S.; Balakrishnan, B.; Guru, B.R.; Mishra, M.K.; Romero, R.; Kannan, R.M.; Kannan, S. Intrinsic Targeting of Inflammatory Cells in the Brain by Polyamidoamine Dendrimers upon Subarachnoid Administration. Nanomedicine 2010, 5, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Kannan, S.; Dai, H.; Navath, R.S.; Balakrishnan, B.; Jyoti, A.; Janisse, J.; Romero, R.; Kannan, R.M. Dendrimer-Based Postnatal Therapy for Neuroinflammation and Cerebral Palsy in a Rabbit Mode. Sci. Transl. Med. 2012, 4, 130ra46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambhampati, S.P.; Clunies-Ross, A.J.M.; Bhutto, I.; Mishra, M.K.; Edwards, M.; McLeod, D.S.; Kannan, R.M.; Lutty, G. Systemic and Intravitreal Delivery of Dendrimers to Activated Microglia/Macrophage in Ischemia/Reperfusion Mouse Retina. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4413–4424. [Google Scholar] [CrossRef] [Green Version]
- Liaw, K.; Zhang, F.; Mangraviti, A.; Kannan, S.; Tyler, B.; Kannan, R.M. Dendrimer Size Effects on the Selective Brain Tumor Targeting in Orthotopic Tumor Models upon Systemic Administration. Bioeng. Transl. Med. 2020, 5, e10160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therapeutics, A. Ashvattha Therapeutics—Developing a New Class of Intracellular Targeted Therapeutics. Available online: https://avttx.com/ (accessed on 26 February 2023).
- Gusdon, A.M.; Faraday, N.; Aita, J.S.; Kumar, S.; Mehta, I.; Choi, H.M.A.; Cleland, J.L.; Robinson, K.; McCullough, L.D.; Ng, D.K.; et al. Dendrimer Nanotherapy for Severe COVID-19 Attenuates Inflammation and Neurological Injury Markers and Improves Outcomes in a Phase2a Clinical Trial. Sci. Transl. Med. 2022, 14, eabo2652. [Google Scholar] [CrossRef]
- Khaitov, M.; Nikonova, A.; Shilovskiy, I.; Kozhikhova, K.; Kofiadi, I.; Vishnyakova, L.; Nikolskii, A.; Gattinger, P.; Kovchina, V.; Barvinskaia, E.; et al. Silencing of SARS-CoV-2 with Modified SiRNA-Peptide Dendrimer Formulation. Allergy Eur. J. Allergy Clin. Immunol. 2021, 76, 2840–2854. [Google Scholar] [CrossRef]
- Therapeutics, A. D-4517.2. Available online: https://avttx.com/pipeline/ophthalmology/ (accessed on 28 February 2023).
- Therapeutics, A. OP-801. Available online: https://avttx.com/pipeline/neurology/ (accessed on 28 February 2023).
- Patterson, C.M.; Balachander, S.B.; Grant, I.; Pop-Damkov, P.; Kelly, B.; McCoull, W.; Parker, J.; Giannis, M.; Hill, K.J.; Gibbons, F.D.; et al. Design and Optimisation of Dendrimer-Conjugated Bcl-2/XL Inhibitor, AZD0466, with Improved Therapeutic Index for Cancer Therapy. Commun. Biol. 2021, 4, 4–6. [Google Scholar] [CrossRef]
- Feeney, O.M.; Ardipradja, K.; Noi, K.F.; Mehta, D.; De Rose, R.; Yuen, D.; Johnston, A.P.R.; Kingston, L.; Ericsson, C.; Elmore, C.S.; et al. Subcutaneous Delivery of a Dendrimer-BH3 Mimetic Improves Lymphatic Uptake and Survival in Lymphoma. J. Control. Release 2022, 348, 420–430. [Google Scholar] [CrossRef]
- Starpharma Ltd. DEP® Docetaxel (Phase 2). Available online: https://starpharma.com/drug_delivery/dep_docetaxel (accessed on 28 February 2023).
- Starpharma Ltd. DEP® Cabazitaxel (Phase 2). Available online: https://starpharma.com/drug_delivery/dep_cabazitaxel (accessed on 28 February 2023).
- Starpharma Ltd. DEP® Irinotecan (Phase 2). Available online: https://starpharma.com/drug_delivery/dep_irinotecan (accessed on 28 February 2023).
- Arulananda, S.; O’Brien, M.; Evangelista, M.; Jenkins, L.J.; Poh, A.R.; Walkiewicz, M.; Leong, T.; Mariadason, J.M.; Cebon, J.; Balachander, S.B.; et al. A Novel BH3-Mimetic, AZD0466, Targeting BCL-XL and BCL-2 Is Effective in Pre-Clinical Models of Malignant Pleural Mesothelioma. Cell Death Discov. 2021, 7, 122. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study of AZD0466 in Patients with Advanced Hematologic or Solid Tumors. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04214093 (accessed on 28 February 2023).
- ClinicalTrials.gov. Study of AZD0466 Monotherapy or in Combination in Patients with Advanced Haematological Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT04865419 (accessed on 15 March 2023).
- ClinicalTrials.gov. A Phase I/II Study of AZD0466 as Monotherapy or in Combination with Anticancer Agents in Advanced Non-Hodgkin Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT05205161 (accessed on 15 March 2023).
- Caminade, A.M.; Yan, D.; Smith, D.K. Dendrimers and Hyperbranched Polymers. Chem. Soc. Rev. 2015, 44, 3870–3873. [Google Scholar] [CrossRef]
- Rades, N.; Licha, K.; Haag, R. Dendritic Polyglycerol Sulfate for Therapy and Diagnostics. Polymers 2018, 10, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supattapone, S.; Nguyen, H.O.B.; Cohen, F.E.; Prusiner, S.B.; Scott, M.R. Elimination of Prions by Branched Polyamines and Implications for Therapeutics. Proc. Natl. Acad. Sci. USA 1999, 96, 14529–14534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfoud, R.; Garmy, N.; Maresca, M.; Yahi, N.; Puigserver, A.; Fantini, J. Identification of a Common Sphingolipid-Binding Domain in Alzheimer, Prion, and HIV-1 Proteins. J. Biol. Chem. 2002, 277, 11292–11296. [Google Scholar] [CrossRef] [Green Version]
- Klajnert, B.; Cortijo-Arellano, M.; Bryszewska, M.; Cladera, J. Influence of Heparin and Dendrimers on the Aggregation of Two Amyloid Peptides Related to Alzheimer’s and Prion Diseases. Biochem. Biophys. Res. Commun. 2006, 339, 577–582. [Google Scholar] [CrossRef]
- Milowska, K.; Malachowska, M.; Gabryelak, T. PAMAM G4 Dendrimers Affect the Aggregation of α-Synuclein. Int. J. Biol. Macromol. 2011, 48, 742–746. [Google Scholar] [CrossRef]
- Rekas, A.; Lo, V.; Gadd, G.E.; Cappai, R.; Yun, S.I. PAMAM Dendrimers as Potentia] Agents against Fibrillation of α-Synuclein, a Parkinson’s Disease Related Protein. Macromol. Biosci. 2009, 9, 230–238. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, X.; Sun, Y. Hydrophobic Modification of Carboxyl-Terminated Polyamidoamine Dendrimer Surface Creates a Potent Inhibitor of Amyloid-β Fibrillation. Langmuir 2018, 34, 14419–14427. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Appelhans, D.; Schwarz, S.; Klajnert, B.; Bryszewska, M.; Voit, B.; Rogers, M. Influence of Surface Functionality of Poly(Propylene Imine) Dendrimers on Protease Resistance and Propagation of the Scrapie Prion Protein. Biomacromolecules 2010, 11, 1314–1325. [Google Scholar] [CrossRef] [Green Version]
- Klajnert, B.; Cortijo-Arellano, M.; Cladera, J.; Majoral, J.P.; Caminade, A.M.; Bryszewska, M. Influence of Phosphorus Dendrimers on the Aggregation of the Prion Peptide PrP 185–208. Biochem. Biophys. Res. Commun. 2007, 364, 20–25. [Google Scholar] [CrossRef]
- Klajnert, B.; Wasiak, T.; Ionov, M.; Fernandez-Villamarin, M.; Sousa-Herves, A.; Correa, J.; Riguera, R.; Fernandez-Megia, E. Dendrimers Reduce Toxicity of Aβ1–28 Peptide during Aggregation and Accelerate Fibril Formation. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 1372–1378. [Google Scholar] [CrossRef]
- Araújo, A.R.; Correa, J.; Dominguez-Arca, V.; Reis, R.L.; Fernandez-Megia, E.; Pires, R.A. Functional Gallic Acid-Based Dendrimers as Synthetic Nanotools to Remodel Amyloid-β-42 into Noncytotoxic Forms. ACS Appl. Mater. Interfaces 2021, 13, 59673–59682. [Google Scholar] [CrossRef]
- Milowska, K.; Szwed, A.; Mutrynowska, M.; Gomez-Ramirez, R.; De La Mata, F.J.; Gabryelak, T.; Bryszewska, M. Carbosilane Dendrimers Inhibit α-Synuclein Fibrillation and Prevent Cells from Rotenone-Induced Damage. Int. J. Pharm. 2015, 484, 268–275. [Google Scholar] [CrossRef]
- Ferrer-Lorente, R.; Lozano-Cruz, T.; Fernández-Carasa, I.; Miłowska, K.; De La Mata, F.J.; Bryszewska, M.; Consiglio, A.; Ortega, P.; Gómez, R.; Raya, A. Cationic Carbosilane Dendrimers Prevent Abnormal α-Synuclein Accumulation in Parkinson’s Disease Patient-Specific Dopamine Neurons. Biomacromolecules 2021, 22, 4582–4591. [Google Scholar] [CrossRef]
- Milowska, K.; Szwed, A.; Zablocka, M.; Caminade, A.M.; Majoral, J.P.; Mignani, S.; Gabryelak, T.; Bryszewska, M. In Vitro PAMAM, Phosphorus and Viologen-Phosphorus Dendrimers Prevent Rotenone-Induced Cell Damage. Int. J. Pharm. 2014, 474, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Milowska, K.; Grochowina, J.; Katir, N.; El Kadib, A.; Majoral, J.P.; Bryszewska, M.; Gabryelak, T. Viologen-Phosphorus Dendrimers Inhibit α-Synuclein Fibrillation. Mol. Pharm. 2013, 10, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Lazniewska, J.; Milowska, K.; Katir, N.; El Kadib, A.; Bryszewska, M.; Majoral, J.P.; Gabryelak, T. Viologen-Phosphorus Dendrimers Exhibit Minor Toxicity against a Murine Neuroblastoma Cell Line. Cell. Mol. Biol. Lett. 2013, 18, 459–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milowska, K.; Grochowina, J.; Katir, N.; El Kadib, A.; Majoral, J.P.; Bryszewska, M.; Gabryelak, T. Interaction between Viologen-Phosphorus Dendrimers and α-Synuclein. J. Lumin. 2013, 134, 132–137. [Google Scholar] [CrossRef]
- Klementieva, O.; Aso, E.; Filippini, D.; Benseny-Cases, N.; Carmona, M.; Juvés, S.; Appelhans, D.; Cladera, J.; Ferrer, I. Effect of Poly(Propylene Imine) Glycodendrimers on β-Amyloid Aggregation in Vitro and in APP/PS1 Transgenic Mice, as a Model of Brain Amyloid Deposition and Alzheimer’s Disease. Biomacromolecules 2013, 14, 3570–3580. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, M.F.; Cangiotti, M.; Fiorani, L.; Fattori, A.; Wasiak, T.; Appelhans, D.; Klajnert, B. Kinetics of Amyloid and Prion Fibril Formation in the Absence and Presence of Dense Shell Sugar-Decorated Dendrimers. Curr. Med. Chem. 2012, 19, 5907–5921. [Google Scholar] [CrossRef]
- Janaszewska, A.; Klajnert-Maculewicz, B.; Marcinkowska, M.; Duchnowicz, P.; Appelhans, D.; Grasso, G.; Deriu, M.A.; Danani, A.; Cangiotti, M.; Ottaviani, M.F. Multivalent Interacting Glycodendrimer to Prevent Amyloid-Peptide Fibril Formation Induced by Cu(II): A Multidisciplinary Approach. Nano Res. 2018, 11, 1204–1226. [Google Scholar] [CrossRef]
- Klajnert, B.; Appelhans, D.; Komber, H.; Morgner, N.; Schwarz, S.; Richter, S.; Brutschy, B.; Ionov, M.; Tonkikh, A.K.; Bryszewska, M.; et al. The Influence of Densely Organized Maltose Shells on the Biological Properties of Poly(Propylene Imine) Dendrimers: New Effects Dependent on Hydrogen Bonding. Chem.—A Eur. J.’ 2008, 14, 7030–7041. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.M.; Rasines Moreno, B.; Filippini, D.; Komber, H.; Maly, M.; Cernescu, M.; Brutschy, B.; Appelhans, D.; Rogers, M.S. Influence of Surface Groups on Poly(Propylene Imine) Dendrimers Antiprion Activity. Biomacromolecules 2013, 14, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Benseny-Cases, N.; Álvarez-Marimon, E.; Aso, E.; Carmona, M.; Klementieva, O.; Appelhans, D.; Ferrer, I.; Cladera, J. In Situ Identification and G4-PPI-His-Mal-Dendrimer-Induced Reduction of Early-Stage Amyloid Aggregates in Alzheimer’s Disease Transgenic Mice Using Synchrotron-Based Infrared Imaging. Sci. Rep. 2021, 11, 18368. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Henry, J.; Good, T. Attenuation of β-Amyloid Induced Toxicity by Sialic Acid-Conjugated Dendrimeric Polymers. Biochim. Biophys. Acta—Gen. Subj. 2006, 1760, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Neelov, I.M.; Janaszewska, A.; Klajnert, B.; Bryszewska, M.; Makova, N.Z.; Hicks, D.; Pearson, H.A.; Vlasov, G.P.; Ilyash, M.Y.; Vasilev, D.S.; et al. Molecular Properties of Lysine Dendrimers and Their Interactions with Aβ-Peptides and Neuronal Cells. Curr. Med. Chem. 2012, 20, 134–143. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, X.; Sun, Y. Mixed Carboxyl and Hydrophobic Dendrimer Surface Inhibits Amyloid-β Fibrillation: New Insight from the Generation Number Effect. Langmuir 2019, 35, 14681–14687. [Google Scholar] [CrossRef]
- Xiang, S.; Wagner, J.; Lückerath, T.; Müllen, K.; Ng, D.Y.W.; Hedrich, J.; Weil, T. Reversing Aβ Fibrillation and Inhibiting Aβ Primary Neuronal Cell Toxicity Using Amphiphilic Polyphenylene Dendrons. Adv. Healthc. Mater. 2022, 11, 2101854. [Google Scholar] [CrossRef]
- Chafekar, S.M.; Malda, H.; Merkx, M.; Meijer, E.W.; Viertl, D.; Lashuel, H.A.; Baas, F.; Scheper, W. Branched KLVFF Tetramers Strongly Potentiate Inhibition of β-Amyloid Aggregation. ChemBioChem 2007, 8, 1857–1864. [Google Scholar] [CrossRef] [Green Version]
- Laumann, K.; Boas, U.; Larsen, H.M.; Heegaard, P.M.H.; Bergström, A.L. Urea and Thiourea Modified Polypropyleneimine Dendrimers Clear Intracellular α-Synuclein Aggregates in a Human Cell Line. Biomacromolecules 2015, 16, 116–124. [Google Scholar] [CrossRef]
- Yoshiike, Y.; Akagi, T.; Takashima, A. Surface Structure of Amyloid-β Fibrils Contributes to Cytotoxicity. Biochemistry 2007, 46, 9805–9812. [Google Scholar] [CrossRef] [PubMed]
- Maysinger, D.; Ji, J.; Moquin, A.; Hossain, S.; Hancock, M.A.; Zhang, I.; Chang, P.K.Y.; Rigby, M.; Anthonisen, M.; Grütter, P.; et al. Dendritic Polyglycerol Sulfates in the Prevention of Synaptic Loss and Mechanism of Action on Glia. ACS Chem. Neurosci. 2018, 9, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Li, X.; Dong, X.Y.; Sun, Y. Insight into the Inhibition Effect of Acidulated Serum Albumin on Amyloid β-Protein Fibrillogenesis and Cytotoxicity. Langmuir 2014, 30, 9789–9796. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Dong, X.; Wang, Y.; Sun, Y. Multifunctionality of Acidulated Serum Albumin on Inhibiting Zn2+-Mediated Amyloid β-Protein Fibrillogenesis and Cytotoxicity. Langmuir 2015, 31, 7374–7380. [Google Scholar] [CrossRef] [PubMed]
- Benseny-Cases, N.; Klementieva, O.; Cladera, J. Dendrimers Antiamyloidogenic Potential in Neurodegenerative Diseases. New J. Chem. 2012, 36, 211–216. [Google Scholar] [CrossRef]
- Lozano-Cruz, T.; Alcarraz-Vizán, G.; de la Mata, F.J.; de Pablo, S.; Ortega, P.; Duarte, Y.; Bravo-Moraga, F.; González-Nilo, F.D.; Novials, A.; Gómez, R. Cationic Carbosilane Dendritic Systems as Promising Anti-Amyloid Agents in Type 2 Diabetes. Chem.—A Eur. J. 2020, 26, 7609–7621. [Google Scholar] [CrossRef]
- Ott, P. Membrane Acetylcholinesterases: Purification, Molecular Properties and Interactions with Amphiphilic Environments. Biochim. Biophys. Acta—Rev. Biomembr. 1985, 822, 375–392. [Google Scholar] [CrossRef]
- Contestabile, A. The History of the Cholinergic Hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef]
- Piasecka, A.; Leyko, W.; Krajewska, E.; Bryszewska, M. Effect of Combined Treatment with Perindoprilat and Low-Power Red Light Laser Irradiation on Human Erythrocyte Membrane Fluidity, Membrane Potential and Acetylcholinesterase Activity. Scand. J. Clin. Lab. Invest. 2000, 60, 395–402. [Google Scholar] [CrossRef]
- Ciepluch, K.; Weber, M.; Katir, N.; Caminade, A.M.; El Kadib, A.; Klajnert, B.; Majoral, J.P.; Bryszewska, M. Effect of Viologen-Phosphorus Dendrimers on Acetylcholinesterase and Butyrylcholinesterase Activities. Int. J. Biol. Macromol. 2013, 54, 119–124. [Google Scholar] [CrossRef]
- Walters, A.; Phillips, E.; Zheng, R.; Biju, M.; Kuruvilla, T. Evidence for Neuroinflammation in Alzheimer’s Disease. Prog. Neurol. Psychiatry 2016, 20, 25–31. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s Disease Locus: Altered Monocyte Function and Amyloid Biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.S.; Gau, B.A.; Welsh, K.A.; Plassman, B.L.; Mc Donald, W.M.; Helms, M.J.; Anthony, J.C. Inverse Association of Anti-Inflammatory Treatments and Alzheimer’s Disease: Initial Results of a Co-Twin Control Study. Neurology 1994, 44, 227–232. [Google Scholar] [CrossRef]
- Rich, J.B.; Rasmusson, D.X.; Folstein, M.F.; Carson, K.A.; Kawas, C.; Brandt, J. Nonsteroidal Anti-Inflammatory Drugs in Alzheimer’s Disease. Neurology 1995, 45, 51–55. [Google Scholar] [CrossRef]
- Hickman, S.E.; El Khoury, J. TREM2 and the Neuroimmunology of Alzheimer’s Disease. Biochem. Pharmacol. 2014, 88, 495–498. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive Glia Not Only Associates with Plaques but Also Parallels Tangles in Alzheimer’s Disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [Green Version]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Carleton Gajdusek, D. Interleukin 1 Regulates Synthesis of Amyloid β-Protein Precursor MRNA in Human Endothelial Cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.S.; Hwang, E.M.; Sim, H.J.; Cho, H.J.; Boo, J.H.; Oh, S.S.; Kim, S.U.; Mook-Jung, I. Interferon γ Stimulates β-Secretase Expression and SAPPβ Production in Astrocytes. Biochem. Biophys. Res. Commun. 2003, 307, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory Effects of Interferon-β and Interleukin-1β or Tumor Necrosis Factor α on the Synthesis of Aβ1–40 and Aβ1–42 by Human Astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Liaoi, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor Necrosis Factor-α, Interleukin-1β, and Interferon-γ Stimulate γ-Secretase-Mediated Cleavage of Amyloid Precursor Protein through a JNK-Dependent MAPK Pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 Induces Alzheimer-Type Phosphorylation of Tau Protein by Deregulating the Cdk5/P35 Pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2022, 12, 5731. [Google Scholar] [CrossRef]
- Qizilbash, N.; Gregson, J.; Johnson, M.E.; Pearce, N.; Douglas, I.; Wing, K.; Evans, S.J.W.; Pocock, S.J. BMI and Risk of Dementia in Two Million People over Two Decades: A Retrospective Cohort Study. Lancet Diabetes Endocrinol. 2015, 3, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Hassing, L.B.; Dahl, A.K.; Pedersen, N.L.; Johansson, B. Overweight in Midlife Is Related to Lower Cognitive Function 30 Years Later: A Prospective Study with Longitudinal Assessments. Dement. Geriatr. Cogn. Disord. 2010, 29, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Spauwen, P.J.J.; Köhler, S.; Verhey, F.R.J.; Stehouwer, C.D.A.; Van Boxtel, M.P.J. Effects of Type 2 Diabetes on 12-Year Cognitive Change: Results from the Maastricht Aging Study. Diabetes Care 2013, 36, 1554–1561. [Google Scholar] [CrossRef] [Green Version]
- McManus, R.M.; Higgins, S.C.; Mills, K.H.G.; Lynch, M.A. Respiratory Infection Promotes T Cell Infiltration and Amyloid-β Deposition in APP/PS1 Mice. Neurobiol. Aging 2014, 35, 109–121. [Google Scholar] [CrossRef]
- Shaunak, S.; Thomas, S.; Gianasi, E.; Godwin, A.; Jones, E.; Teo, I.; Mireskandari, K.; Luthert, P.; Duncan, R.; Patterson, S.; et al. Polyvalent Dendrimer Glucosamine Conjugates Prevent Scar Tissue Formation. Nat. Biotechnol. 2004, 22, 977–984. [Google Scholar] [CrossRef]
- Chauhan, A.S.; Diwan, P.V.; Jain, N.K.; Tomalia, D.A. Unexpected in Vivo Anti-Inflammatory Activity Observed for Simple, Surface Functionalized Poly(Amidoamine) Dendrimers. Biomacromolecules 2009, 10, 1195–1202. [Google Scholar] [CrossRef]
- Fruchon, S.; Poupot, R. The ABP Dendrimer, a Drug-Candidate against Inflammatory Diseases That Triggers the Activation of Interleukin-10 Producing Immune Cells. Molecules 2018, 23, 1272. [Google Scholar] [CrossRef] [Green Version]
- Caminade, A.M.; Turrin, C.O.; Poupot, R. Curing Inflammatory Diseases Using Phosphorous Dendrimers. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1783. [Google Scholar] [CrossRef]
- Dernedde, J.; Rausch, A.; Weinhart, M.; Enders, S.; Tauber, R.; Licha, K.; Schirner, M.; Zügel, U.; von Bonin, A.; Haag, R. Dendritic Polyglycerol Sulfates as Multivalent Inhibitors of Inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 19679–19684. [Google Scholar] [CrossRef] [Green Version]
- Maysinger, D.; Gröger, D.; Lake, A.; Licha, K.; Weinhart, M.; Chang, P.K.Y.; Mulvey, R.; Haag, R.; McKinney, R.A. Dendritic Polyglycerol Sulfate Inhibits Microglial Activation and Reduces Hippocampal CA1 Dendritic Spine Morphology Deficits. Biomacromolecules 2015, 16, 3073–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruchon, S.; Poupot, M.; Martinet, L.; Turrin, C.-O.; Majoral, J.-P.; Fournié, J.-J.; Caminade, A.-M.; Poupot, R. Anti-Inflammatory and Immunosuppressive Activation of Human Monocytes by a Bioactive Dendrimer. J. Leukoc. Biol. 2009, 85, 553–562. [Google Scholar] [CrossRef]
- Degboé, Y.; Fruchon, S.; Baron, M.; Nigon, D.; Turrin, C.O.; Caminade, A.M.; Poupot, R.; Cantagrel, A.; Davignon, J.L. Modulation of Pro-Inflammatory Activation of Monocytes and Dendritic Cells by Aza-Bis-Phosphonate Dendrimer as an Experimental Therapeutic Agent. Arthritis Res. Ther. 2014, 16, R98. [Google Scholar] [CrossRef] [Green Version]
- Portevin, D.; Poupot, M.; Rolland, O.; Turrin, C.O.; Fournié, J.J.; Majoral, J.P.; Caminade, A.M.; Poupot, R. Regulatory Activity of Azabisphosphonate-Capped Dendrimers on Human CD4+ T Cell Proliferation Enhances Ex-Vivo Expansion of NK Cells from PBMCs for Immunotherapy. J. Transl. Med. 2009, 7, 82. [Google Scholar] [CrossRef]
- Hayder, M.; Poupot, M.; Baron, M.; Turrin, C.-O.; Caminade, A.-M.; Majoral, J.-P.; Eisenberg, R.A.; Fournié, J.-J.; Cantagrel, A.; Poupot, R.; et al. Frequency and Route of Administration in the Treatment of Experimental Arthritis by Phosphorus-Based Dendrimer. Ann. Rheum. Dis. 2012, 71, A8. [Google Scholar] [CrossRef] [Green Version]
- Hayder, M.; Poupot, M.; Baron, M.; Nigon, D.; Turrin, C.O.; Caminade, A.M.; Majoral, J.P.; Eisenberg, R.A.; Fournié, J.J.; Cantagrel, A.; et al. A Phosphorus-Based Dendrimer Targets Inflammation and Osteoclastogenesis in Experimental Arthritis. Sci. Transl. Med. 2011, 3, 81ra35. [Google Scholar] [CrossRef] [PubMed]
- Posadas, I.; Romero-Castillo, L.; El Brahmi, N.; Manzanares, D.; Mignani, S.; Majoral, J.P.; Ceña, V. Neutral High-Generation Phosphorus Dendrimers Inhibit Macrophage-Mediated Inflammatory Response in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E7660–E7669. [Google Scholar] [CrossRef] [Green Version]
- Türk, H.; Haag, R.; Alban, S. Dendritic Polyglycerol Sulfates as New Heparin Analogues and Potent Inhibitors of the Complement System. Bioconjug. Chem. 2004, 15, 162–167. [Google Scholar] [CrossRef]
- Silberreis, K.; Niesler, N.; Rades, N.; Haag, R.; Dernedde, J. Sulfated Dendritic Polyglycerol Is a Potent Complement Inhibitor. Biomacromolecules 2019, 20, 3809–3818. [Google Scholar] [CrossRef] [PubMed]
- Reimann, S.; Gröger, D.; Kühne, C.; Riese, S.B.; Dernedde, J.; Haag, R. Shell Cleavable Dendritic Polyglycerol Sulfates Show High Anti-Inflammatory Properties by Inhibiting L-Selectin Binding and Complement Activation. Adv. Healthc. Mater. 2015, 4, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; Silberreis, K.; Mohammadifar, E.; Neumann, F.; Dernedde, J.; Haag, R. Biodegradable Polyglycerol Sulfates Exhibit Promising Features for Anti-Inflammatory Applications. Biomacromolecules 2018, 19, 4524–4533. [Google Scholar] [CrossRef] [PubMed]
- Nance, E.; Zhang, F.; Mishra, M.K.; Zhang, Z.; Kambhampati, S.P.; Kannan, R.M.; Kannan, S. Nanoscale Effects in Dendrimer-Mediated Targeting of Neuroinflammation. Biomaterials 2016, 101, 96–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaw, K.; Gök, O.; DeRidder, L.B.; Kannan, S.; Kannan, R.M. Quantitative Assessment of Surface Functionality Effects on Microglial Uptake and Retention of PAMAM Dendrimers. J. Nanoparticle Res. 2018, 20, 111. [Google Scholar] [CrossRef]
- Lesniak, W.G.; Mishra, M.K.; Jyoti, A.; Balakrishnan, B.; Zhang, F.; Nance, E.; Romero, R.; Kannan, S.; Kannan, R.M. Biodistribution of Fluorescently Labeled PAMAM Dendrimers in Neonatal Rabbits: Effect of Neuroinflammation. Mol. Pharm. 2013, 10, 4560–4571. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, R.; Zhang, Z.; Liaw, K.; Kambhampati, S.P.; Porterfield, J.E.; Lin, K.C.; DeRidder, L.B.; Kannan, S.; Kannan, R.M. Dense Hydroxyl Polyethylene Glycol Dendrimer Targets Activated Glia in Multiple CNS Disorders. Sci. Adv. 2020, 6, eaay8514. [Google Scholar] [CrossRef] [Green Version]
- Neibert, K.; Gosein, V.; Sharma, A.; Khan, M.; Whitehead, M.A.; Maysinger, D.; Kakkar, A. “Click” Dendrimers as Anti-Inflammatory Agents: With Insights into Their Binding from Molecular Modeling Studies. Mol. Pharm. 2013, 10, 2502–2508. [Google Scholar] [CrossRef] [Green Version]
- Maysinger, D.; Lalancette-Hébert, M.; Ji, J.; Jabbour, K.; Dernedde, J.; Silberreis, K.; Haag, R.; Kriz, J. Dendritic Polyglycerols Are Modulators of Microglia-Astrocyte Crosstalk. Future Neurol. 2019, 14, FNL31. [Google Scholar] [CrossRef] [Green Version]
- Pant, K.; Pufe, J.; Zarschler, K.; Bergmann, R.; Steinbach, J.; Reimann, S.; Haag, R.; Pietzsch, J.; Stephan, H. Surface Charge and Particle Size Determine the Metabolic Fate of Dendritic Polyglycerols. Nanoscale 2017, 9, 8723–8739. [Google Scholar] [CrossRef]
- Poupot, M.; Griffe, L.; Marchand, P.; Maraval, A.; Rolland, O.; Martinet, L.; L’Faqihi-Olive, F.; Turrin, C.; Caminade, A.; Fournié, J.; et al. Design of Phosphorylated Dendritic Architectures to Promote Human Monocyte Activation. FASEB J. 2006, 20, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- Rolland, O.; Griffe, L.; Poupot, M.; Maraval, A.; Ouali, A.; Coppel, Y.; Fournié, J.J.; Bacquet, G.; Turrin, C.O.; Caminade, A.M.; et al. Tailored Control and Optimisation of the Number of Phosphonic Acid Termini on Phosphorus-Containing Dendrimers for the Ex-Vivo Activation of Human Monocytes. Chem.—A Eur. J. 2008, 14, 4836–4850. [Google Scholar] [CrossRef] [PubMed]
- Hayder, M.; Garzoni, M.; Bochicchio, D.; Caminade, A.M.; Couderc, F.; Ong-Meang, V.; Davignon, J.L.; Turrin, C.O.; Pavan, G.M.; Poupot, R. Three-Dimensional Directionality Is a Pivotal Structural Feature for the Bioactivity of Azabisphosphonate-Capped Poly(PhosphorHydrazone) Nanodrug Dendrimers. Biomacromolecules 2018, 19, 712–720. [Google Scholar] [CrossRef]
- Caminade, A.M.; Fruchon, S.; Turrin, C.O.; Poupot, M.; Ouali, A.; Maraval, A.; Garzoni, M.; Maly, M.; Furer, V.L.; Kovalenko, V.; et al. The Key Role of the Scaffold on the Efficiency of Dendrimer Nanodrugs. Nat. Commun. 2015, 6, 7722. [Google Scholar] [CrossRef] [Green Version]
- Iezzi, R.; Guru, B.R.; Glybina, I.V.; Mishra, M.K.; Kennedy, A.; Kannan, R.M. Dendrimer-Based Targeted Intravitreal Therapy for Sustained Attenuation of Neuroinflammation in Retinal Degeneration. Biomaterials 2012, 33, 979–988. [Google Scholar] [CrossRef]
- Mishra, M.K.; Beaty, C.A.; Lesniak, W.G.; Kambhampati, S.P.; Zhang, F.; Wilson, M.A.; Blue, M.E.; Troncoso, J.C.; Kannan, S.; Johnston, M.V.; et al. Dendrimer Brain Uptake and Targeted Therapy for Brain Injury in a Large Animal Model of Hypothermic Circulatory Arrest. ACS Nano 2014, 8, 2134–2147. [Google Scholar] [CrossRef]
- Zhang, F.; Trent Magruder, J.; Lin, Y.A.; Crawford, T.C.; Grimm, J.C.; Sciortino, C.M.; Wilson, M.A.; Blue, M.E.; Kannan, S.; Johnston, M.V.; et al. Generation-6 Hydroxyl PAMAM Dendrimers Improve CNS Penetration from Intravenous Administration in a Large Animal Brain Injury Model. J. Control. Release 2017, 249, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Nance, E.; Porambo, M.; Zhang, F.; Mishra, M.K.; Buelow, M.; Getzenberg, R.; Johnston, M.; Kannan, R.M.; Fatemi, A.; Kannan, S. Systemic Dendrimer-Drug Treatment of Ischemia-Induced Neonatal White Matter Injury. J. Control. Release 2015, 214, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, C.L.; Drummond, G.T.; Mishra, M.K.; Zhang, F.; Carr, P.; Garcia, M.S.; Doman, S.; Fatemi, A.; Johnston, M.V.; Kannan, R.M.; et al. Uptake of Dendrimer-Drug by Different Cell Types in the Hippocampus after Hypoxic–Ischemic Insult in Neonatal Mice: Effects of Injury, Microglial Activation and Hypothermia. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2359–2369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Nance, E.; Alnasser, Y.; Kannan, R.; Kannan, S. Microglial Migration and Interactions with Dendrimer Nanoparticles Are Altered in the Presence of Neuroinflammation. J. Neuroinflamm. 2016, 13, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alnasser, Y.; Kambhampati, S.P.; Nance, E.; Rajbhandari, L.; Shrestha, S.; Venkatesan, A.; Kannan, R.M.; Kannan, S. Preferential and Increased Uptake of Hydroxyl-Terminated PAMAM Dendrimers by Activated Microglia in Rabbit Brain Mixed Glial Culture. Molecules 2018, 23, 1025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lin, Y.A.; Kim, S.Y.; Su, L.; Liu, J.; Kannan, R.M.; Kannan, S. Systemic Dendrimer-Drug Nanomedicines for Long-Term Treatment of Mild-Moderate Cerebral Palsy in a Rabbit Model. J. Neuroinflamm. 2020, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Deridder, L.; Sharma, A.; Liaw, K.; Sharma, R.; John, J.; Kannan, S.; Kannan, R.M. Dendrimer-Tesaglitazar Conjugate Induces a Phenotype Shift of Microglia and Enhances β-Amyloid Phagocytosis. Nanoscale 2021, 13, 939–952. [Google Scholar] [CrossRef]
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the Peroxisome Proliferator-Activated Receptor-α and the Retinoid X Receptor Inhibit Inflammatory Responses of Microglia. J. Neurosci. Res. 2005, 81, 403–411. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, X.; Yan, S.; Xie, X.; Fan, Y.; Zhang, J.; Peng, C.; You, Z. The Antidepressant-like Effects of Pioglitazone in a Chronic Mild Stress Mouse Model Are Associated with PPARγ-Mediated Alteration of Microglial Activation Phenotypes. J. Neuroinflamm. 2016, 13, 259. [Google Scholar] [CrossRef] [Green Version]
- Bays, H.; McElhattan, J.; Bryzinski, B.S. A Double-Blind, Randomised Trial of Tesaglitazar versus Pioglitazone in Patients with Type 2 Diabetes Mellitus. Diabetes Vasc. Dis. Res. 2007, 4, 181–193. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Study to Evaluate OP-101 (Dendrimer N-Acetyl-Cysteine) in Severe Coronavirus Disease 2019 (COVID-19) Patients (PRANA). Available online: https://clinicaltrials.gov/ct2/show/NCT04458298 (accessed on 15 March 2023).
- Maršić, Ž.S.; Maysinger, D.; Bonačić-Kouteckỳ, V. Insights into Interactions between Interleukin-6 and Dendritic Polyglycerols. Int. J. Mol. Sci. 2021, 22, 2415. [Google Scholar] [CrossRef]
- Durocher, I.; Girard, D. In Vivo Proinflammatory Activity of Generations 0–3 (G0–G3) Polyamidoamine (PAMAM) Nanoparticles. Inflamm. Res. 2016, 65, 745–755. [Google Scholar] [CrossRef]
- Fruchon, S.; Mouriot, S.; Thiollier, T.; Grandin, C.; Caminade, A.M.; Turrin, C.O.; Contamin, H.; Poupot, R. Repeated Intravenous Injections in Non-Human Primates Demonstrate Preclinical Safety of an Anti-Inflammatory Phosphorus-Based Dendrimer. Nanotoxicology 2015, 9, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; Mcguire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of Cytokines as a Double-Edged Sword in Sepsis. In Vivo 2013, 27, 669–684. [Google Scholar] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(Ethylene Glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chemie Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; De Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated Protein-Bound Levels of the Lipid Peroxidation Product, 4-Hydroxy-2-Nonenal, in Brain from Persons with Mild Cognitive Impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased Levels of 4-Hydroxynonenal and Acrolein, Neurotoxic Markers of Lipid Peroxidation, in the Brain in Mild Cognitive Impairment and Early Alzheimer’s Disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef]
- Ramassamy, C.; Averill, D.; Beffert, U.; Bastianetto, S.; Theroux, L.; Lussier-Cacan, S.; Cohn, J.S.; Christen, Y.; Davignon, J.; Quirion, R.; et al. Oxidative Damage and Protection by Antioxidants in the Frontal Cortex of Alzheimer’s Disease Is Related to the Apolipoprotein E Genotype. Free Radic. Biol. Med. 1999, 27, 544–553. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased Peroxidation and Reduced Antioxidant Enzyme Activity in Alzheimer’s Disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated Thiobarbituric Acid-Reactive Substances and Antioxidant Enzyme Activity in the Brain in Alzheimer’s Disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef]
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread Peroxynitrite-Mediated Damage in Alzheimer’s Disease. J. Neurosci. 1997, 17, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated Levels of 3-Nitrotyrosine in Brain from Subjects with Amnestic Mild Cognitive Impairment: Implications for the Role of Nitration in the Progression of Alzheimer’s Disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA Damage in Mild Cognitive Impairment and Late-Stage Alzheimer’s Disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colurso, G.J.; Nilson, J.E.; Vervoort, L.G. Quantitative Assessment of DNA Fragmentation and Beta-Amyloid Deposition in Insular Cortex and Midfrontal Gyrus from Patients with Alzheimer’s Disease. Life Sci. 2003, 73, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid Beta Peptide and NMDA Induce ROS from NADPH Oxidase and AA Release from Cytosolic Phospholipase A2 in Cortical Neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef]
- Ferreiro, E.; Baldeiras, I.; Ferreira, I.L.; Costa, R.O.; Rego, A.C.; Pereira, C.F.; Oliveira, C.R. Mitochondrial- and Endoplasmic Reticulum-Associated Oxidative Stress in Alzheimers Disease: From Pathogenesis to Biomarkers. Int. J. Cell Biol. 2012, 2012, 735206. [Google Scholar] [CrossRef] [Green Version]
- Labieniec, M.; Ulicna, O.; Vancova, O.; Kucharska, J.; Gabryelak, T.; Watala, C. Effect of Poly(Amido)Amine (Pamam) G4 Dendrimer on Heart and Liver Mitochondria in Animal Model of Diabetes. Cell Biol. Int. 2009, 34, 89–97. [Google Scholar] [CrossRef]
- Kannan, A.; Saravanan, V.; Rajakumar, P. Synthesis, Photophysical, Electrochemical Studies, and Antioxidant Properties of Fluorescein-Linked Glycodendrimers. Asian J. Org. Chem. 2016, 5, 1155–1163. [Google Scholar] [CrossRef]
- Vankoten, H.W.; Moore, R.S.; Cloninger, M.J. Nanoparticles to Study Lectins in Caenorhabditis Elegans: Multivalent Galactose Β1-4 Fucose-Functionalized Dendrimers Provide Protection from Oxidative Stress. Biomacromolecules 2021, 22, 4720–4729. [Google Scholar] [CrossRef]
- Rajakumar, P.; Venkatesan, N.; Sekar, K.; Nagaraj, S.; Rengasamy, R. Synthesis, Optical, and Antioxidant Studies of Anthraquinone-Core-Based Dendrimers with N-Phenylcarbazole as Surface Group. Aust. J. Chem. 2014, 67, 636–643. [Google Scholar] [CrossRef]
- Mieriņa, I.; Peipiņa, E.; Aišpure, K.; Jure, M. 1st Generation Dendrimeric Antioxidants Containing Meldrum’s Acid Moieties as Surface Groups. New J. Chem. 2022, 46, 607–620. [Google Scholar] [CrossRef]
- Xue, J.; Luo, Y.; Balasubramanian, B.; Upadhyay, A.; Li, Z.; Luo, Y. Development of Novel Biopolymer-Based Dendritic Nanocomplexes for Encapsulation of Phenolic Bioactive Compounds: A Proof-of-Concept Study. Food Hydrocoll. 2021, 120, 106987. [Google Scholar] [CrossRef]
- Lee, C.Y.; Sharma, A.; Uzarski, R.L.; Cheong, J.E.; Xu, H.; Held, R.A.; Upadhaya, S.K.; Nelson, J.L. Potent Antioxidant Dendrimers Lacking Pro-Oxidant Activity. Free Radic. Biol. Med. 2011, 50, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Halkes, S.B.A.; Vrasidas, I.; Rooijer, G.R.; Van den Berg, A.J.J.; Liskamp, R.M.J.; Pieters, R.J. Synthesis and Biological Activity of Polygalloyl-Dendrimers as Stable Tannic Acid Mimics. Bioorganic Med. Chem. Lett. 2002, 12, 1567–1570. [Google Scholar] [CrossRef]
- Balu, P.; Asharani, I.V.; Thirumalai, D. Synthesis of Melamine Core Starburst Polyamide G1 Dendrimer and Its Antibacterial and Antioxidant Activities. Asian J. Chem. 2021, 33, 185–189. [Google Scholar] [CrossRef]
- Sathiyaraj, S.; Shanavas, A.; Kumar, K.A.; Sathiyaseelan, A.; Senthilselvan, J.; Kalaichelvan, P.T.; Nasar, A.S. The First Example of Bis(Indolyl)Methane Based Hyperbranched Polyurethanes: Synthesis, Solar Cell Application and Anti-Bacterial and Anti-Oxidant Properties. Eur. Polym. J. 2017, 95, 216–231. [Google Scholar] [CrossRef]
- Rajakumar, P.; Venkatesan, N.; Sekar, K.; Nagaraj, S.; Rengasamy, R. Synthesis and Antioxidant Properties of Enone Core Based Dendrimers with Carbazole as Surface Group. Eur. J. Med. Chem. 2010, 45, 1220–1224. [Google Scholar] [CrossRef]
- Rajavelu, K.; Subaraja, M.; Rajakumar, P. Synthesis, Optical Properties, and Antioxidant and Anticancer Activity of Benzoheterazole Dendrimers with Triazole Bridging Unit. New J. Chem. 2018, 42, 3282–3292. [Google Scholar] [CrossRef]
- Del Olmo, N.S.; González, C.E.P.; Rojas, J.D.; Gómez, R.; Ortega, P.; Escarpa, A.; de la Mata, F.J. Antioxidant and Antibacterial Properties of Carbosilane Dendrimers Functionalized with Polyphenolic Moieties. Pharmaceutics 2020, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Oliveri, P.; Malegori, C. Assessment of the Efficiency of a Nanospherical Gallic Acid Dendrimer for Long-Term Preservation of Essential Oils: An Integrated Chemometric-Assisted FTIR Study. ChemistrySelect 2019, 4, 8891–8901. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Zuccari, G.; Turrini, F.; Domenicotti, C. Dendrimer Nanodevices and Gallic Acid as Novel Strategies to Fight Chemoresistance in Neuroblastoma Cells. Nanomaterials 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Signorello, M.G.; Schito, A.; Catena, S.; Turrini, F. Reshaped as Polyester-Based Nanoparticles, Gallic Acid Inhibits Platelet Aggregation, Reactive Oxygen Species Production and Multi-Resistant Gram-Positive Bacteria with an Efficiency Never Obtained. Nanoscale Adv. 2019, 1, 4148–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Catena, S.; Turrini, F. Biodegradable and Biocompatible Spherical Dendrimer Nanoparticles with a Gallic Acid Shell and a Double-Acting Strong Antioxidant Activity as Potential Device to Fight Diseases from “Oxidative Stress”. Drug Deliv. Transl. Res. 2020, 10, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ma, L.; Zhao, Y.; Zhao, J.; Ouyang, L.; Guo, L. Design and Synthesis of Novel Janus Dendrimers as Lipophilized Antioxidants. Synlett 2013, 24, 1011–1015. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wei, Y.; Shi, W.; Wang, J.; Wang, B. Antioxidant Capacity and Kinetics of Dendritic Hindered Phenols Using DPPH Assay. Prog. React. Kinet. Mech. 2015, 40, 279–290. [Google Scholar] [CrossRef]
- Sowinska, M.; Morawiak, M.; Bochyńska-Czyż, M.; Lipkowski, A.W.; Ńska, E.Z.; Zabłocka, B.; Urbanczyk-Lipkowska, Z. Molecular Antioxidant Properties and in Vitro Cell Toxicity of the P-Aminobenzoic Acid (PABA) Functionalized Peptide Dendrimers. Biomolecules 2019, 9, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Q.; Guo, S.Y.; Wang, J.; Shi, W.G.; Zhang, Z.Q.; Wang, P.X. Kinetics and Structure-Activity Relationship of Dendritic Bridged Hindered Phenol Antioxidants to Protect Styrene against Free Radical Induced Peroxidation. Russ. J. Phys. Chem. A 2017, 91, 2350–2360. [Google Scholar] [CrossRef]
- Lee, C.Y.; Sharma, A.; Semenya, J.; Anamoah, C.; Chapman, K.N.; Barone, V. Computational Study of Ortho-Substituent Effects on Antioxidant Activities of Phenolic Dendritic Antioxidants. Antioxidants 2020, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Olszowy, M. What Is Responsible for Antioxidant Properties of Polyphenolic Compounds from Plants? Plant Physiol. Biochem. 2019, 144, 135–143. [Google Scholar] [CrossRef]
- Katsumi, H.; Nishikawa, M.; Hirosaki, R.; Okuda, T.; Kawakami, S.; Yamashita, F.; Hashida, M.; Sakane, T.; Yamamoto, A. Development of PEGylated Cysteine-Modified Lysine Dendrimers with Multiple Reduced Thiols to Prevent Hepatic Ischemia/Reperfusion Injury. Mol. Pharm. 2016, 13, 2867–2873. [Google Scholar] [CrossRef]
- Matsuura, S.; Katsumi, H.; Suzuki, H.; Hirai, N.; Takashima, R.; Morishita, M.; Sakane, T.; Yamamoto, A. L-Cysteine and l-Serine Modified Dendrimer with Multiple Reduced Thiols as a Kidney-Targeting Reactive Oxygen Species Scavenger to Prevent Renal Ischemia/Reperfusion Injury. Pharmaceutics 2018, 10, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Qu, Y.; Wang, X. Amyloid β-Targeted Metal Complexes for Potential Applications in Alzheimer’s Disease. Future Med. Chem. 2018, 10, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, M.F.; Montalti, F.; Turro, N.J.; Tomalia, D.A. Characterization of Starburst Dendrimers by the EPR Technique. Copper(II) Ions Binding Full-Generation Dendrimers. J. Phys. Chem. B 1997, 101, 158–166. [Google Scholar] [CrossRef]
- Furlan, S.; La Penna, G.; Appelhans, D.; Cangiotti, M.; Ottaviani, M.F.; Danani, A. Combined EPR and Molecular Modeling Study of PPI Dendrimers Interacting with Copper Ions: Effect of Generation and Maltose Decoration. J. Phys. Chem. B 2014, 118, 12098–12111. [Google Scholar] [CrossRef]
- Appelhans, D.; Oertel, U.; Mazzeo, R.; Komber, H.; Hoffmann, J.; Weidner, S.; Brutschy, B.; Voit, B.; Ottaviani, M.F. Dense-Shell Glycodendrimers: UV/Vis and Electron Paramagnetic Resonance Study of Metal Ion Complexation. Proc. R. Soc. A Math. Phys. Eng. Sci. 2010, 466, 1489–1513. [Google Scholar] [CrossRef]
- Canonico, B.; Carloni, R.; Sanz Del Olmo, N.; Papa, S.; Nasoni, M.G.; Fattori, A.; Cangiotti, M.; De La Mata, F.J.; Ottaviani, M.F.; García-Gallego, S. Fine-Tuning the Interaction and Therapeutic Effect of Cu(II) Carbosilane Metallodendrimers in Cancer Cells: An in Vitro Electron Paramagnetic Resonance Study. Mol. Pharm. 2020, 17, 2691–2702. [Google Scholar] [CrossRef]
- Gothwal, A.; Singh, H.; Jain, S.K.; Dutta, A.; Borah, A.; Gupta, U. Behavioral and Biochemical Implications of Dendrimeric Rivastigmine in Memory-Deficit and Alzheimer’s Induced Rodents. ACS Chem. Neurosci. 2019, 10, 3789–3795. [Google Scholar] [CrossRef]
- Gothwal, A.; Nakhate, K.T.; Alexander, A.; Ajazuddin, A.; Gupta, U. Boosted Memory and Improved Brain Bioavailability of Rivastigmine: Targeting Effort to the Brain Using Covalently Tethered Lower Generation PAMAM Dendrimers with Lactoferrin. Mol. Pharm. 2018, 15, 4538–4549. [Google Scholar] [CrossRef] [PubMed]
- Gothwal, A.; Kumar, H.; Nakhate, K.T.; Ajazuddin; Dutta, A.; Borah, A.; Gupta, U. Lactoferrin Coupled Lower Generation PAMAM Dendrimers for Brain Targeted Delivery of Memantine in Aluminum-Chloride-Induced Alzheimer’s Disease in Mice. Bioconjug. Chem. 2019, 30, 2573–2583. [Google Scholar] [CrossRef]
- Igartúa, D.E.; Martinez, C.S.; Alonso, S.D.V.; Prieto, M.J. Combined Therapy for Alzheimer’s Disease: Tacrine and PAMAM Dendrimers Co-Administration Reduces the Side Effects of the Drug without Modifying Its Activity. AAPS PharmSciTech 2020, 21, 110. [Google Scholar] [CrossRef]
- Singh, A.K.; Gothwal, A.; Rani, S.; Rana, M.; Sharma, A.K.; Yadav, A.K.; Gupta, U. Dendrimer Donepezil Conjugates for Improved Brain Delivery and Better in Vivo Pharmacokinetics. ACS Omega 2019, 4, 4519–4528. [Google Scholar] [CrossRef]
- Navas Guimaraes, M.E.; Lopez-Blanco, R.; Correa, J.; Fernandez-Villamarin, M.; Bistué, M.B.; Martino-Adami, P.; Morelli, L.; Kumar, V.; Wempe, M.F.; Cuello, A.C.; et al. Liver X Receptor Activation with an Intranasal Polymer Therapeutic Prevents Cognitive Decline without Altering Lipid Levels. ACS Nano 2021, 15, 4678–4687. [Google Scholar] [CrossRef]
- Inoue, M.; Higashi, T.; Hayashi, Y.; Onodera, R.; Fujisawa, K.; Taharabaru, T.; Yokoyama, R.; Ouchi, K.; Misumi, Y.; Ueda, M.; et al. Multifunctional Therapeutic Cyclodextrin-Appended Dendrimer Complex for Treatment of Systemic and Localized Amyloidosis. ACS Appl. Mater. Interfaces 2022, 14, 40599–40611. [Google Scholar] [CrossRef]
- Tallon, C.; Bell, B.J.; Sharma, A.; Pal, A.; Malvankar, M.M.; Thomas, A.G.; Yoo, S.W.; Hollinger, K.R.; Coleman, K.; Wilkinson, E.L.; et al. Dendrimer-Conjugated NSMase2 Inhibitor Reduces Tau Propagation in Mice. Pharmaceutics 2022, 14, 2066. [Google Scholar] [CrossRef]
- Zhong, G.; Long, H.; Zhou, T.; Liu, Y.; Zhao, J.; Han, J.; Yang, X.; Yu, Y.; Chen, F.; Shi, S. Blood-Brain Barrier Permeable Nanoparticles for Alzheimer’s Disease Treatment by Selective Mitophagy of Microglia. Biomaterials 2022, 288, 121690. [Google Scholar] [CrossRef]
- Singh, A.; Mhaske, A.; Shukla, R. Fabrication of TPGS-Grafted Polyamidoamine Dendrimer for Enhanced Piperine Brain Delivery and Pharmacokinetics. AAPS PharmSciTech 2022, 23, 236. [Google Scholar] [CrossRef]
- Singh, A.; Ujjwal, R.R.; Naqvi, S.; Verma, R.K.; Tiwari, S.; Kesharwani, P.; Shukla, R. Formulation Development of Tocopherol Polyethylene Glycol Nanoengineered Polyamidoamine Dendrimer for Neuroprotection and Treatment of Alzheimer Disease. J. Drug Target. 2022, 30, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzawi, S.; Masheta, D.; Guildford, A.L.; Phillips, G.; Santin, M. Dendrimeric Poly(Epsilon-Lysine) Delivery Systems for the Enhanced Permeability of Flurbiprofen across the Blood-Brain Barrier in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Zhang, T.; Chen, Q.; Li, C.; Chu, Y.; Guo, Q.; Zhang, Y.; Zhou, W.; Chen, H.; Zhou, Z.; et al. Biomimetic Dendrimer–Peptide Conjugates for Early Multi-Target Therapy of Alzheimer’s Disease by Inflammatory Microenvironment Modulation. Adv. Mater. 2021, 33, 2100746. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel Tacrine-Related Drugs as Potential Candidates for the Treatment of Alzheimer’s Disease. Bioorganic Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Liver X Receptors as Integrators of Metabolic and Inflammatory Signaling. J. Clin. Invest. 2006, 116, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Hu, S.; Yin, J.J.; He, W.; Lu, W.; Ma, M.; Gu, N.; Zhang, Y. Prussian Blue Nanoparticles as Multienzyme Mimetics and Reactive Oxygen Species Scavengers. J. Am. Chem. Soc. 2016, 138, 5860–5865. [Google Scholar] [CrossRef] [PubMed]
- Sowinska, M.; Urbanczyk-Lipkowska, Z. Advances in the Chemistry of Dendrimers. New J. Chem. 2014, 38, 2168–2203. [Google Scholar] [CrossRef]
- Maraval, V.; Laurent, R.; Marchand, P.; Caminade, A.M.; Majoral, J.P. Accelerated Methods of Synthesis of Phosphorus-Containing Dendrimers. J. Organomet. Chem. 2005, 690, 2458–2471. [Google Scholar] [CrossRef]
- Patel, P.; Patel, V.; Patel, P.M. Synthetic Strategy of Dendrimers: A Review. J. Indian Chem. Soc. 2022, 99, 100514. [Google Scholar] [CrossRef]
- Brauge, L.; Magro, G.; Caminade, A.M.; Majoral, J.P. First Divergent Strategy Using Two AB2 Unprotected Monomers for the Rapid Synthesis of Dendrimers. J. Am. Chem. Soc. 2001, 123, 6698–6699. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chemie Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Gothwal, A.; Malik, S.; Gupta, U.; Jain, N.K. Toxicity and Biocompatibility Aspects of Dendrimers; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128145272. [Google Scholar]
- Aurelia Chis, A.; Dobrea, C.; Morgovan, C.; Arseniu, A.M.; Rus, L.L.; Butuca, A.; Juncan, A.M.; Totan, M.; Vonica-Tincu, A.L.; Cormos, G.; et al. Applications and Limitations of Dendrimers in Biomedicine. Molecules 2020, 25, 3982. [Google Scholar] [CrossRef]
- Jones, C.F.; Campbell, R.A.; Brooks, A.E.; Assemi, S.; Tadjiki, S.; Thiagarajan, G.; Mulcock, C.; Weyrich, A.S.; Brooks, B.D.; Ghandehari, H.; et al. Cationic PAMAM Dendrimers Aggressively Initiate Blood Clot Formation. ACS Nano 2012, 6, 9900–9910. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.F.; Campbell, R.A.; Franks, Z.; Gibson, C.C.; Thiagarajan, G.; Vieira-De-Abreu, A.; Sukavaneshvar, S.; Mohammad, S.F.; Li, D.Y.; Ghandehari, H.; et al. Cationic PAMAM Dendrimers Disrupt Key Platelet Functions. Mol. Pharm. 2012, 9, 1599–1611. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Patri, A.K.; Simak, J.; Hall, J.B.; Semberova, J.; De Paoli Lacerda, S.H.; McNeil, S.E. Nanoparticle Size and Surface Charge Determine Effects of PAMAM Dendrimers on Human Platelets in Vitro. Mol. Pharm. 2012, 9, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Agashe, H.B.; Dutta, T.; Garg, M.; Jain, N.K. Investigations on the Toxicological Profile of Functionalized Fifth-Generation Poly(Propylene Imine) Dendrimer. J. Pharm. Pharmacol. 2010, 58, 1491–1498. [Google Scholar] [CrossRef]
- Bhadra, D.; Yadav, A.K.; Bhadra, S.; Jain, N.K. Glycodendrimeric Nanoparticulate Carriers of Primaquine Phosphate for Liver Targeting. Int. J. Pharm. 2005, 295, 221–233. [Google Scholar] [CrossRef]
- Malik, N.; Wiwattanapatapee, R.; Klopsch, R.; Lorenz, K.; Frey, H.; Weener, J.W.; Meijer, E.W.; Paulus, W.; Duncan, R. Dendrimers: Relationship between Structure and Biocompatibility in Vitro, and Preliminary Studies on the Biodistribution of 125I-Labelled Polyamidoamine Dendrimers in vivo. J. Control. Release 2000, 65, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Vidal, F.; Vásquez, P.; Cayumán, F.R.; Díaz, C.; Fuentealba, J.; Aguayo, L.G.; Yévenes, G.E.; Alderete, J.; Guzmán, L. Prevention of Synaptic Alterations and Neurotoxic Effects of PAMAM Dendrimers by Surface Functionalization. Nanomaterials 2017, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhu, H.; Wang, S.; Qian, X.; Fan, J.; Wang, Z.; Song, P.; Zhang, X.; Lu, W.; Ju, D. Interplay of Oxidative Stress and Autophagy in PAMAM Dendrimers-Induced Neuronal Cell Death. Theranostics 2015, 5, 1363–1377. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Kurokawa, Y.; Zeng, Q.; Win-Shwe, T.T.; Nansai, H.; Zhang, Z.; Sone, H. Effects of Polyamidoamine Dendrimers on a 3-D Neurosphere System Using Human Neural Progenitor Cells. Toxicol. Sci. 2016, 152, 128–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, S.; Fan, J.; Zhang, X.; Qian, X.; Zhang, X.; Luan, J.; Song, P.; Wang, Z.; Chen, Q.; et al. Targeting TNFα Ameliorated Cationic PAMAM Dendrimer-Induced Hepatotoxicity via Regulating NLRP3 Inflammasomes Pathway. ACS Biomater. Sci. Eng. 2017, 3, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, X.; Wang, S.; Sun, Y.; Wang, Z.; Fan, J.; Song, P.; Ju, D. Inhibition of Autophagy Protects against PAMAM Dendrimers-Induced Hepatotoxicity. Nanotoxicology 2015, 9, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Al-Zaid, B.; El-Hashim, A.Z.; Chandrasekhar, B.; Attur, S.; Benter, I.F. Impact of PAMAM Delivery Systems on Signal Transduction Pathways in Vivo: Modulation of ERK1/2 and P38 MAP Kinase Signaling in the Normal and Diabetic Kidney. Int. J. Pharm. 2016, 514, 353–363. [Google Scholar] [CrossRef]
- Li, C.; Liu, H.; Sun, Y.; Wang, H.; Guo, F.; Rao, S.; Deng, J.; Zhang, Y.; Miao, Y.; Guo, C.; et al. PAMAM Nanoparticles Promote Acute Lung Injury by Inducing Autophagic Cell Death through the Akt-TSC2-MTOR Signaling Pathway. J. Mol. Cell Biol. 2009, 1, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.P.; Davoren, M.; Byrne, H.J. In Vitro Mammalian Cytotoxicological Study of PAMAM Dendrimers—Towards Quantitative Structure Activity Relationships. Toxicol. Vitr. 2010, 24, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.P.; Lyng, F.M.; Garcia, A.; Davoren, M.; Byrne, H.J. Mechanistic Studies of in Vitro Cytotoxicity of Poly(Amidoamine) Dendrimers in Mammalian Cells. Toxicol. Appl. Pharmacol. 2010, 248, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, J.B.; Harper, B.J.; Harper, S.L. Comparative Toxicological Assessment of PAMAM and Thiophosphoryl Dendrimers Using Embryonic Zebrafish. Int. J. Nanomed. 2014, 9, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Naha, P.C.; Davoren, M.; Casey, A.; Byrne, H.J. An Ecotoxicological Study of Poly(Amidoamine) Dendrimers-toward Quantitative Structure Activity Relationships. Environ. Sci. Technol. 2009, 43, 6864–6869. [Google Scholar] [CrossRef] [PubMed]
- Janaszewska, A.; McZyńska, K.; Matuszko, G.; Appelhans, D.; Voit, B.; Klajnert, B.; Bryszewska, M. Cytotoxicity of PAMAM, PPI and Maltose Modified PPI Dendrimers in Chinese Hamster Ovary (CHO) and Human Ovarian Carcinoma (SKOV3) Cells. New J. Chem. 2012, 36, 428–437. [Google Scholar] [CrossRef]
- Felczak, A.; Wrońska, N.; Janaszewska, A.; Klajnert, B.; Bryszewska, M.; Appelhans, D.; Voit, B.; Rózalska, S.; Lisowska, K. Antimicrobial Activity of Poly(Propylene Imine) Dendrimers. New J. Chem. 2012, 36, 2215–2222. [Google Scholar] [CrossRef]
- Lazniewska, J.; Milowska, K.; Zablocka, M.; Mignani, S.; Caminade, A.M.; Majoral, J.P.; Bryszewska, M.; Gabryelak, T. Mechanism of Cationic Phosphorus Dendrimer Toxicity against Murine Neural Cell Lines. Mol. Pharm. 2013, 10, 3484–3496. [Google Scholar] [CrossRef]
- Gomulak, P.; Klajnert, B.; Bryszewska, M.; Majoral, J.P.; Caminade, A.M.; Blasiak, J. Cytotoxicity and Genotoxicity of Cationic Phosphorus-Containing Dendrimers. Curr. Med. Chem. 2015, 19, 6233–6240. [Google Scholar] [CrossRef]
- El Brahmi, N.; El Kazzouli, S.; Mignani, S.M.; Essassi, E.M.; Aubert, G.; Laurent, R.; Caminade, A.M.; Bousmina, M.M.; Cresteil, T.; Majoral, J.P. Original Multivalent Copper(II)-Conjugated Phosphorus Dendrimers and Corresponding Mononuclear Copper(II) Complexes with Antitumoral Activities. Mol. Pharm. 2013, 10, 1459–1464. [Google Scholar] [CrossRef]
- Khandare, J.; Kumar, S. Biodegradable Dendrimers and Dendritic Polymers. In Handbook of Biodegradable Polymers: Isolation, Synthesis, Characterization and Applications; Lendkein, A., Sisson, A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 237–262. ISBN 9783527324415. [Google Scholar]
- Leiro, V.; Garcia, J.P.; Moreno, P.M.D.; Spencer, A.P.; Fernandez-Villamarin, M.; Riguera, R.; Fernandez-Megia, E.; Paula Pêgo, A. Biodegradable PEG-Dendritic Block Copolymers: Synthesis and Biofunctionality Assessment as Vectors of SiRNA. J. Mater. Chem. B 2017, 5, 4901–4917. [Google Scholar] [CrossRef] [Green Version]
- Pêgo, A.P.; Leiro, V.; Fernandez-Megia, E.; Riguera, R. Biodegradable Dendritic Structure, Methods and Uses Thereof. WO2017203437A1, 30 November 2017. (US10858484B2, 8 December 2020). [Google Scholar]
- Leiro, V.; Spencer, A.P.; Magalhães, N.; Pêgo, A.P. Versatile Fully Biodegradable Dendritic Nanotherapeutics. Biomaterials 2022, 281, 121356. [Google Scholar] [CrossRef] [PubMed]
- Padilla De Jesús, O.L.; Ihre, H.R.; Gagne, L.; Fréchet, J.M.J.; Szoka, F.C. Polyester Dendritic Systems for Drug Delivery Applications: In Vitro and in Vivo Evaluation. Bioconjug. Chem. 2002, 13, 453–461. [Google Scholar] [CrossRef]
- Lundberg, P.; Walter, M.V.; Montañez, M.I.; Hult, D.; Hult, A.; Nyström, A.; Malkoch, M. Linear Dendritic Polymeric Amphiphiles with Intrinsic Biocompatibility: Synthesis and Characterization to Fabrication of Micelles and Honeycomb Membranes. Polym. Chem. 2011, 2, 394–402. [Google Scholar] [CrossRef]
- Lee, C.C.; Gillies, E.R.; Fox, M.E.; Guillaudeu, S.J.; Fréchet, J.M.J.; Dy, E.E.; Szoka, F.C. A Single Dose of Doxorubicin-Functionalized Bow-Tie Dendrimer Cures Mice Bearing C-26 Colon Carcinomas. Proc. Natl. Acad. Sci. USA 2006, 103, 16649–16654. [Google Scholar] [CrossRef] [Green Version]
- Dhanikula, R.S.; Hildgen, P. Influence of Molecular Architecture of Polyether-Co-Polyester Dendrimers on the Encapsulation and Release of Methotrexate. Biomaterials 2007, 28, 3140–3152. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K. α-Synuclein Structure, Posttranslational Modification and Alternative Splicing as Aggregation Enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef]
- Klajnert, B.; Cangiotti, M.; Calici, S.; Majoral, J.P.; Caminade, A.M.; Cladera, J.; Bryszewska, M.; Ottaviani, M.F. EPR Study of the Interactions between Dendrimers and Peptides Involved in Alzheimer’s and Prion Diseases. Macromol. Biosci. 2007, 7, 1065–1074. [Google Scholar] [CrossRef]
- Martins, P.M.; Navarro, S.; Silva, A.; Pinto, M.F.; Sárkány, Z.; Figueiredo, F.; Pereira, P.J.B.; Pinheiro, F.; Bednarikova, Z.; Burdukiewicz, M.; et al. MIRRAGGE—Minimum Information Required for Reproducible AGGregation Experiments. Front. Mol. Neurosci. 2020, 13, 582488. [Google Scholar] [CrossRef]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A Primer on the Use of Thioflavin T to Investigate Amyloid Formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mignani, S.; Shi, X.; Rodrigues, J.; Tomas, H.; Karpus, A.; Majoral, J.P. First-in-Class and Best-in-Class Dendrimer Nanoplatforms from Concept to Clinic: Lessons Learned Moving Forward. Eur. J. Med. Chem. 2021, 219, 113456. [Google Scholar] [CrossRef]
- Bodur, O.C.; Hasanoğlu Özkan, E.; Çolak, Ö.; Arslan, H.; Sarı, N.; Dişli, A.; Arslan, F. Preparation of Acetylcholine Biosensor for the Diagnosis of Alzheimer’s Disease. J. Mol. Struct. 2021, 1223, 129168. [Google Scholar] [CrossRef]
- Razzino, C.A.; Serafín, V.; Gamella, M.; Pedrero, M.; Montero-Calle, A.; Barderas, R.; Calero, M.; Lobo, A.O.; Yáñez-Sedeño, P.; Campuzano, S.; et al. An Electrochemical Immunosensor Using Gold Nanoparticles-PAMAM-Nanostructured Screen-Printed Carbon Electrodes for Tau Protein Determination in Plasma and Brain Tissues from Alzheimer Patients. Biosens. Bioelectron. 2020, 163, 112238. [Google Scholar] [CrossRef] [PubMed]








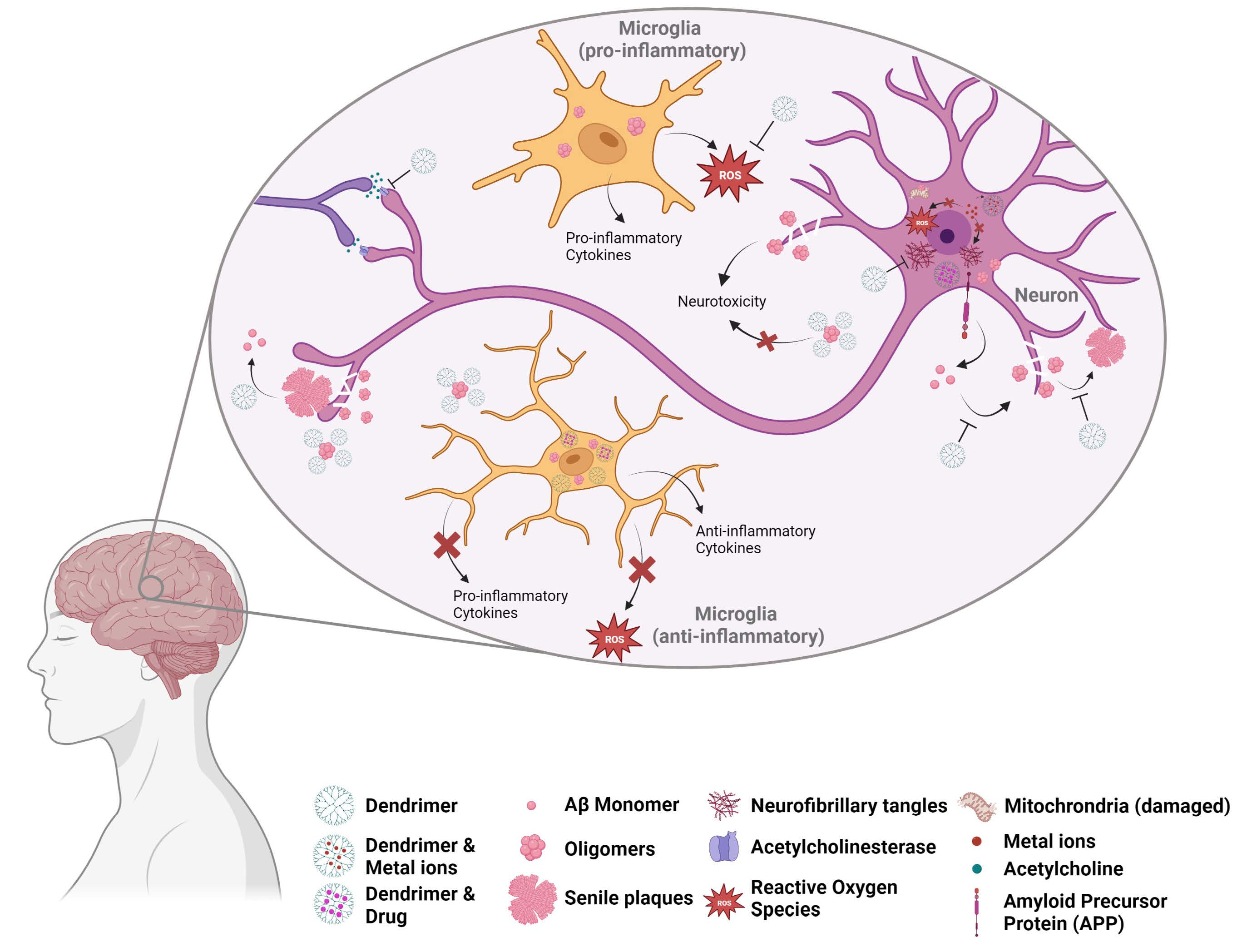{kind=link}
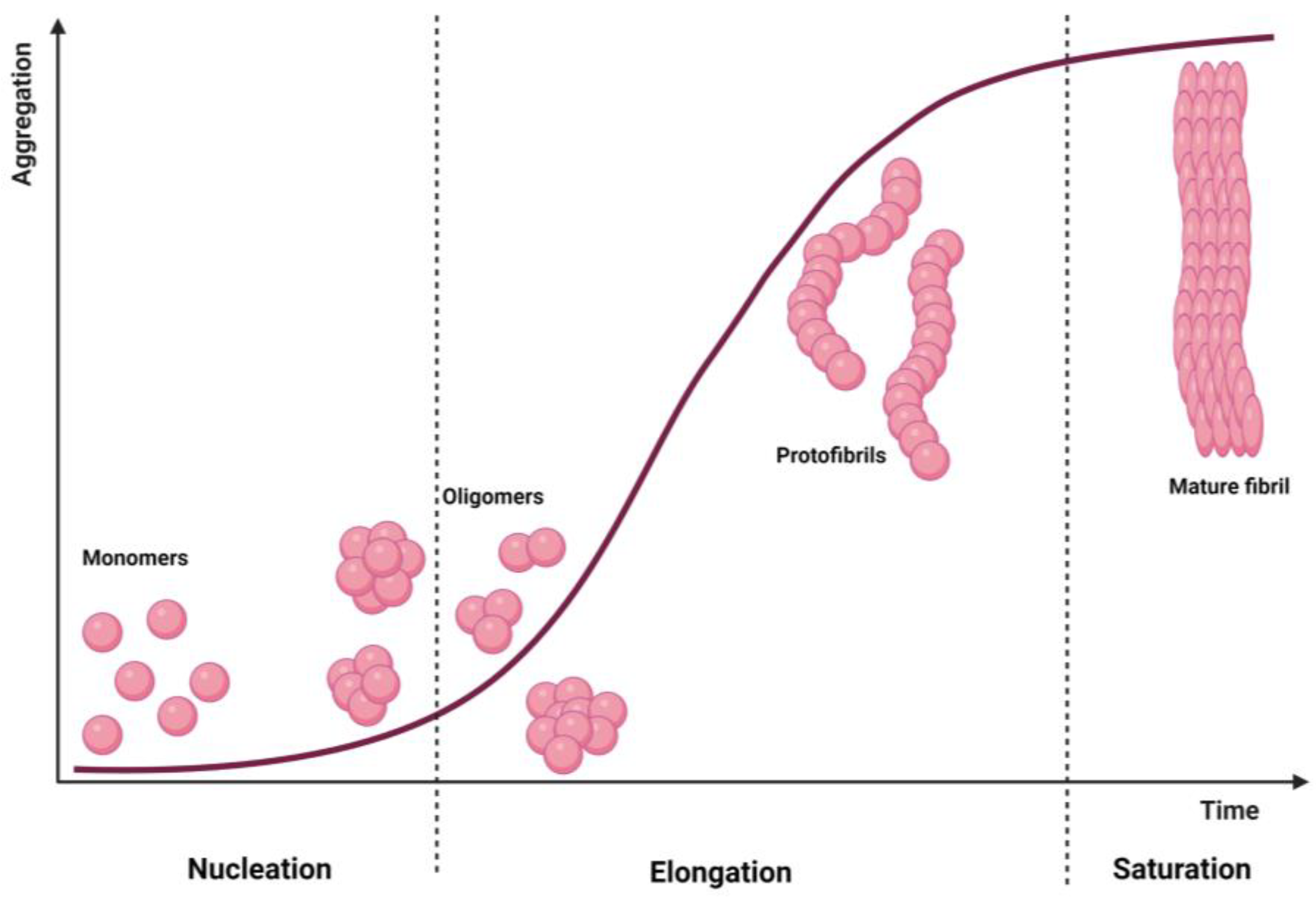{kind=link}
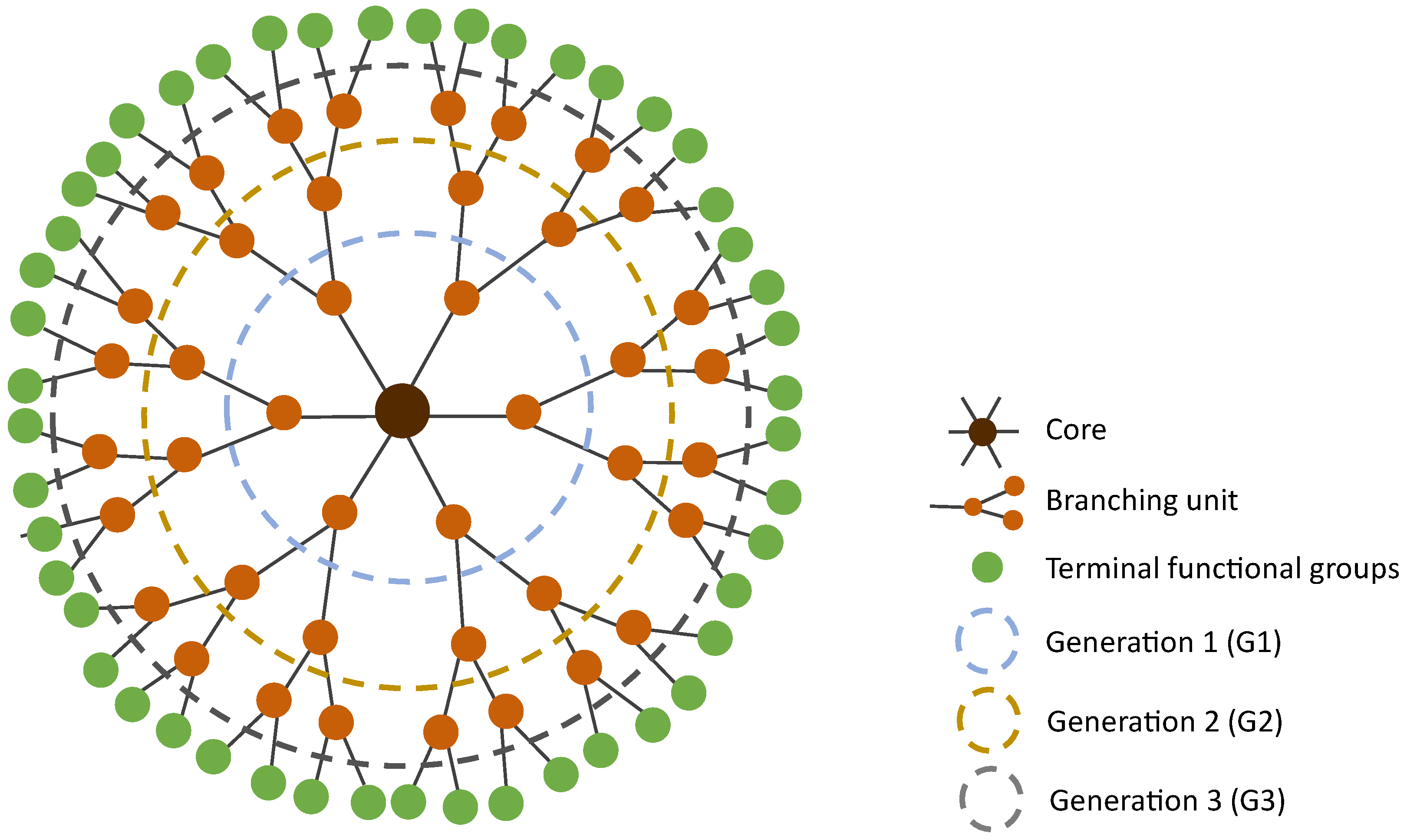{kind=link}
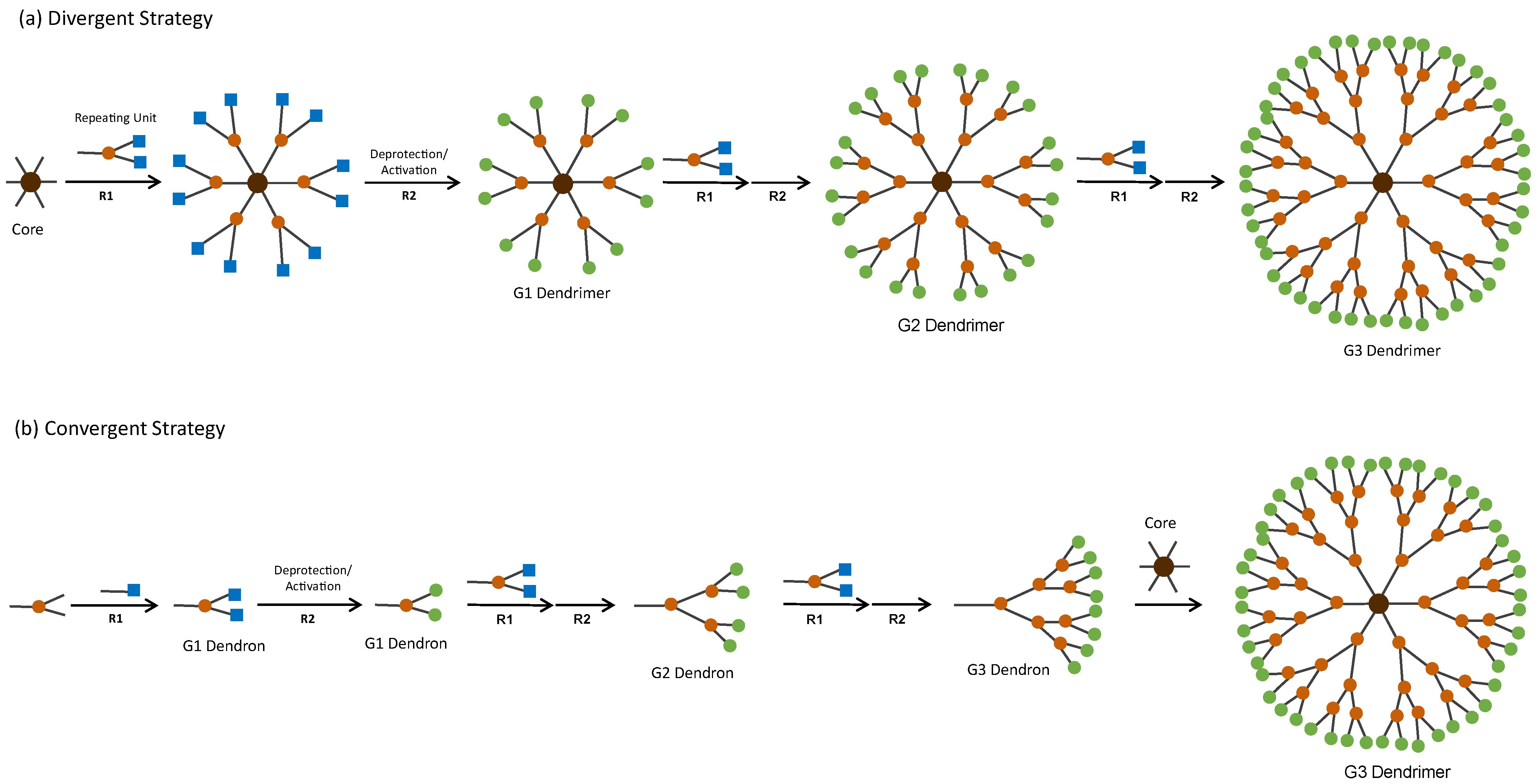{kind=link}
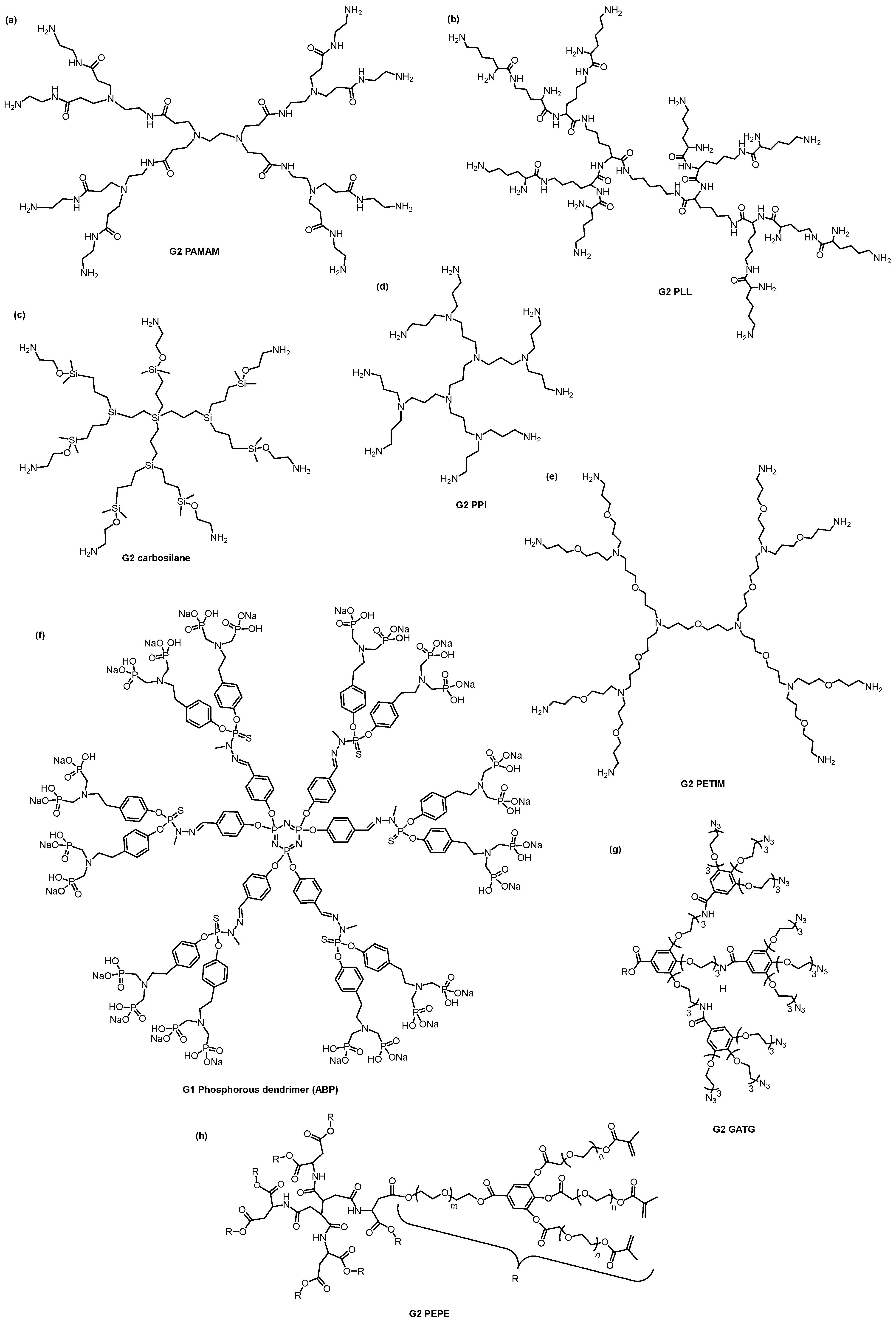{kind=link}
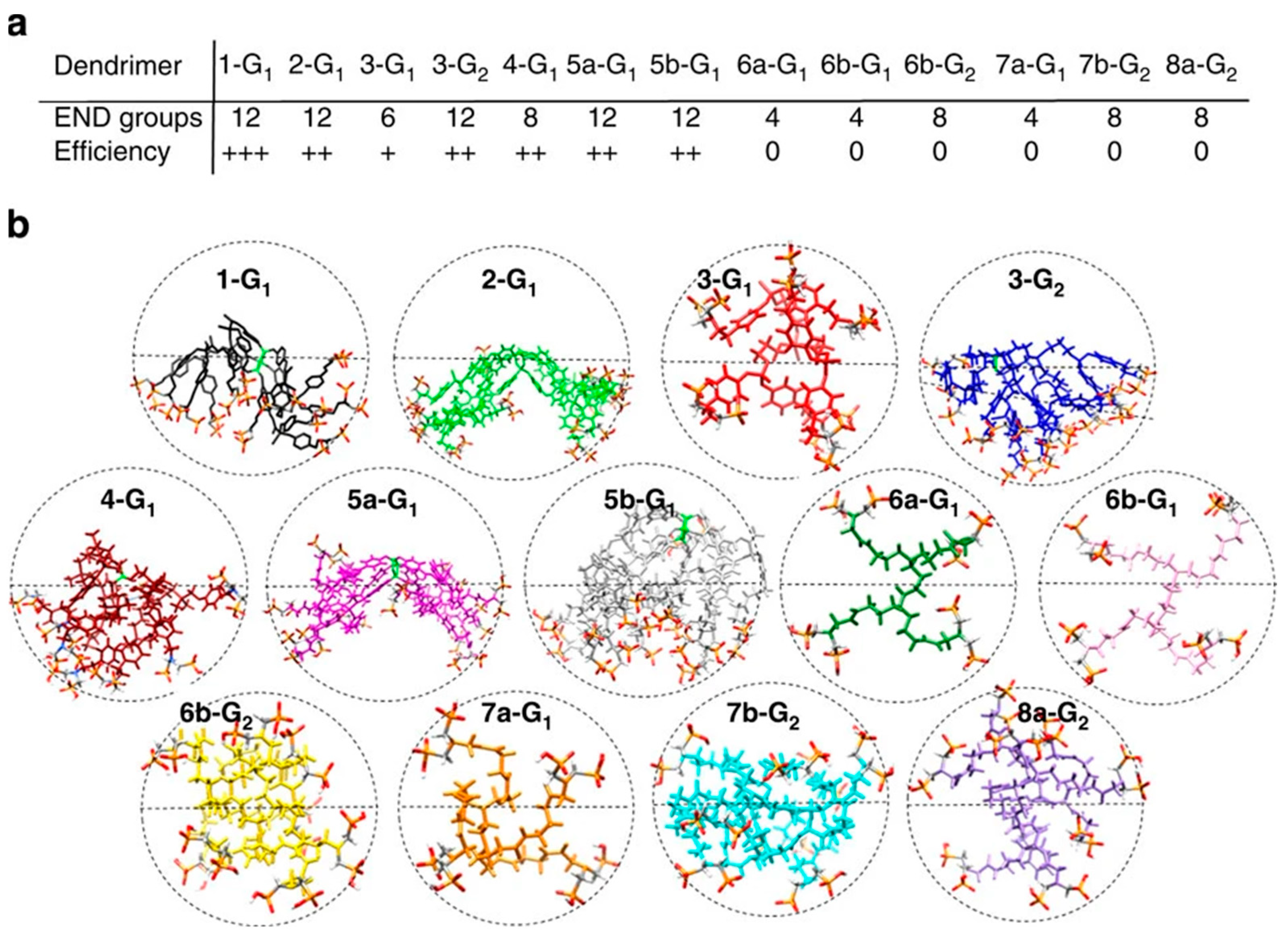{kind=link}
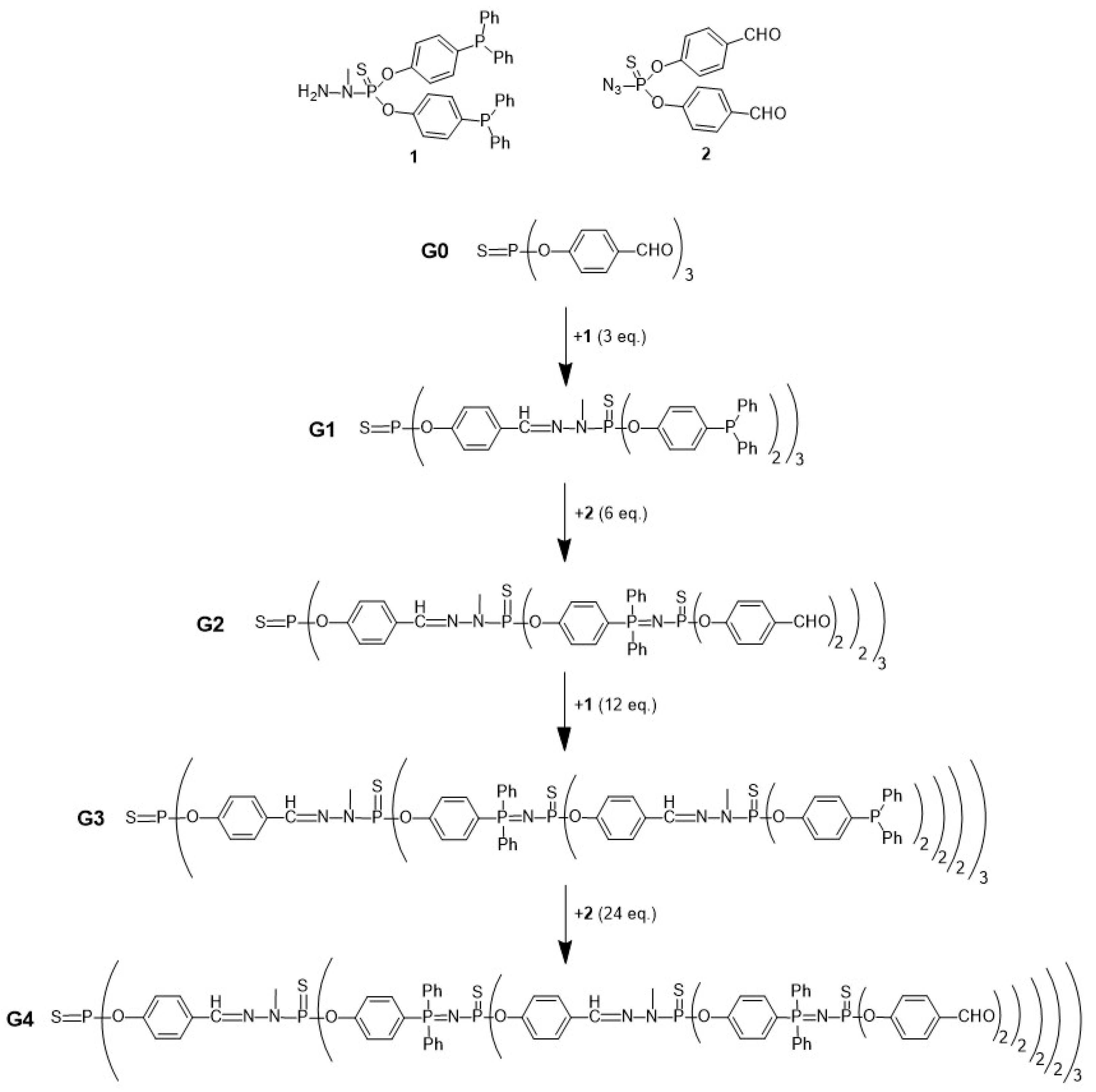{kind=link}
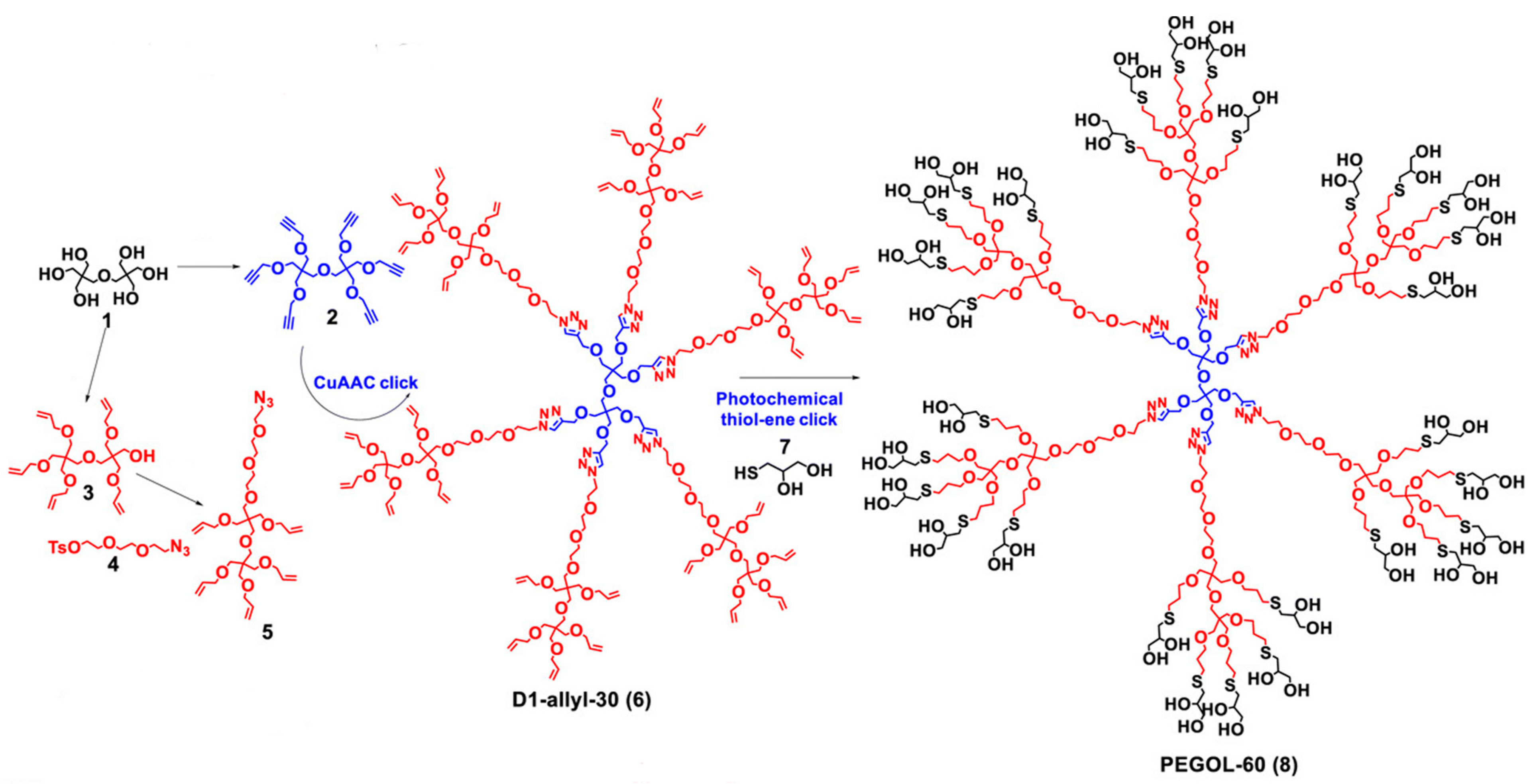{kind=link}
| Dendrimer (No. Terminal Groups (1)) | Terminal Group (Charge) | Dendrimer/Peptide Ratio | Effect on Fibrillation (to Peptide Alone) | Morphology and Secondary Structure of Fibrils | Disaggregation Ability? | Attenuation of Aβ Cytotoxicity? (Cell Type) | Refs. |
|---|---|---|---|---|---|---|---|
| G3 PAMAM (32) | -NH2 (+) | 0.0002 | ↑ elongation rate; | Clumps | Yes * | Yes (SH-SY5Y) | [19,104,124] |
| 0.002 | ↓ elongation rate and fibril amount (~30%) | ||||||
| 0.02 | ↓ elongation rate and fibril amount (~ 50%) | ||||||
| 0.10 | Complete inhibition of aggregation | ||||||
| G4 PAMAM (64) | -NH2 (+) | 0.0002 | ↑ elongation rate; | Yes * | Yes (SH-SY5Y) | [104,124] | |
| 0.002 | ↓ fibril amount (~30%) | ||||||
| 0.02 | ↓ elongation rate and fibril amount (~65%) | ||||||
| G5 PAMAM (128) | -NH2 (+) | 0.0002 | ↑ elongation rate | Amorphous aggregates | Yes * | [104,124] | |
| 0.002 | ↓ elongation rate and fibril amount (25%) | ||||||
| 0.02 | Complete inhibition of aggregation | ||||||
| G3 CPD (48) | -NHEt (+) | 0.0002 | ↑ elongation rate and fibril amount (~60%) | Long fibrils. Accelerated the conformational transition to β-sheet | Yes (N2a) | [21] | |
| 0.0002 | No effect | Accelerated the conformational transition to β-sheet | |||||
| 0.02–0.2 | Complete inhibition of aggregation | No fibrils. Inhibition on transition to β-sheet. | |||||
| G4 CPD (96) | -NHEt (+) | 0.0002 | ↑ elongation rate and fibril amount (~60%) | Long fibrils. Accelerated the conformational transition to β-sheet | Yes | Yes (N2a) | [21] |
| 0.0002 | ↑ elongation rate and fibril amount (~20%) | ||||||
| 0.02–0.2 | Complete inhibition of aggregation | No fibrils. Inhibition on transition to β-sheet. | |||||
| G3 PPI (16) | -NH2 (+) | 0.02 | ↑ elongation rate; ↓ fibril amount (↓ 12%) | Yes * | [22] | ||
| 0.03 | ↑ elongation rate; ↓ fibril amount (↓ 56%) | ||||||
| 0.04 | ↓ fibril amount (↓ 56%) | ||||||
| 0.08 | Complete inhibition of aggregation | ||||||
| G4 mPPI (32 (64)) | Maltose (0) | 0.1 | ↑ elongation rate | Clumped fibrils | Yes * | Yes (PC12/SH-SY5Y) | [20,118,122] |
| 1 | ↑ elongation rate | ||||||
| 5 | ↓ elongation rate and fibril amount (↓ ~50%) | ||||||
| 10 | ↓ elongation rate and fibril amount (↓ ~80%) | ||||||
| G4 m-IIIPPI OS (32 (64)) | -NH (36%)/-NH2 (28%)/Maltotriose (36%) (+) | 0.01 | Complete inhibition of aggregation | Granular aggregates | No | [118] | |
| G4 mPPI OS (32 (64)) | -NH (37.5%)/ -NH2 (25%)/Maltose (37.5%) (+) | 0.01 | ↑ nucleation rate, elongation rate and fibril amount (↑ 100%) | Fibrillar. No oligomers | Yes (SH-SY5Y) | [118] | |
| >0.01 | Complete inhibition of aggregation | ||||||
| G5 mPPI (64 (128)) | -NH (10%)/Maltose (90%) (0) | 0.005 | ↑ nucleation and elongation rate | Fibrillar. No oligomers | Yes (PC12/SH-SY5Y) | [20,118] | |
| 0.1–10 | Complete inhibition of aggregation | Amorphous aggregates | |||||
| G5 mPPI (64 (128)) | Maltose (0) | 0.002 | ↑ elongation rate and fibril amount (↑ 120%) | Acceleration of the conformational transition to β-sheet | [119] | ||
| 0.02 | ↑ elongation rate and fibril amount (↑ 100%) | ||||||
| 0.2 | Complete inhibition of aggregation | Inhibition of conformational transition to β-sheet. | |||||
| G5 m-IIIPPI (64 (128)) | -NH (6%)/Maltotriose (94%) (0) | 0.002 | ↑ elongation rate and fibril amount (↑ 100%) | Acceleration of the conformational transition to β-sheet | [119] | ||
| 0.02 | ↑ fibril amount (↑ ~20%) | ||||||
| 0.2 | Complete inhibition of aggregation | Inhibition of conformational transition to β-sheet. | |||||
| G5 mPPI SO3 (64 (128)) | Maltose/ -O-SO3− (0.76 equivalent of OH units) (−) | 0.0002 | ↓ fibril amount (↓ ~25%) | Yes (mHippoE-18) | [120] | ||
| 0.002 | ↓ fibril amount (↓ ~45%) | ||||||
| 0.02 | ↓ fibril amount (↓ ~45%) | Slows down the conformational transition to β-sheet | |||||
| G4HisMal (64 (128)) | -NH (32%)/Maltose (48%)/Histine (20%) (+) | 0.10 | ↓ elongation rate and fibril amount (↓ ~65%) | No oligomers. Mesh like fibrils | Yes (SH−SY5Y) | [26] | |
| 1 | ↓ elongation rate and fibril amount (↓ ~75%) | No oligomers. More irregular aggregates | |||||
| GATG-Mor (27) | Morpholine (0) | 0.0002 | No effect | Yes (B14) | [110] | ||
| 0.002 | ↑ fibril amount (↑ 25%) | ||||||
| 0.02 | ↑ elongation and fibril amount (↑ 116%) | Long fibrils; ↑ fibrils; Faster conformational transition to β-sheet | |||||
| 2G0-GaOH (2) | Gallic acid (-OH) (0) | 0.5 | Complete inhibition of aggregation | Yes | Yes (SH-SY5Y) | [111] | |
| 1 | Complete inhibition of aggregation. ↓ final amount of ThT+ aggregates in cell culture (~50%). | ↓ of non-fibrillar structures (~20%); ↓ Elongated fibrils. ↑ unstructured aggregates (condensed and less organized) and/or shorter fibrils | |||||
| 2 | Complete inhibition of aggregation | ||||||
| 2G1-Ga-OH (6) | Gallic acid (-OH) (0) | 0.5 | Complete inhibition of aggregation | Yes | Yes (SH-SY5Y) | [111] | |
| 1 | Complete inhibition of aggregation. ↓ final amount of ThT+ aggregates in cell culture (~60%). | ↓ of non-fibrillar structures (~43%); ↓ small aggregates (~10%); ↓ Elongated fibrils; ↑ unstructured aggregates (condensed and less organized) and/or shorter fibrils | |||||
| 2 | Complete inhibition of aggregation | ||||||
| 3G1-Ga-OH (9) | Gallic acid (-OH) (0) | 0.5 | Complete inhibition of aggregation | Yes | Yes (SH-SY5Y) | [111] | |
| 1 | Complete inhibition of aggregation. ↓ the final amount of ThT+ aggregates in cell culture (~40%). | ↓ of non-fibrillar structures (~32%); ↓ Elongated fibrils; ↑ unstructured aggregates (condensed and less organized) and/or shorter fibrils | |||||
| 2 | Complete inhibition of aggregation | ||||||
| G3 Lysine dendrimer (8) | (CH2)4NH3+(+) | 0.02 | ↑ nucleation rate, elongation rate and fibril amount (↑ ~20%) | Yes (SH-SY5Y) | [125] | ||
| 0.1 | ↑ nucleation rate and elongation rate | ||||||
| G5 Lysine dendrimer (26) | (CH2)4NH3+ (+) | 0.02 | ↓ nucleation rate, elongation rate and fibril amount (↓ ~100%) | [125] | |||
| 0.1 | ↑ fibril amount (↑ ~50%) | ||||||
| G3/G4 PAMAM-COOH (32/64) | -COOH (−) | 0.1–25 | No effect | Fibrillar. No change in the secondary structure of the peptides | [126] | ||
| G5 PAMAM-COOH (128) | -COOH (−) | 0.1–10 | No effect | Fibrillar. No change in the secondary structure of the peptides | No (SH-SY5Y) | [107] | |
| 25 | ↓ fibril amount (20%) | ||||||
| G6 PAMAM-COOH (256) | -COOH (−) | 0.1–25 | No effect | Fibrillar. No change in the secondary structure of the peptides | [126] | ||
| G3 PAMP (32) | -COOH/Phenyl groups (28.8%) (−) | 0.01 | No effect | Fibrillar. No change in the secondary structure of the peptides | [126] | ||
| 0.1–1 | ↑ elongation rate and fibril amount (up to 30%) | ||||||
| G4 PAMP (64) | -COOH/ Phenyl groups (28.2%) (−) | 0.01–0.5 | ↑ elongation rate and fibril amount (up to 30%) | Fibrillar. No change in the secondary structure of the peptides | [126] | ||
| 1 | ↑ elongation rate | ||||||
| G5 PAMP (128) | -COOH/ Phenyl groups (7.2%) (−) | 0.01 | ↓ fibril amount (↓ ~10%) | Fibrillar. No change in the secondary structure of the peptides | Yes (SH-SY5Y) | [107] | |
| 0.1 | ↓ fibril amount (↓ ~15%) | ||||||
| 0.5 | ↓ fibril amount (↓ ~20%) | ||||||
| 1 | ↓ elongation rate and fibril amount (↓ ~28%) | ||||||
| -COOH/ Phenyl groups (21.2%) (−) | 0.01 | ↓ fibril amount (↓ ~15%) | Fibrillar. No change in the secondary structure of the peptides | Yes (SH-SY5Y) | [107] | ||
| 0.1 | ↓ fibril amount (↓ ~20%) | ||||||
| 0.5 | ↓ fibril amount (↓ ~30%) | ||||||
| 1 | ↓ elongation rate and fibril amount (↓ ~38%) | ||||||
| -COOH/ Phenyl groups (30.5%) (−) | 0.01 | ↓ fibril amount (↓ ~25%) | Irregular aggregates. Inhibition on the conformational transition to β-sheet at the equimolar ratio of peptide/dendrimer | Yes (SH-SY5Y) | [107,126] | ||
| 0.1 | ↓ fibril amount (↓ ~35%) | ||||||
| 0.5 | ↓ fibril amount (↓ ~40%) | ||||||
| 1 | ↓ elongation rate and fibril amount (↓ ~70%) | ||||||
| -COOH/ Phenyl groups (42.3%) (−) | 0.01 | ↓ fibril amount (↓ ~25%) | Irregular aggregates. Inhibition on the conformational transition to β-sheet at the equimolar ratio of peptide/dendrimer | Yes (SH-SY5Y) | [107] | ||
| 0.1 | ↓ fibril amount (↓ ~40%) | ||||||
| 0.5 | ↓ fibril amount (↓ ~50%) | ||||||
| 1 | ↓ elongation rate and fibril amount (↓ ~70%) | ||||||
| G6 PAMP (256) | -COOH/ Phenyl groups (28.8%) (−) | 0.01–1 | ↓ elongation rate and fibril amount in a concentration-dependent manner. At peptide/dendrimer ratio 1:0.5 ↓ fibril amount by 70% | Irregular aggregates. Inhibition on the conformational transition to β-sheet at the equimolar ratio of peptide/dendrimer | [126] | ||
| G5 PAMP-OH (128) | -OH/Phenyl groups (28.8%) (+) | 0.1–1 | No effect | No effect at different peptide/dendrimer ratios | [107] | ||
| APD (8) | -SO3− (50%)/CH2CH2CH3 (50%) (−) | 0.125 | ↓ elongation rate and fibril amount to 66.5% | Fibrillar structures | Yes | [127] | |
| 0.2 | ↓ elongation rate and fibril amount to ~60% | Fibrillar structures | |||||
| 1 | ↓ elongation rate and fibril amount to ~10% | Amorphous aggregates and fibrils | Yes (primary murine neuronal cells) | ||||
| 0.75 | ↓ elongation rate and fibril amount to 52% | Amorphous aggregates and few fibrils | |||||
| 4 | Complete inhibition of fibrillation | Amorphous aggregates; Inhibition of transition to β-sheet | Yes (primary murine neuronal cells) | ||||
| SA-D (32) | -SO3− (50%)/CH2CH2CH3 (50%) (−) | 0.125 | ↓ elongation rate and fibril amount to 66.3% | Amorphous aggregates and fibrils | Yes | [127] | |
| 0.333333333 | ↓ elongation rate and fibril amount to 50.4% | Amorphous aggregates and few fibrils | |||||
| 0.5–1 | Complete inhibition of fibrillation | Amorphous aggregates | Yes (primary murine neuronal cells) |
| Dendrimer (No. Terminal Groups) | Terminal Group (Charge) | Type of Assay | Biodistribution & Cellular Uptake | Effect on Inflammation | Refs. |
|---|---|---|---|---|---|
| G4 PAMAM (64) | -NH2 (+) | in vivo/ in vitro | Biodistribution: CP newborn rabbit model • i.p.: only found at the injection site. • i.v.: only present inside blood vessels Cellular uptake: BV2 microglial cells • Dendrimer uptake: 0.093 ± 0.01 pg/cell (resting state) & 0.517 ± 0.01 pg/cell (LPS-activated) • 2-fold lower uptake than neutral G4 PAMAM-OH | In vivo Carrageenan-induced paw edema in Rats • 35.50 ± 1.64% inhibitory activity of paw edema at 16 mg/kg dose, 4 h post-administration Cotton Pellet Test in Rats • Exhibited higher inflammatory inhibition than indomethacin alone Adjuvant-induced Arthritis in Rats • Exhibited higher inflammatory activity than indomethacin alone (25 ± 2.1 vs. 18 ± 0.7, p < 0.05) In vitro • Exhibited COX-1 and COX-2 inhibition and ~50% reduction of the NO production on LPS-activated rat peritoneal macrophages | [160,175,176] |
| -OH (0) | in vivo/ in vitro | Biodistribution: CP newborn rabbit model • i.p.: found within the cells several millimeters. • i.v.: Co-localization with activated microglia in CP kits (but not in healthy age-matched) 4 h post-injection. Extent of dendrimer uptake correlated with the extent of disease Cellular uptake: BV2 microglial cells • Maximal Dendrimer uptake (resting): 0.201 ± 0.02 (pg/cell) • 4.05-fold increase in maximal dendrimer uptake by LPS stimulation (0.814 ± 0.08 pg/cell) | In vivo Carrageenan-induced paw edema in Rats • 31.22 ± 1.58% inhibitory activity towards paw edema at 16 mg/kg dose, 4 h post-administration In vitro • COX-2 inhibition (not COX-1) and ~50% reduction of NO production on LPS-activated rat peritoneal macrophages | [83,160,175,176,177] | |
| G3.5 PAMAM (64) | -COOH (−) | in vivo | Biodistribution: CP newborn rabbit model (i.v.) • i.v.: co-localized with microglial cells 24h post-injection Cellular uptake: BV2 microglial cells • Maximal Dendrimer uptake (resting): 0.234 ± 0.01 pg/cell • 1.72-fold increase in maximal dendrimer uptake by LPS stimulation (0.404 ± 0.01 pg/cell) | In vivo Carrageenan-induced paw edema in Rats • 11.00 ± 1.60% inhibitory activity towards paw edema at 16 mg/kg dose, 4 h post-administration In vitro • ~43% reduction of the NO production on LPS-activated rat peritoneal macrophages. Inhibitory activity was significantly lower than G4 PAMAM-NH2 and G4 PAMAM-OH (p < 0.05) • Did not exhibit COX inhibition on LPS-activated rat peritoneal macrophages | [160,175,176] |
| -COOH/Glucosamine (14%) (−) | in vitro | in vitro • Significant reduction in the release of the pro-inflammatory chemokines (MIP-1α, MIP-1β, IL-8, TNF-α, IL-1β and IL-6) in LPS-activated human PDMC • Significant reduction in the release of the pro-inflammatory chemokines (MIP-1β and TNF-α in LPS-activated human DC and MDM • Inhibited lymphocyte proliferation in the mixed leukocyte reaction and prevent INF-γ production | [159] | ||
| G6 PAMAM (256) | -OH (0) | in vivo/ in vitro | Biodistribution: CP newborn rabbit model (i.v.) • Increased circulation time comparing to G4 • Brain uptake higher than G4 • Extent of dendrimer uptake correlated with the extent of disease Cellular uptake: BV2 microglial cells • Maximal Dendrimer uptake (resting): 0.038 ± 0.03 pg/cell • 6.25-fold increase in maximal dendrimer uptake by LPS stimulation (0.377 ± 0.01 pg/cell) | [175,176] | |
| G3/G4 CCPD (48/96) | -NHEt (+) | in vitro | In vitro • Decreased TNF-α release in LPS-activated BV-2 cells, to the same extent as NAC. • No differences in the anti-inflammatory properties between G3 and G4 • Scavenger capacity of DPPH free radicals • Exhibit reduction capacity of ferric ions. G4 had a stronger reduction capacity than G3 | [24] | |
| PEGOL-60 (60) | -OH (0) | in vivo/ in vitro | Biodistribution: CP newborn rabbit model (i.v.) • 10-fold higher brain uptake than age-matched healthy kits • Co-localization with activated microglia within 1h post-administration • No co-localization with activated microglia in healthy controls | In vitro • Co-treatment with LPS led to a significant downregulation in TNF-α, IL-6, IL-10, and iNOS, and upregulation in CD206, Arg1, IL-4 • Significant reduction in excreted TNF-α and NO in LPS-activated BV2 microglial • Pre-treatment resulted in a significant improvement in cellular viability upon a H2O2 challenge in BV2 microglial cells. | [178] |
| G0/G1 “Click dendrimers” (3 or 6) | -OH (0) | in vitro | In vitro • Reduction of NO release in LPS-activated N9 microglial cells • Inhibition of PGE2 in LPS-activated N9 microglial cells • Higher anti-inflammatory effect of G1 than G0 | [179] | |
| -Acetylene (0) | in vitro | In vitro • Reduction of NO release in LPS-activated N9 microglial cells only at the highest concentration (50 µM) • Inhibition of PGE2 in LPS-activated N9 microglial cells • Lower anti-inflammatory effect of G1 than G0 | [179] | ||
| dPG (64) | -OH (0) | in vivo/ in vitro | Cellular uptake • Increased cellular uptake with increased size in Macrophage-differentiated THP-1 cells • Lower particle uptake than negative dPGS for similar sized particles in Macrophage-differentiated THP-1 cells | in vivo TLR2-/GFAP-luciferase transgenic mice—i.n. with LPS • dPG could not suppress the LPS-activated effects on microglia and astrocytes Mouse Organotypic Hippocampal Brain Slices • Did not prevent spine loss caused by an Aβ insult in vitro • Three-hour pre-treatment did not change the mitochondrial activity, NO nor the cytokine release in LPS-activated N9 neonatal murine microglia cells | [131,164,180,181] |
| -OSO3− (−) | in vivo/ in vitro | Cellular uptake • Selectively taken up by microglial cells and not astrocytes or neurons in Mouse Organotypic Hippocampal Brain Slices • Higher uptake by microglia than astrocytes in mixed mouse cortical cultures • Higher particle uptake than neutral dPG for similar sized particles in macrophage-differentiated THP-1 cells • Increased cellular uptake with increased size in macrophage-differentiated THP-1 cells | in vivo TLR2-/GFAP-luciferase transgenic mice—i.n. with LPS • Reduction of microglial activation (but not astrocyte activation) in a concentration and time-dependent manner Mouse model of complement activation • Pre-treatment reduced C5a generation in a LPS-stimulated mice Mouse contact dermatitis model • Reduced leukocyte extravasation to inflamed tissue in vitro • Act as a scavenger for IL-6 and LCN2 • Directly bind to L-selectin, P-selectin, complement factor C3 and C5 • Three-hour pre-treatment inhibited the reduction of mitochondrial activity of LPS-activated N9 cells in ~25% • Three-hour pre-treatment decreased the release of NO, TNF-α and IL-6, and reduced the nitrosylated proteins in LPS-activated N9cells • Decreased the amount of Aβ internalized by neuroglia in mixed mouse cortical cultures Mouse Organotypic Hippocampal Brain Slices • Three-hour pre-treatment decreased the release of NO, TNF-α and IL-6 in LPS-activated slices • Three-hour pre-treatment prevented spine loss caused by LPS stimulation. • Avoided the morphological changes on postsynaptic dendritic spines of an Aβ (1-42) insult. • Reduced LCN2 production in Aβ-exposed slice, comparing to Aβ (1-42) incubation alone | [131,163,164,180,181] | |
| G1 PPH (10/12) | -N(CH2P(O)(OH)(ONa))2 (ABP) (−) | in vivo/ in vitro | In vivo Mouse arthritis model (IL-1-ra−/−)—i.v. • Completely inhibited inflammation and arthritis at doses of 1 and 10 mg/kg • Decreased serum concentrations of IL-1β, TNF-α, IL-6, and IL-17 and the amount of MMP3 and MMP9 K/BxN serum transfer mouse model—i.v. • Prevented inflammation and arthritis In vitro • Activate human monocytes in an alternative pathway • Human monocytes remained viable longer than control monocytes, underwent phenotypical changes and increased NF-kB. • Skew mice splenocytes towards an anti-inflammatory phenotype, as it increased their production of anti-inflammatory cytokines (IL-4 and IL-10) and reduced the production of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-17, IL-2, INF-γ) | [165,169,182,183,184] | |
| -CH(NHMe)(P(O)(OH)(ONa)) (AMP) (−) | in vivo | In vivo Mouse arthritis model (IL-1-ra−/−)—i.v. • Did not change paw swelling and in arthritis score | [169] | ||
| -COONa | in vitro | In vitro • Lower activation of human monocytes than dendrimers ending with phosphonic acid groups | [182] | ||
| G1 PPH (2,4,6,8,16) | -N(CH2P(O)(OH)(ONa))2 (ABP) (-) | in vivo/ in vitro | In vivo K/BxN serum transfer mouse model—i.v. • Could not prevent inflammation and arthritis. In vitro • G1 PPH (2,4,6): Lower activity towards human monocytes than ABP-12 • G1 PPH (8): Most bioactive dendrimer to activate human monocytes • G1 PPH (16): Similar activity towards human monocytes to ABP-12 | [183,184] | |
| G3/G4 PPH (48/96) | -N(CH2P(O)(OCH3)2)2 (0) | In vivo Mouse air pouch injected with zymosan—i.v. • Reduced the number of migrating cells into the pouch • Decreased NO levels and reduced the iNOS and CD86 expression in infiltrating cells and cells lining the air pouch cavity. CD163 expression was restored in these cells. • Able to modulate M1/M2 ratio in vivo In vitro • Reduced NO, TNF-α and IL-1β release and prevent IL-4 decrease in LPS-activated mouse peritoneal macrophages. G4 > G3. • Reduced iNOS and CD86 expression in LPS-activated mouse peritoneal macrophages G4 > G3 • Prevent macrophages polarization to M1 state and return the M1/M2 balance. • Similar anti-inflammatory properties on monocyte-derived human macrophages | [170] | ||
| G1 Carbosilane (8) | -N(CH2P(O)(OH)(ONa))2 (ABP) (−) | in vitro | In vitro • Good activity in alternatively-activation of human monocytes | [185] | |
| G2 PPI (8) | -N(CH2P(O)(OH)(ONa))2 (ABP) (−) | in vivo/ in vitro | In vivo Mouse arthritis model (IL-1-ra−/−)—i.v. • Did not change paw swelling and in arthritis score In vitro • Did not activate human monocytes | [169,185] | |
| G1/G2 PAMAM (4/8) | -N(CH2P(O)(OH)(ONa))2 (ABP) (−) | in vitro | In vitro • Did not activate human monocytes | [185] | |
| G2 PLL (8) | -N(CH2P(O)(OH)(ONa))2 (ABP) (−) | in vitro | In vitro • Did not activate human monocytes | [185] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, D.A.; Santos, S.D.; Leiro, V.; Pêgo, A.P. Dendrimers and Derivatives as Multifunctional Nanotherapeutics for Alzheimer’s Disease. Pharmaceutics 2023, 15, 1054. https://doi.org/10.3390/pharmaceutics15041054
Moreira DA, Santos SD, Leiro V, Pêgo AP. Dendrimers and Derivatives as Multifunctional Nanotherapeutics for Alzheimer’s Disease. Pharmaceutics. 2023; 15(4):1054. https://doi.org/10.3390/pharmaceutics15041054
Chicago/Turabian StyleMoreira, Débora A., Sofia D. Santos, Victoria Leiro, and Ana P. Pêgo. 2023. "Dendrimers and Derivatives as Multifunctional Nanotherapeutics for Alzheimer’s Disease" Pharmaceutics 15, no. 4: 1054. https://doi.org/10.3390/pharmaceutics15041054