
Developing a Formulation Strategy Coupled with PBPK Modeling and Simulation for the Weakly Basic Drug Albendazole

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Measurement
2.3. Particle Size Distribution Analysis
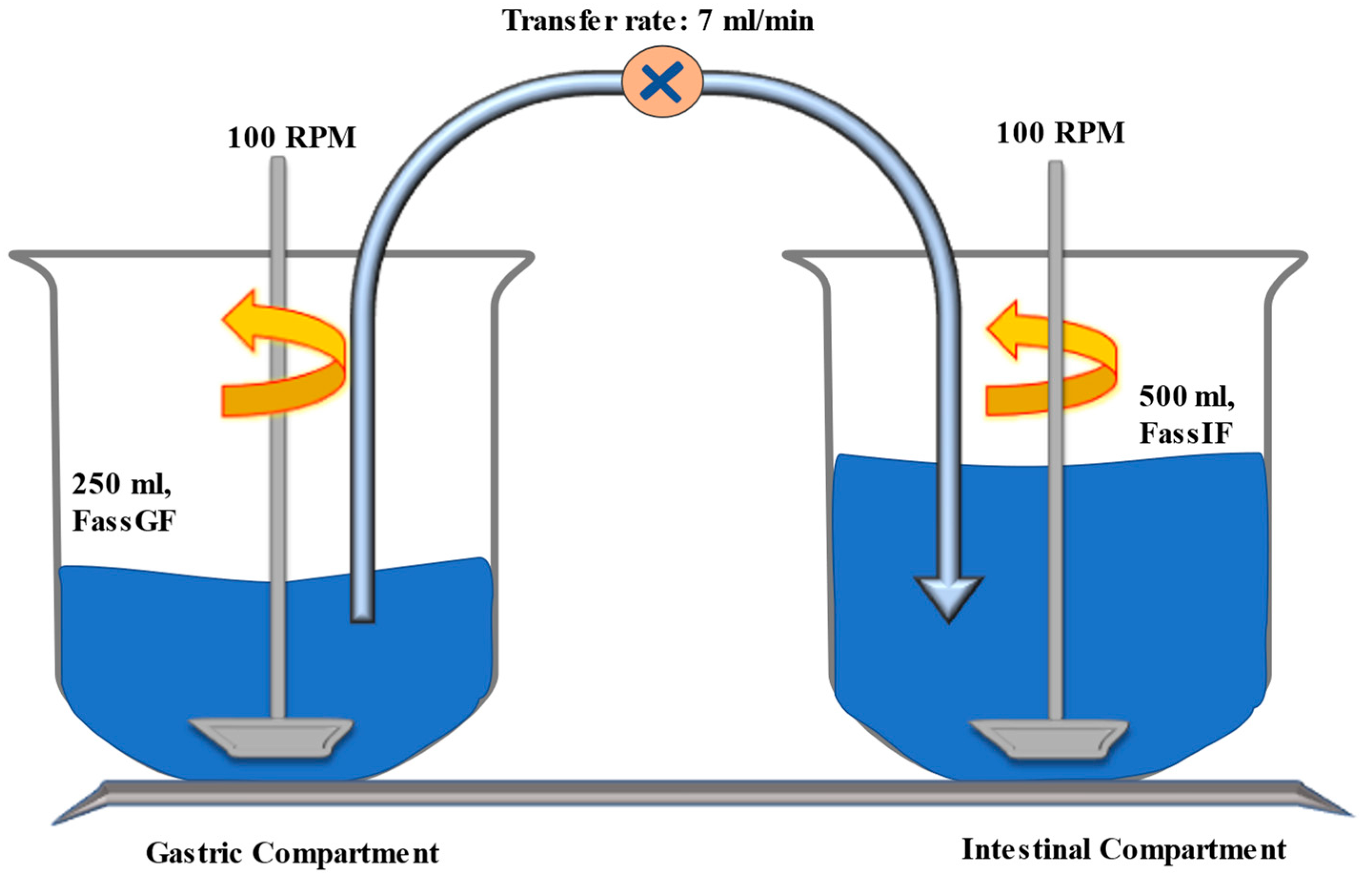
2.4. Transfer System Experiment to Determine Precipitation Kinetic Parameters
2.5. High-Performance Liquid Chromatography (HPLC) Analysis
2.6. PBPK Modeling
2.6.1. Model Development
2.6.2. Model Verification in Healthy Human Volunteer Population
2.6.3. Sensitivity Analysis
3. Results
3.1. pH Solubility Profile of Albendazole
3.2. Particle Size Distribution
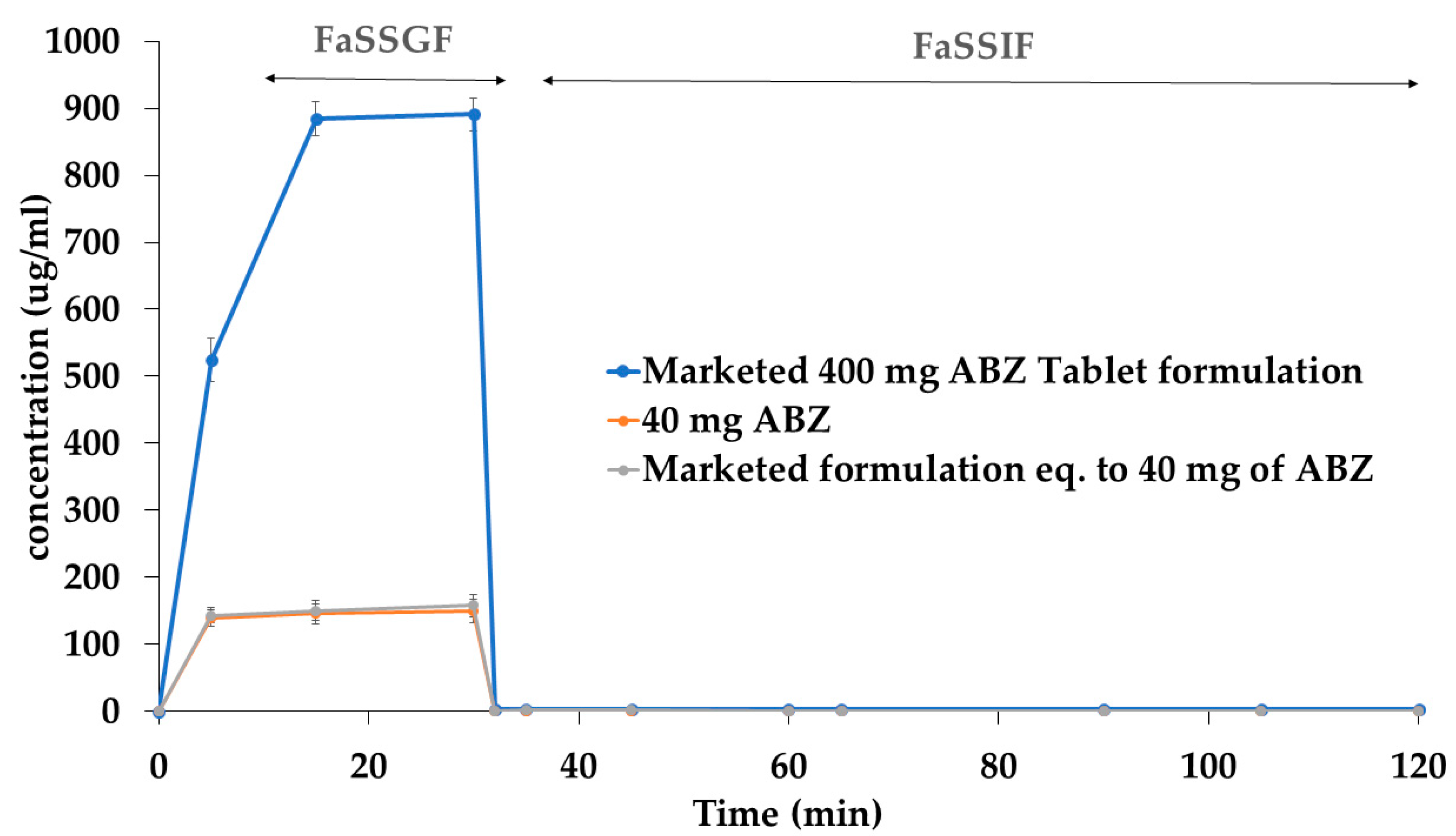
3.3. Precipitation Kinetics
3.4. PBPK Model Verification
3.5. Sensitivity Analysis
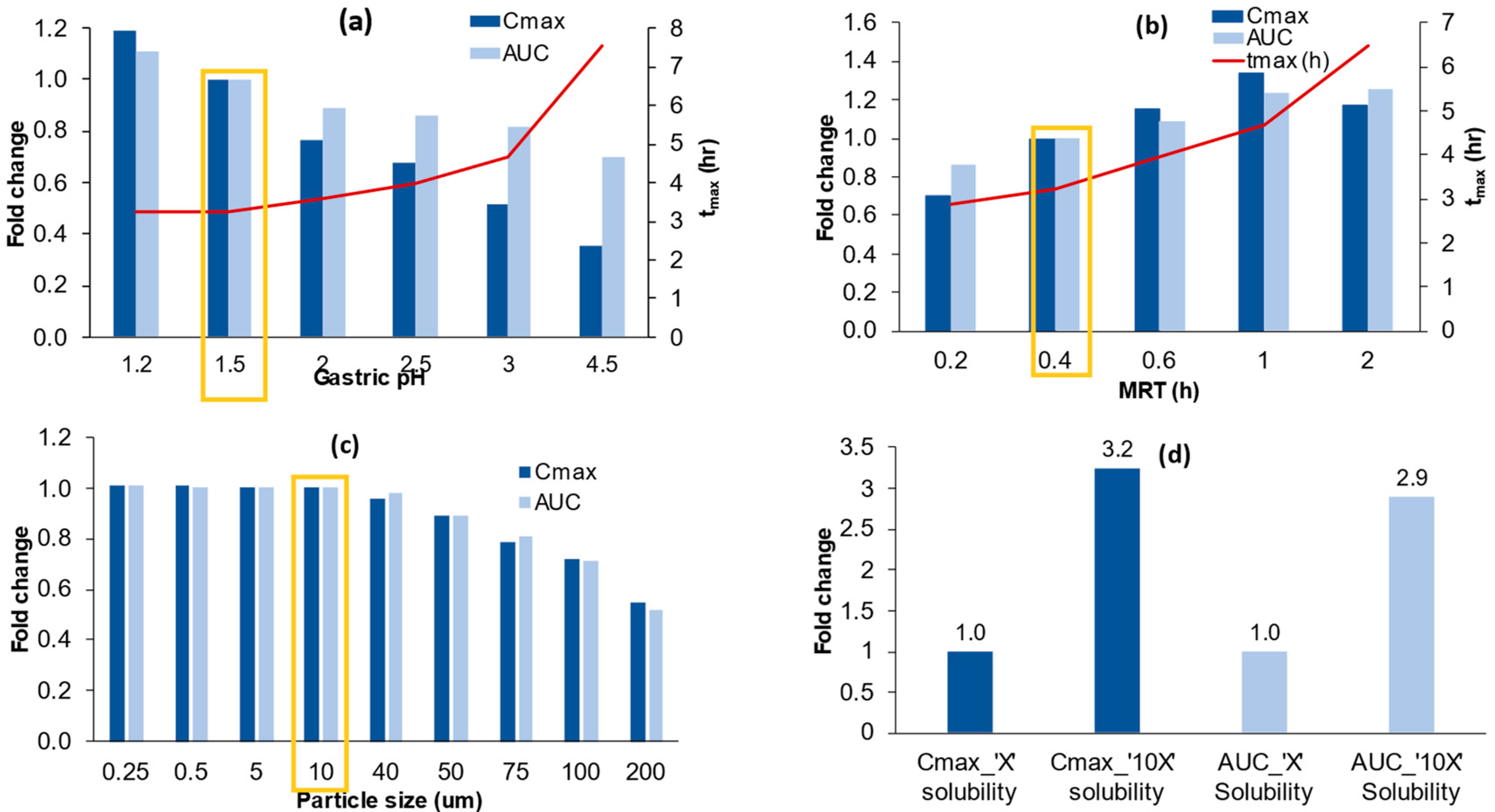
3.5.1. Model Sensitivity to Gastric PH
3.5.2. Model Sensitivity to Gastric Mean Residence Time (MRT)
3.5.3. Model Sensitivity to Particle Size
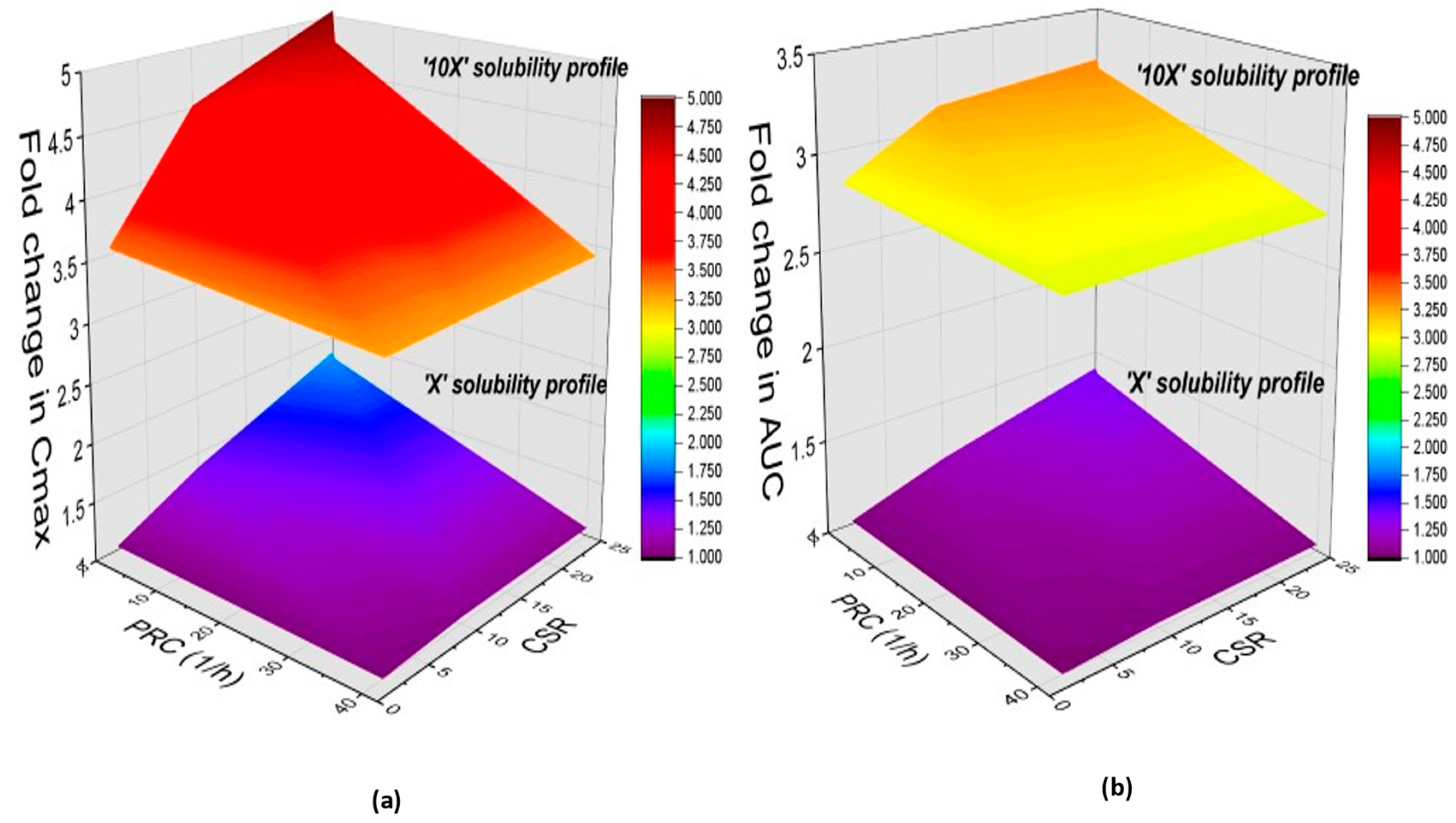
3.5.4. Model Sensitivity to Solubility and the Interplay between Degree of Supersaturation and Precipitation Rate
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovacević, I.; Parojcić, J.; Homsek, I.; Tubić-Grozdanis, M.; Langguth, P. Justification of biowaiver for carbamazepine, a low soluble high permeable compound, in solid dosage forms based on IVIVC and gastrointestinal simulation. Mol. Pharm. 2009, 6, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Idkaidek, N.M.; Najib, N.; Salem, I.; Jilani, J. Physiologically-based IVIVC of azithromycin. Am. J. Pharmacol. Sci. 2014, 2, 100–102. [Google Scholar] [CrossRef] [Green Version]
- Kesisoglou, F.; Xia, B.; Agrawal, N.G. Comparison of Deconvolution-Based and Absorption Modeling IVIVC for Extended Release Formulations of a BCS III Drug Development Candidate. AAPS J. 2015, 17, 1492–1500. [Google Scholar] [CrossRef] [Green Version]
- Balan, G.; Timmins, P.; Greene, D.S.; Marathe, P.H. In vitro-in vivo correlation (IVIVC) models for metformin after administration of modified-release (MR) oral dosage forms to healthy human volunteers. J. Pharm. Sci. 2001, 90, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Min, J.S.; Bae, S.K. Prediction of drug-drug interaction potential using physiologically based pharmacokinetic modeling. Arch. Pharm. Res. 2017, 40, 1356–1379. [Google Scholar] [CrossRef]
- Varma, M.V.; Lai, Y.; Feng, B.; Litchfield, J.; Goosen, T.C.; Bergman, A. Physiologically based modeling of pravastatin transporter-mediated hepatobiliary disposition and drug-drug interactions. Pharm. Res. 2012, 29, 2860–2873. [Google Scholar] [CrossRef]
- Sinha, V.K.; Snoeys, J.; Osselaer, N.V.; Peer, A.V.; Mackie, C.; Heald, D. From preclinical to human--prediction of oral absorption and drug-drug interaction potential using physiologically based pharmacokinetic (PBPK) modeling approach in an industrial setting: A workflow by using case example. Biopharm. Drug Dispos. 2012, 33, 111–121. [Google Scholar] [CrossRef] [Green Version]
- De Buck, S.S.; Mackie, C.E. Physiologically based approaches towards the prediction of pharmacokinetics: In vitro-in vivo extrapolation. Expert. Opin. Drug. Metab. Toxicol. 2007, 3, 865–878. [Google Scholar] [CrossRef]
- Patel, S.; Zhu, W.; Xia, B.; Sharma, N.; Hermans, A.; Ehrick, J.D.; Kesisoglou, F.; Pennington, J. Integration of Precipitation Kinetics from an In Vitro, Multicompartment Transfer System and Mechanistic Oral Absorption Modeling for Pharmacokinetic Prediction of Weakly Basic Drugs. J. Pharm. Sci. 2019, 108, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jin, J.Y.; Mukadam, S.; Malhi, V.; Kenny, J.R. Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: Strategy and approach during the drug discovery phase with four case studies. Biopharm. Drug. Dispos. 2012, 33, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Cristofoletti, R.; Hens, B.; Patel, N.; Esteban, V.V.; Schmidt, S.; Dressman, J. Integrating Drug- and Formulation-Related Properties With Gastrointestinal Tract Variability Using a Product-Specific Particle Size Approach: Case Example Ibuprofen. J. Pharm. Sci. 2019, 108, 3842–3847. [Google Scholar] [CrossRef] [Green Version]
- Thelen, K.; Jantratid, E.; Dressman, J.B.; Lippert, J.; Willmann, S. Analysis of nifedipine absorption from soft gelatin capsules using PBPK modeling and biorelevant dissolution testing. J. Pharm. Sci. 2010, 99, 2899–2904. [Google Scholar] [CrossRef]
- Effinger, A.; O’Driscoll, C.M.; McAllister, M.; Fotaki, N. Predicting budesonide performance in healthy subjects and patients with Crohn’s disease using biorelevant in vitro dissolution testing and PBPK modeling. Eur. J. Pharm. Sci. 2021, 157, 105617. [Google Scholar] [CrossRef]
- Gajera, B.Y.; Shah, D.A.; Dave, R.H. Development of an amorphous nanosuspension by sonoprecipitation-formulation and process optimization using design of experiment methodology. Int. J. Pharm. 2019, 559, 348–359. [Google Scholar] [CrossRef]
- Brough, C.; Williams, R.O., 3rd. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int. J. Pharm. 2013, 453, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Dai, W.G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Kurkov, S.V.; Thorsteinn, L. Cyclodextrins. Int. J. Pharm. 2013, 453, 167–180. [Google Scholar]
- Wagner, C.; Jantratid, E.; Kesisoglou, F.; Vertzoni, M.; Reppas, C.; BDressman, J. Predicting the oral absorption of a poorly soluble, poorly permeable weak base using biorelevant dissolution and transfer model tests coupled with a physiologically based pharmacokinetic model. Eur. J. Pharm. Biopharm. 2012, 82, 127–138. [Google Scholar] [CrossRef]
- Pathak, S.M.; Schaefer, K.J.; Jamei, M.; Turner, D.B. Biopharmaceutic IVIVE-Mechanistic Modeling of Single- and Two-Phase In Vitro Experiments to Obtain Drug-Specific Parameters for Incorporation into PBPK Models. J. Pharm. Sci. 2019, 108, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Pathak, S.M.; Ruff, A.; Kostewicz, E.S.; Patel, N.; Turner, D.B.; Jamei, M. Model-Based Analysis of Biopharmaceutic Experiments to Improve Mechanistic Oral Absorption Modeling: An Integrated in Vitro in Vivo Extrapolation Perspective Using Ketoconazole as a Model Drug. Mol. Pharm. 2017, 14, 4305–4320. [Google Scholar] [CrossRef] [Green Version]
- Kuentz, M. Drug absorption modeling as a tool to define the strategy in clinical formulation development. AAPS J. 2008, 10, 473–479, Erratum in AAPS J. 2009, 11, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesisoglou, F.; Mitra, A. Application of Absorption Modeling in Rational Design of Drug Product Under Quality-by-Design Paradigm. AAPS J. 2015, 17, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Model List of Essential Medicines: 21st List 2019; World Health Organization: Geneva, Switzerland, 2019; License: CC BY-NC-SA 3.0 IGO; Available online: https://apps.who.int/iris/handle/10665/325771 (accessed on 20 November 2022).
- Kourentas, A.; Vertzoni, M.; Symillides, M.; Goumas, K.; Gibbon, R.; Butler, J.; Reppas, C. Effectiveness of supersaturation promoting excipients on albendazole concentrations in upper gastrointestinal lumen of fasted healthy adults. Eur. J. Pharm. Sci. 2016, 91, 11–19. [Google Scholar] [CrossRef]
- Baka, E.; Comer, J.E.; Takács-Novák, K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J. Pharm. Biomed. Anal. 2008, 46, 335–341. [Google Scholar] [CrossRef]
- Zhigang, F.; Patterson, B.R.; Turner, M.E. Modeling particle size distributions by the Weibull distribution function. Mater. Charact. 1993, 31, 177–182. [Google Scholar] [CrossRef]
- Kaur, N.; Thakur, P.S.; Shete, G.; Gangwal, R.; Sangamwar, A.T.; Bansal, A.K. Understanding the Oral Absorption of Irbesartan Using Biorelevant Dissolution Testing and PBPK Modeling. AAPS PharmSciTech 2020, 21, 102. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Wunderlich, M.; Brauns, U.; Becker, R.; Bock, T.; Dressman, J.B. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J. Pharm. Pharmacol. 2004, 56, 43–51. [Google Scholar] [CrossRef]
- Mathias, N.R.; Xu, Y.; Patel, D.; Grass, M.; Caldwell, B.; Jager, C.; Mullin, J.; Hansen, L.; Crison, J.; Saari, A.; et al. Assessing the risk of pH-dependent absorption for new molecular entities: A novel in vitro dissolution test, physicochemical analysis, and risk assessment strategy. Mol. Pharm. 2013, 10, 4063–4073. [Google Scholar] [CrossRef]
- Taupitz, T.; Dressman, J.B.; Klein, S. In vitro tools for evaluating novel dosage forms of poorly soluble, weakly basic drugs: Case example ketoconazole. J. Pharm. Sci. 2013, 102, 3645–3652. [Google Scholar] [CrossRef]
- US Pharmacopeia. National Formulary. In The United States Pharmacopeial Convention; US Pharmacopeia: North Bethesda, MD, USA, 1 May 2012; Volume USP 35-NF 30. [Google Scholar]
- Jamei, M.; Dickinson, G.L.; Rostami-Hodjegan, A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: A tale of ‘bottom-up’ vs ‘top-down’ recognition of covariates. Drug Metab. Pharmacokinet. 2009, 24, 53–75, Erratum in Drug Metab. Pharmacokinet. 2009, 24, 488. [Google Scholar] [CrossRef]
- PubChem. Albendazole Chemical and Physical Properties. 2019. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Albendazole#section=Computed-Properties (accessed on 23 November 2022).
- DrugBank. Albendazole Properties, Albendazole. Available online: http://www.drugbank.ca/drugs/DB00518 (accessed on 23 November 2022).
- Jung, H.L.; Medina, L.; Garcia IFuentes, I.; Moreno-Esparza, R. Biopharmaceutics: Absorption studies of albendazole and some physicochemical properties of the drug and its metabolite albendazole sulphoxide. J. Pharm. Pharmacol. 1998, 50, 43–48. [Google Scholar]
- Rawden, H.C.; Kokwaro, G.O.; Ward, S.A.; Edwards, G. Relative contribution of cytochromes P–450 and flavin–containing monoxygenases to the metabolism of albendazole by human liver microsomes. Br. J. Clin. Pharm. 2000, 49, 313–322. [Google Scholar]
- National Center for Biotechnology Information. 2004–PubChem Compound Summary for CID 2082, Albendazole; PubChem [Internet]; National Library of Medicine (US): Bethesda, MD, USA, 2004. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Albendazole (accessed on 3 January 2023).
- Drugbank. Albendazole Sulfoxide Predicted Properties. Available online: https://www.drugbank.ca/metabolites/DBMET00791 (accessed on 23 November 2022).
- Yamashita, S. Oral Drug Absorption. Ph.D. Thesis, Shetsunan University, Osaka, Japan, 2006. Available online: https://kuscholarworks.ku.edu/handle/1808/1061 (accessed on 23 November 2022).
- Xu, M.; Bhatt, D.K.; Yeung, C.K.; Claw, K.G.; Chaudhry, A.S.; Gaedigk, A.; Pearce, R.E.; Broeckel, U.; Gaedigk, R.; Nickerson, D.A.; et al. Genetic and Nongenetic Factors Associated with Protein Abundance of Flavin-Containing Monooxygenase 3 in Human Liver. J. Pharmacol. Exp. Ther. 2017, 363, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Rathod, D.M.; Patel, K.R.; Mistri, H.N.; Jangid, A.G.; Shrivastav, P.S.; Sanyal, M. Liquid chromatography--tandem mass spectrometry method for simultaneous determination of Rohatgialbendazole and albendazole sulfoxide in human plasma for bioequivalence studies. J. Pharm. Anal. 2016, 6, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, A. WebPlotDigitizer, Version 4.6, Pacifica, CA, USA. Available online: https://automeris.io/WebPlotDigitizer (accessed on 23 November 2022).
- Villaverde, C.; Alvarez, A.I.; Redondo, P.; Voces, J.; Del Estal, J.L.; Prieto, J.G. Small intestinal sulphoxidation of albendazole. Xenobiotica 1995, 25, 433–441. [Google Scholar] [CrossRef]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Lennernäs, H.; Aarons, L.; Augustijns, P.; Beato, S.; Bolger, M.; Box, K.; Brewster, M.; Butler, J.; Dressman, J.; Holm, R.; et al. Oral biopharmaceutics tools–time for a new initiative–an introduction to the IMI project OrBiTo. Eur. J. Pharm. Sci. 2014, 57, 292–299. [Google Scholar] [CrossRef]
- Tomilo, D.L.; Smith, P.F.; Ogundele, A.B.; Difrancesco, R.; Berenson, C.S.; Eberhardt, E.; Bednarczyk, E.; Morse, G.D. Inhibition of atazanavir oral absorption by lansoprazole gastric acid suppression in healthy volunteers. Pharmacotherapy 2006, 26, 341–346. [Google Scholar] [CrossRef]
- Zhuang, X.; Lu, C. PBPK modeling and simulation in drug research and development. Acta Pharm. Sin. B 2016, 6, 430–440. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, P.J.; Litou, C.; Box, K.J.; Dressman, J.B.; Kostewicz, E.S.; Kuentz, M.; Reppas, C. In vitro methods to assess drug precipitation in the fasted small intestine—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 536–556. [Google Scholar] [CrossRef] [Green Version]
- Russell, T.L.; Berardi, R.R.; Barnett, J.L.; O’Sullivan, T.L.; Wagner, J.G.; Dressman, J.B. pH-related changes in the absorption of dipyridamole in the elderly. Pharm. Res. 1994, 11, 136–143. [Google Scholar] [CrossRef]
- Krishna, G.; Moton, A.; Ma, L.; Medlock, M.M.; McLeod, J. Pharmacokinetics and absorption of posaconazole oral suspension under various gastric conditions in healthy volunteers. Antimicrob. Agents Chemother. 2009, 53, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Po, H.N.; Senozan, N.M. The Henderson-Hasselbalch equation: Its history and limitations. J. Chem. Educ. 2001, 78, 1499. [Google Scholar] [CrossRef]
- Badawy, S.I.; Gray, D.B.; Zhao, F.; Sun, D.; Schuster, A.E.; Hussain, M.A. Formulation of solid dosage forms to overcome gastric pH interaction of the factor Xa inhibitor, BMS-561389. Pharm. Res. 2006, 23, 989–996. [Google Scholar] [CrossRef]
- Mitra, A.; Kesisoglou, F. Impaired drug absorption due to high stomach pH: A review of strategies for mitigation of such effect to enable pharmaceutical product development. Mol. Pharm. 2013, 10, 3970–3979. [Google Scholar] [CrossRef]
- Chavanpatil, M.D.; Jain, P.; Chaudhari, S.; Shear, R.; Vavia, P.R. Novel sustained release, swellable and bioadhesive gastroretentive drug delivery system for ofloxacin. Int. J. Pharm. 2006, 316, 86–92. [Google Scholar] [CrossRef]
- Pawar, H.A.; Gharat, P.R.; Dhavale, R.V.; Joshi, P.R.; Rakshit, P.P. Development and evaluation of gastroretentive floating tablets of an antihypertensive drug using hydrogenated cottonseed oil. ISRN Pharm. 2013, 2013, 137238. [Google Scholar] [CrossRef]
- Sawicki, W. Pharmacokinetics of verapamil and norverapamil from controlled release floating pellets in humans. Eur. J. Pharm. Biopharm. 2002, 53, 29–35. [Google Scholar] [CrossRef]
- Li, Z.; Xu, H.; Li, S.; Li, Q.; Zhang, W.; Ye, T.; Yang, X.; Pan, W. A novel gastro-floating multiparticulate system for dipyridamole (DIP) based on a porous and low-density matrix core: In vitro and in vivo evaluation. Int. J. Pharm. 2014, 461, 540–548. [Google Scholar] [CrossRef]
- Patil, G.B.; Singh, S.S.; Ramani, K.P.; Chatap, V.K.; Deshmukh, P.K. Design and development of novel dual-compartment capsule for improved gastroretention. ISRN Pharm. 2013, 2013, 752471. [Google Scholar] [CrossRef]
- Shah, H.G.; Rathod, V.; Basim, P.; Gajera, B.; Dave, R.H. Understanding the Impact of Multi-factorial Composition on Efficient Loading of the Stable Ketoprofen Nanoparticles on Orodispersible Films Using Box-Behnken Design. J. Pharm. Sci. 2022, 111, 1451–1462. [Google Scholar] [CrossRef]
- Stillhart, C.; Parrott, N.J.; Lindenberg, M.; Chalus, P.; Bentley, D.; Szepes, A. Characterising Drug Release from Immediate-Release Formulations of a Poorly Soluble Compound, Basmisanil, Through Absorption Modelling and Dissolution Testing. AAPS J. 2017, 19, 827–836. [Google Scholar] [CrossRef]
- Wang, J.; Flanagan, D.R. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory J. Pharm. Sci. 1999, 88, 731–738. [Google Scholar] [CrossRef]
- Hintz, R.J.; Johnson, K.C. The effect of particle size distribution on dissolution rate and oral absorption. Int. J. Pharm. 1989, 51, 9–17. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Wu, Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008, 10, 516–525, Erratum in AAPS J. 2009, 11, 31. [Google Scholar] [CrossRef]
- Parrott, N.; Hainzl, D.; Scheubel, E.; Krimmer, S.; Boetsch, C.; Guerini, E.; Martin-Facklam, M. Physiologically based absorption modelling to predict the impact of drug properties on pharmacokinetics of bitopertin. AAPS J. 2014, 16, 1077–1084. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Pollock-Dove, C.; Dong, L.C.; Chen, J.; Creasey, A.A.; Dai, W.G. Enhanced bioavailability of a poorly water-soluble weakly basic compound using a combination approach of solubilization agents and precipitation inhibitors: A case study. Mol. Pharm. 2012, 9, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Vora, C.; Patadia, R.; Mittal, K.; Mashru, R. Preparation and characterization of dipyridamole solid dispersions for stabilization of supersaturation: Effect of precipitation inhibitors type and molecular weight. Pharm. Dev. Technol. 2016, 21, 847–855. [Google Scholar] [CrossRef]
- Sugawara, M.; Kadomura, S.; He, X.; Takekuma, Y.; Kohri, N.; Miyazaki, K. The use of an in vitro dissolution and absorption system to evaluate oral absorption of two weak bases in pH-independent controlled-release formulations. Eur. J. Pharm. Sci. 2005, 26, 1–8. [Google Scholar] [CrossRef]
- Rathod, V.R.; Shah, D.A.; Dave, R.H. Systematic implementation of quality-by-design (QbD) to develop NSAID-loaded nanostructured lipid carriers for ocular application: Preformulation screening studies and statistical hybrid-design for optimization of variables. Drug Dev. Ind. Pharm. 2020, 46, 443–455. [Google Scholar] [CrossRef]
- Patel, V.D.; Rathod, V.; Haware, R.V.; Stagner, W.C. Optimized L-SNEDDS and spray-dried S-SNEDDS using a linked QbD-DM3 rational design for model compound ketoprofen. Int. J. Pharm. 2023, 631, 122494. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Albendazole (Substrate) | |||
|---|---|---|---|
| Physiochemical Properties | Value | Ref/Comments | |
| Molecular weight (g/mol) | 265.33 | [33] | |
| Log Po: w | 2.7 | [34] | |
| pKa (Ampholyte) | |||
| pKa1 | 10.26 | [35] | |
| pKa2 | 2.8 | [35] | |
| B/P (User input) | 0.55 | Default | |
| fu | 0.156 | Predicted | |
| Absorption | |||
| Model (ADAM) | |||
| fuGut | 1 | Predicted | |
| Permeability parameters | |||
| Peff,man (10–4 cm/s) | 8.03 | Predicted | |
| Formulation parameters | |||
| Solid Formulation (Diffusion layer Model) | |||
| Include bile salt-medicated component (Km:w) | Neutral: 2.924 | Experimental (SIVA) | |
| Ion: 5.423 | |||
| Particle size distribution (polydispersed) (µm) | Weibull Distribution | Alpha: 1.014 Beta: 8.732 | Experimental |
| Mean Radius (µm) | 7.52 | ||
| Intrinsic solubility (mg/mL) | 0.012 | Experimental (SIVA) | |
| Critical Supersaturation Ratio | 1 | Experimental | |
| Precipitation rate constant | 1000 (max. value) | ||
| Distribution | |||
| Full PBPK model Vss (L/kg) | 2.59 | Predicted | |
| Elimination | |||
| Enzyme Kinetics CYP3A4 | |||
| Vmax (pmol/min/pmol) | 369 | ||
| km (µM) | 10.1 | [36] | |
| FMO3 | |||
| Vmax (pmol/min/pmol) | 1103 | ||
| km (µM) | 9.6 | [36] | |
| Gut lumen Clint (µL/h/g) of the total luminal content | 2000 | Optimized | |
| Albendazole sulfoxide (Primary metabolite) | |||
| Molecular weight (g/mol) | 284.35 | [37] | |
| Log Po: w | 1.17 | [38] | |
| pka (Ampholyte) | |||
| pka1 | 9.79 | [35] | |
| pka2 | 0.2 | ||
| B/P (User input) | 0.55 | Default | |
| fu | 0.3 | Literature | |
| Absorption | |||
| Model (ADAM) | |||
| fuGut | 0.36 | Predicted | |
| Peff,man (10–4 cm/s) | 4.13 | Predicted | |
| Distribution | |||
| Minimal PBPK Vss (L/kg) | 2.3 | Optimized | |
| Elimination | |||
| Oral clearance (L/h) | 40 | Optimized | |
| pH/Media | Solubility (mg/mL) |
|---|---|
| pH 1.2 HCl buffer | 0.515 ± 0.045 |
| pH 2.0 HCl buffer | 0.025 ± 0.005 |
| pH 4.5 Acetate buffer | 0.008 ± 0.002 |
| pH 6.8 Phosphate buffer | 0.005 ± 0.001 |
| FaSSGF (pH 1.6) | 0.146 ± 0.012 |
| FaSSIF (pH 6.5) | 0.008 ± 0.001 |
| FeSSIF (pH 5.0) | 0.021 ± 0.004 |
| Albendazole | Albendazole Sulfoxide | |
|---|---|---|
| Cmax (Observed) (ng/mL) | 9.55 | 193.5 |
| Cmax (Predicted) (ng/mL) | 11.2 | 195.1 |
| Fold error * | 1.16 | 1.01 |
| AUC0–∞ (observed) (ng/mL × h) | 53.0 | 3475 |
| AUC0–∞ (predicted) (ng/mL × h) | 59.7 | 3405 |
| Fold error * | 1.13 | 0.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, H.; Shah, K.; Gajera, B.; Dave, R.H.; Taft, D.R. Developing a Formulation Strategy Coupled with PBPK Modeling and Simulation for the Weakly Basic Drug Albendazole. Pharmaceutics 2023, 15, 1040. https://doi.org/10.3390/pharmaceutics15041040
Shah H, Shah K, Gajera B, Dave RH, Taft DR. Developing a Formulation Strategy Coupled with PBPK Modeling and Simulation for the Weakly Basic Drug Albendazole. Pharmaceutics. 2023; 15(4):1040. https://doi.org/10.3390/pharmaceutics15041040
Chicago/Turabian StyleShah, Harsh, Kushal Shah, Bhavin Gajera, Rutesh H. Dave, and David R. Taft. 2023. "Developing a Formulation Strategy Coupled with PBPK Modeling and Simulation for the Weakly Basic Drug Albendazole" Pharmaceutics 15, no. 4: 1040. https://doi.org/10.3390/pharmaceutics15041040