
Does Green Exfoliation of Graphene Produce More Biocompatible Structures?
 ,
,  ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Synthesis of Bio-Graphene (bG)
2.3. Synthesis of Chemical-Graphene (cG)
2.4. Cell Lines
2.5. Cell Viability Assay
2.6. Clonogenic Assay
2.7. Measurement of Reactive Oxygen Species (ROS) Production
2.8. Detection of Apoptosis
2.9. Cell Cycle Analysis
2.10. Western Blotting Analysis
2.11. Statistical Analysis
3. Results and Discussion
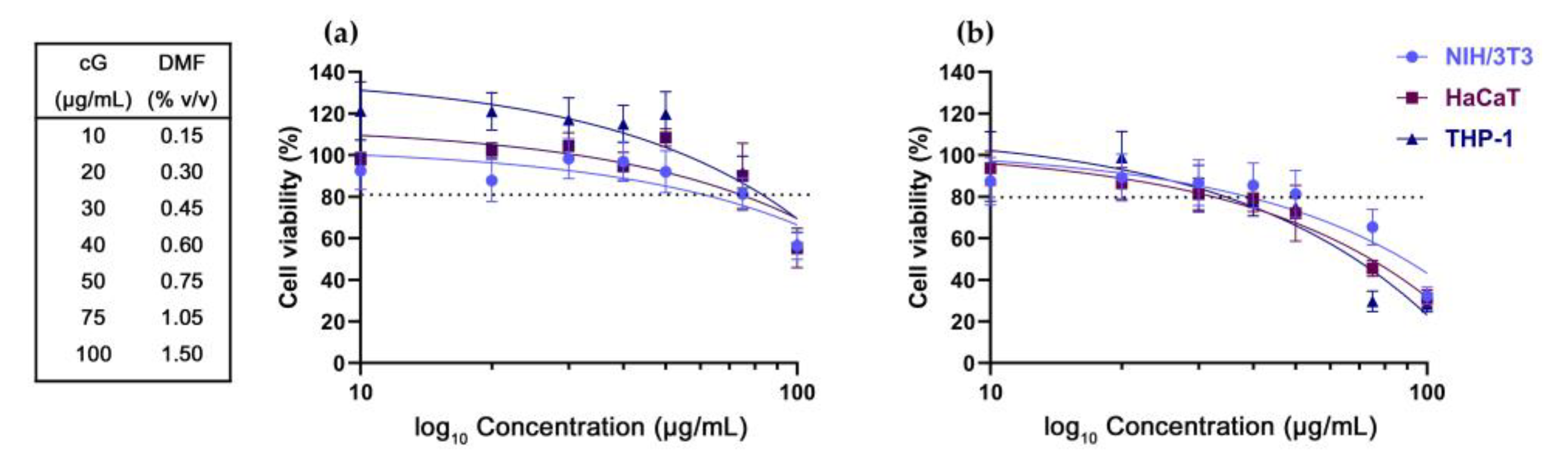
3.1. DMF Toxicity
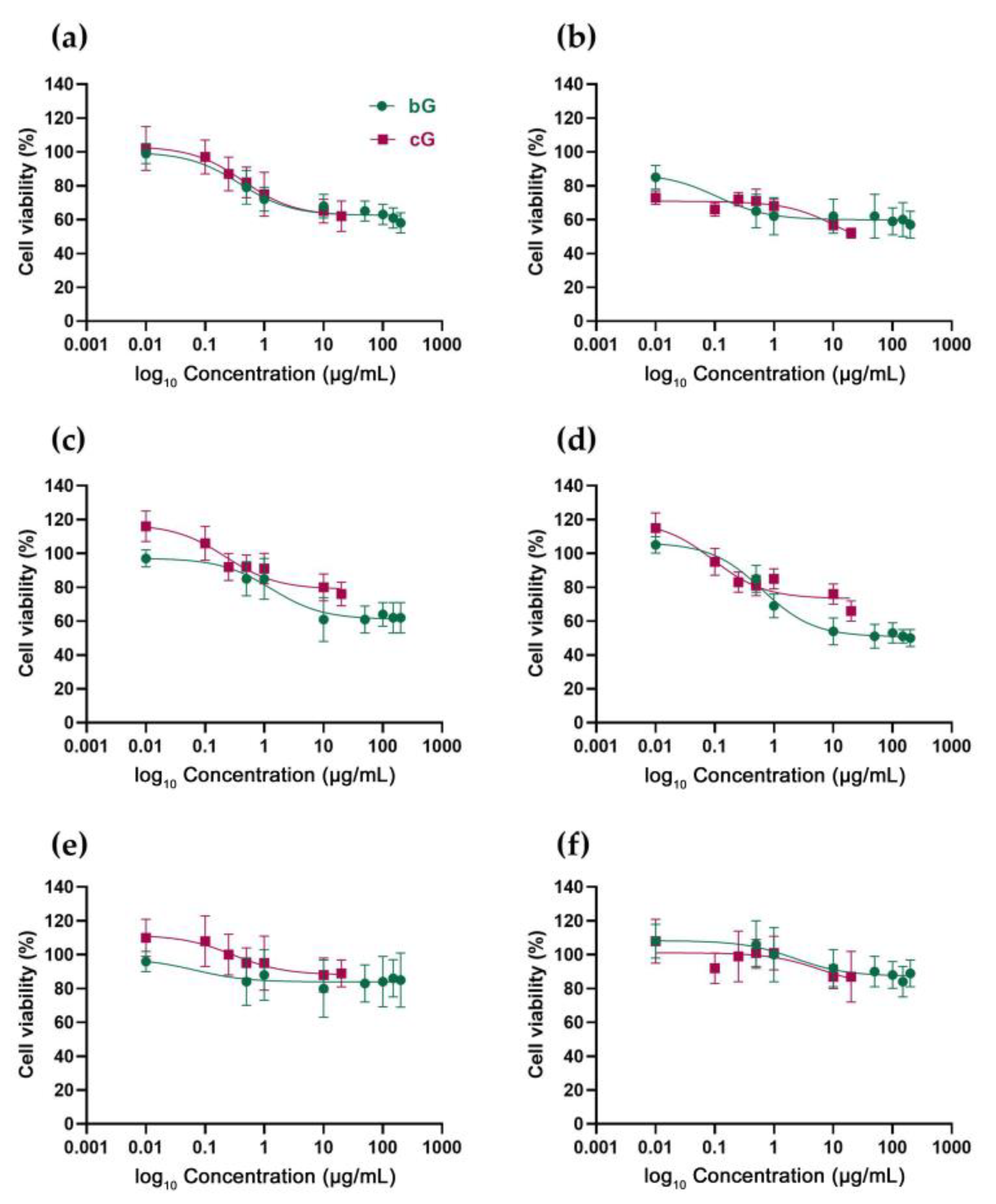
3.2. In Vitro Toxicity against NIH/3T3, HaCaT and THP-1 Cells
3.3. Ability of NIH/3T3 and HaCat Cells to Form Colonies
3.4. Intracellular ROS Production in NIH/3T3, HaCaT and THP-1 Cells
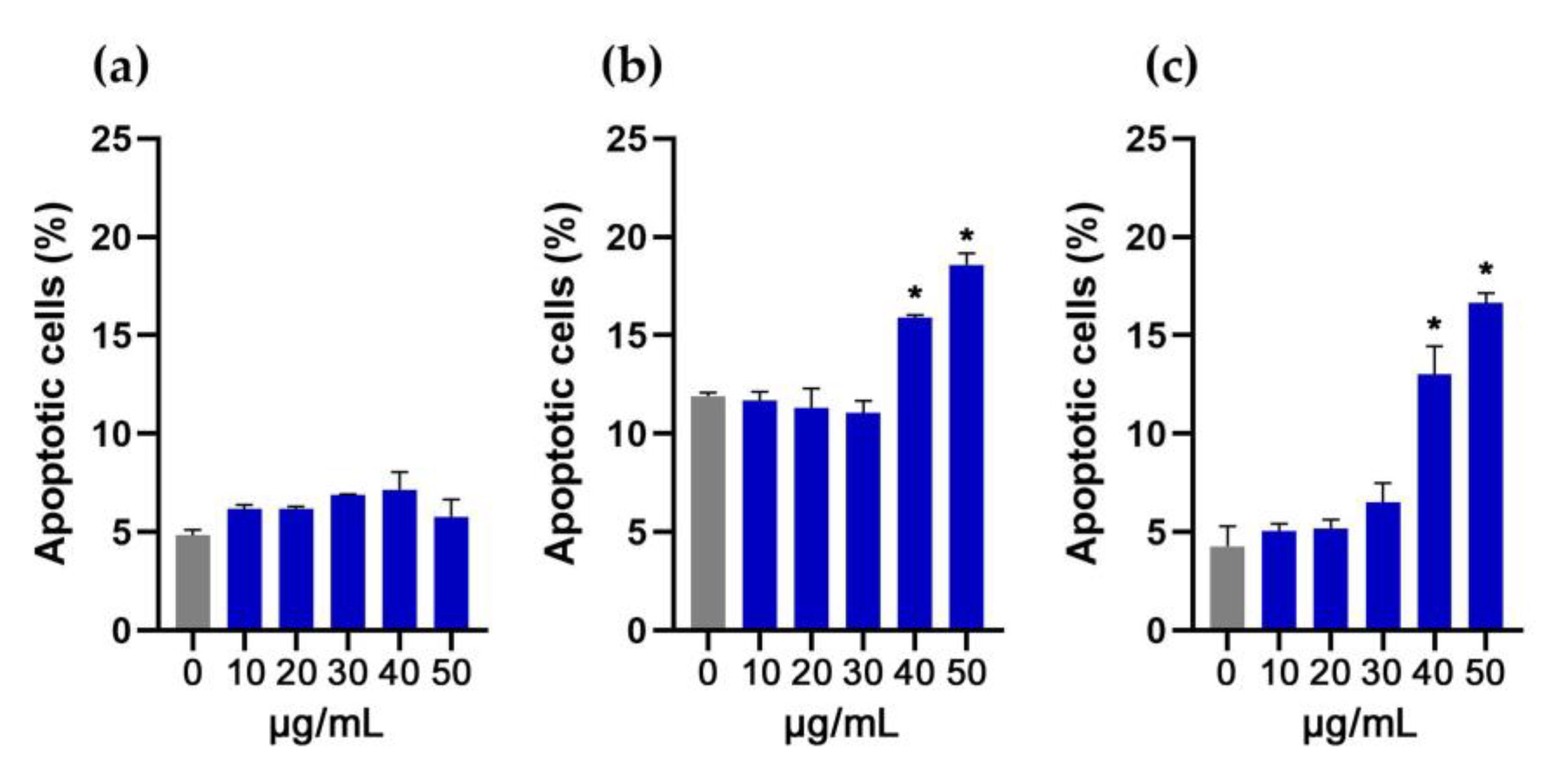
3.5. Evaluation of Apoptosis in NIH/3T3, HaCaT and THP-1 Cells
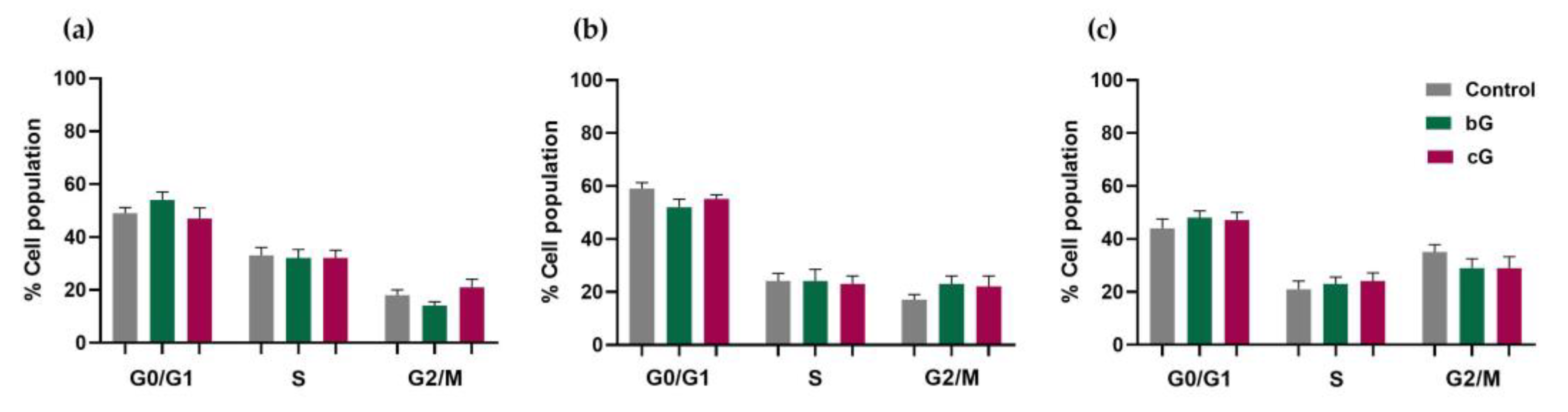
3.6. Cell Cycle Arrest
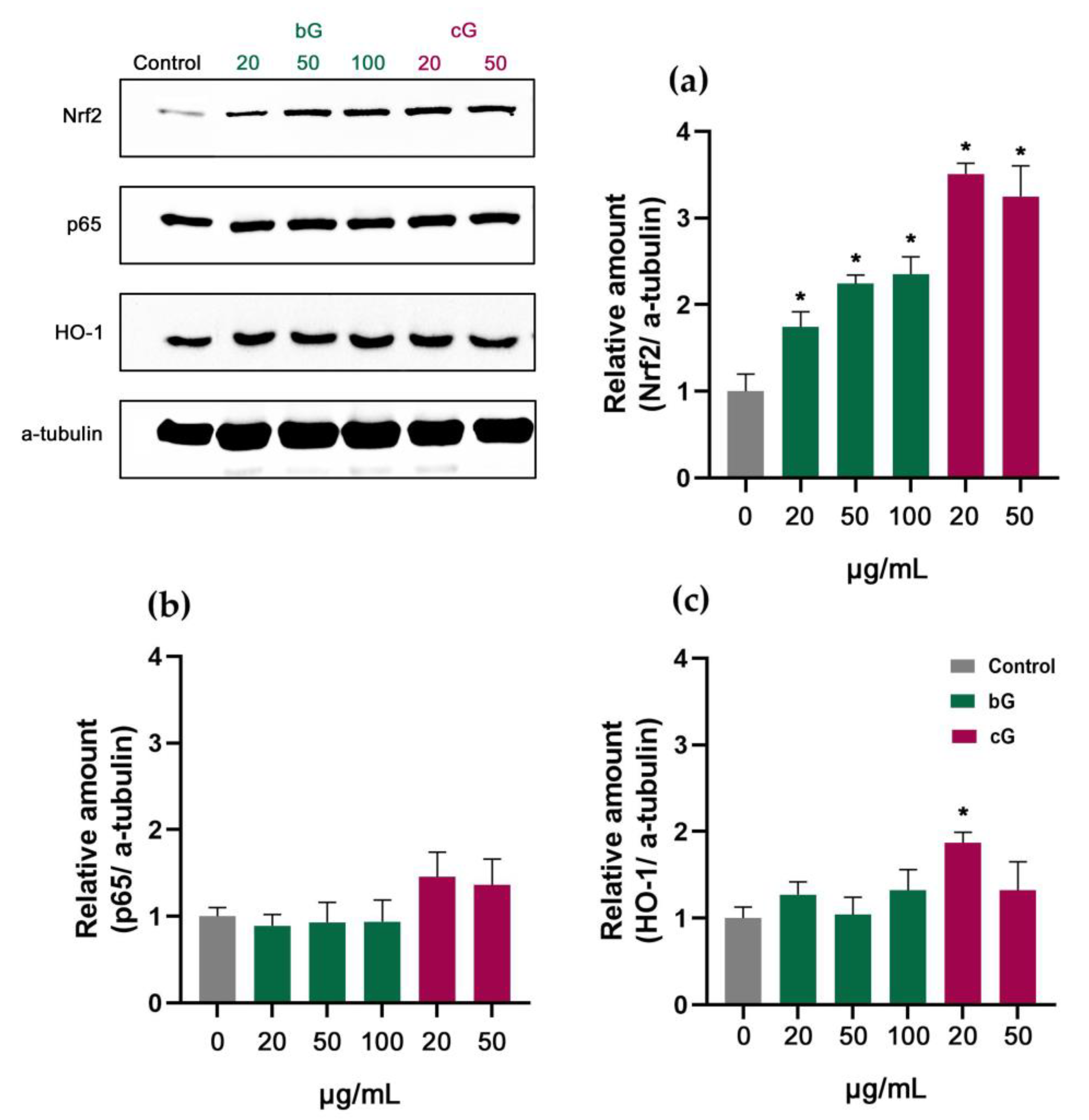
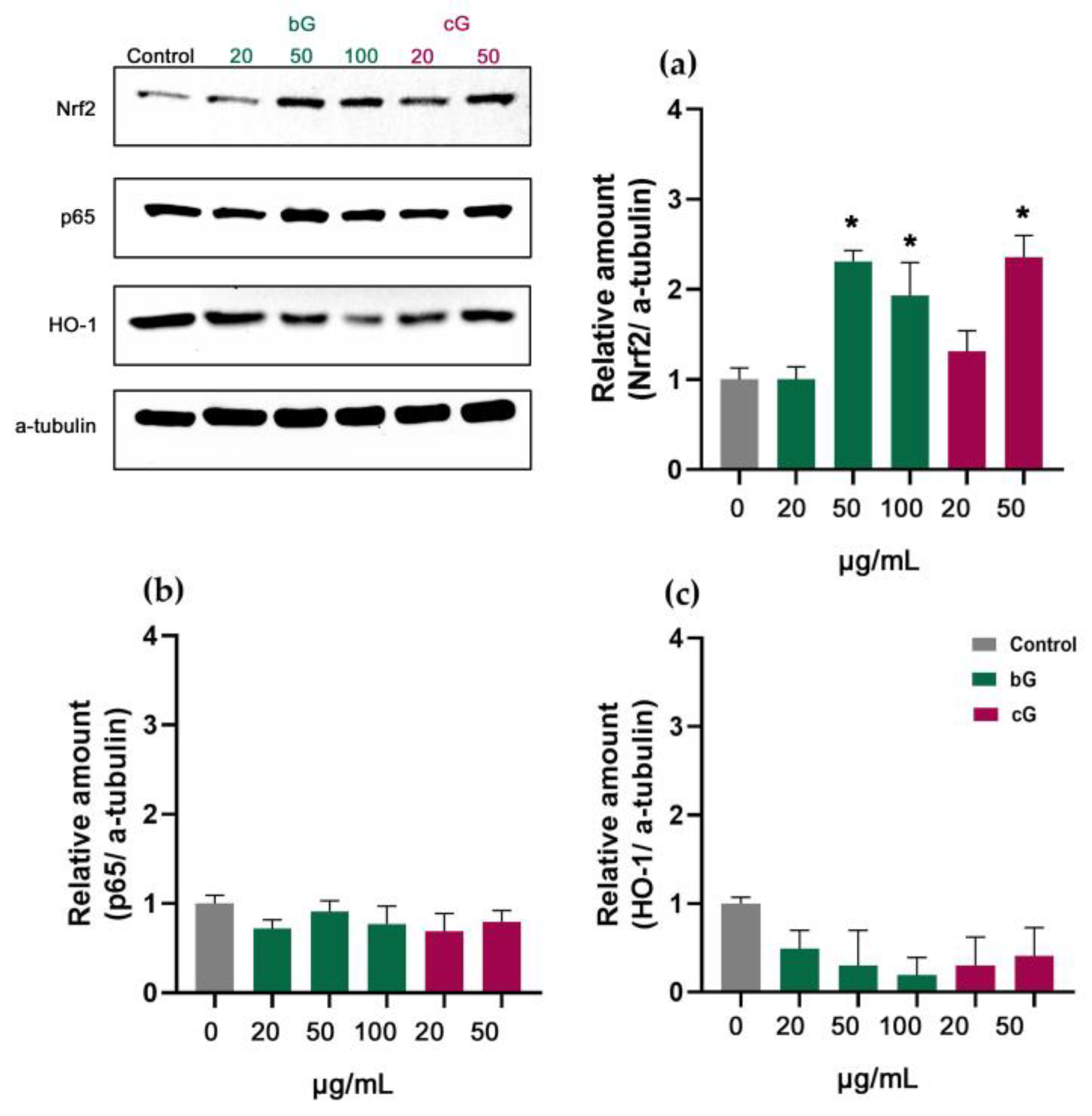
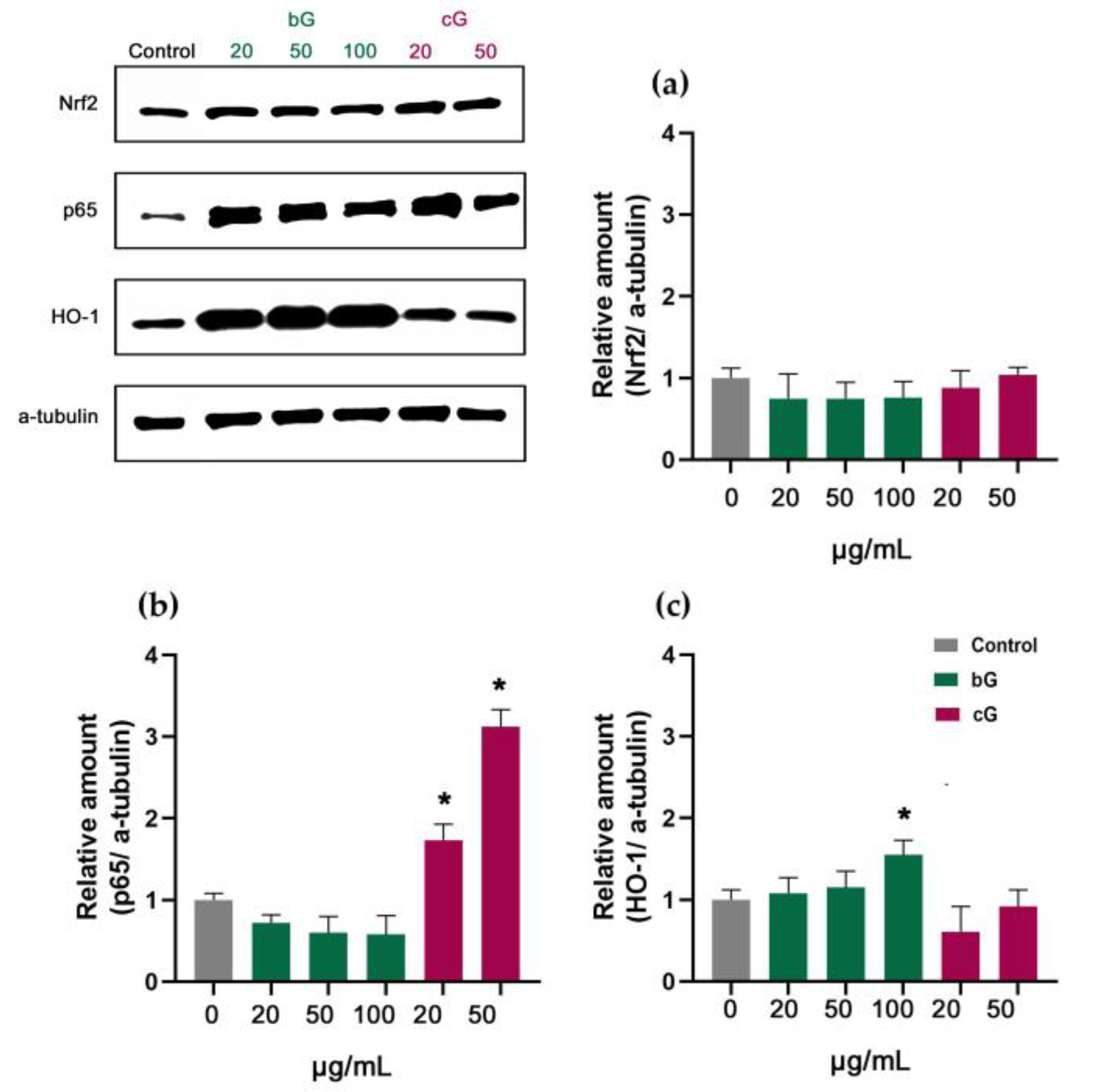
3.7. Nrf2/HO-1 Signaling Pathway in NIH/3T3, HaCaT and THP-1 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, R.; Militky, J. Carbon-Based Nanomaterials. Nanotechnol. Text. Theory Appl. 2018, 163–179. [Google Scholar] [CrossRef]
- Maiti, D.; Tong, X.; Mou, X.; Yang, K. Carbon-Based Nanomaterials for Biomedical Applications: A Recent Study. Front. Pharmacol. 2019, 9, 1401. [Google Scholar] [CrossRef] [Green Version]
- Magne, T.M.; de Oliveira Vieira, T.; Alencar, L.M.R.; Junior, F.F.M.; Gemini-Piperni, S.; Carneiro, S.V.; Fechine, L.M.U.D.; Freire, R.M.; Golokhvast, K.; Metrangolo, P.; et al. Graphene and Its Derivatives: Understanding the Main Chemical and Medicinal Chemistry Roles for Biomedical Applications; Springer: Berlin/Heidelberg, Germany, 2022; Volume 12, ISBN 0123456789. [Google Scholar]
- Jampilek, J.; Kralova, K. Advances in Drug Delivery Nanosystems Using Graphene-based Materials and Carbon Nanotubes. Materials 2021, 14, 1059. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jeong, B. Graphene-Based Nanomaterials and Their Applications in Biosensors. Adv. Exp. Med. Biol. 2018, 1064, 61–71. [Google Scholar] [CrossRef]
- Papanikolaou, E.; Simos, Y.V.; Spyrou, K.; Tzianni, E.I.; Vezyraki, P.; Tsamis, K.; Patila, M.; Tigas, S.; Prodromidis, M.I.; Gournis, D.P.; et al. Is Graphene the Rock upon Which New Era Continuous Glucose Monitors Could Be Built? Exp. Biol. Med. 2022, 1–12. [Google Scholar] [CrossRef]
- Mandal, T.K.; Lee, Y.R.; Parvin, N. Red Phosphorus Decorated Graphene Oxide Nanosheets: Label-Free DNA Detection. Biomater. Sci. 2020, 8, 125–131. [Google Scholar] [CrossRef]
- Ghanbarzadeh, S.; Hamishehkar, H. Application of Graphene and its Derivatives in Cancer Diagnosis and Treatment. Drug Res. Georg. Thieme Verlag. 2017, 67, 681–687. [Google Scholar] [CrossRef] [PubMed]
- de Melo-Diogo, D.; Lima-Sousa, R.; Alves, C.G.; Costa, E.C.; Louro, R.O.; Correia, I.J. Functionalization of Graphene Family Nanomaterials for Application in Cancer Therapy. Colloids Surf. B Biointerfaces 2018, 171, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xi, Y.; Weng, Y. Progress in the Development of Graphene-Based Biomaterials for Tissue Engineering and Regeneration. Materials 2022, 15, 2164. [Google Scholar] [CrossRef]
- Lin, J.; Chen, X.; Huang, P. Graphene-Based Nanomaterials for Bioimaging Graphical Abstract HHS Public Access. Adv. Drug Deliv. Rev. 2016, 105, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.; Geim, A.; Morozov, S.; Jiang, D.; Zhang, Y.; Dubonos, S.; Grigorieva, I.; Firsov, A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Parvin, N.; Kumar, V.; Joo, S.W.; Park, S.S.; Mandal, T.K. Recent Advances in the Characterized Identification of Mono-to-Multi-Layer Graphene and Its Biomedical Applications: A Review. Electronics 2022, 11, 3345. [Google Scholar] [CrossRef]
- Xu, Y.; Cao, H.; Xue, Y.; Li, B.; Cai, W. Liquid-Phase Exfoliation of Graphene: An Overview on Exfoliation Media, Techniques, and Challenges. Nanomaterials 2018, 8, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooil, S.P.; Song, F.; Williams, G.T.; Roberts, O.R.; Langstaff, D.P.; Jørgensen, B.; Høydalsvik, K.; Breiby, D.W.; Wahlström, E.; Evans, D.A.; et al. Iron-Mediated Growth of Epitaxial Graphene on SiC and Diamond. Carbon 2012, 50, 5099–5105. [Google Scholar] [CrossRef]
- Fernandes, J.; Nemala, S.S.; De Bellis, G.; Capasso, A. Green Solvents for the Liquid Phase Exfoliation Production of Graphene: The Promising Case of Cyrene. Front. Chem. 2022, 10, 878799. [Google Scholar] [CrossRef]
- Ahadian, S.; Estili, M.; Surya, V.J.; Ramón-Azcón, J.; Liang, X.; Shiku, H.; Ramalingam, M.; Matsue, T.; Sakka, Y.; Bae, H.; et al. Facile and Green Production of Aqueous Graphene Dispersions for Biomedical Applications. Nanoscale 2015, 7, 6436–6443. [Google Scholar] [CrossRef]
- Williams, D.F. A Model for Biocompatibility. J. Biomed. Eng. 1989, 11, 185–191. [Google Scholar] [CrossRef]
- Yao, J.; Wang, H.; Chen, M.; Yang, M. Recent Advances in Graphene-Based Nanomaterials: Properties, Toxicity and Applications in Chemistry, Biology and Medicine. Microchim. Acta 2019, 186, 395. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Li, Y.; Tjong, S.C. Graphene Nanomaterials: Synthesis, Biocompatibility, and Cytotoxicity. Int. J. Mol. Sci. 2018, 19, 3564. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Bao, S.; Wang, X. Applications of Graphene-Based Materials in Sensors: A Review. Micromachines 2022, 13, 184. [Google Scholar] [CrossRef]
- Zhang, M.; Liao, C.; Mak, C.H.; You, P.; Mak, C.L.; Yan, F. Highly Sensitive Glucose Sensors Based on Enzyme-Modified Whole-Graphene Solution-Gated Transistors. Sci. Rep. 2015, 5, 8311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatzoglou, C.; Patila, M.; Giannakopoulou, A.; Spyrou, K.; Yan, F.; Li, W.; Chalmpes, N.; Polydera, A.C.; Rudolf, P.; Gournis, D.; et al. Development of a Multi-Enzymatic Biocatalytic System through Immobilization on High Quality Few-Layer Bio-Graphene. Nanomaterials 2023, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Gengler, R.Y.N.; Spyrou, K.; Rudolf, P. A Roadmap to High Quality Chemically Prepared Graphene. J. Phys. D Appl. Phys. 2010, 43, 374015. [Google Scholar] [CrossRef] [Green Version]
- Staudenmaier, L. Verfahren zur Darstellung der Graphitsäure. Ber. Dtsch. Chem. Ges. 1898, 31, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Bourlinos, A.B.; Gournis, D.; Petridis, D.; Szabó, T.; Szeri, A.; Dékány, I. Graphite Oxide: Chemical Reduction to Graphite and Surface Modification with Primary Aliphatic Amines and Amino Acids. Langmuir 2003, 19, 6050–6055. [Google Scholar] [CrossRef]
- Cai, D.; Song, M.; Xu, C. Highly Conductive Carbon-Nanotube/Graphite-Oxide Hybrid Films. Adv. Mater. 2008, 20, 1706–1709. [Google Scholar] [CrossRef]
- Loeffler, H.; Jonitz-Heincke, A.; Peters, K.; Mueller-Hilke, B.; Fiedler, T.; Bader, R.; Klinder, A. Comparison of Inflammatory Effects in THP-1 Monocytes and Macrophages after Exposure to Metal Ions. Materials 2020, 13, 1150. [Google Scholar] [CrossRef] [Green Version]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices—Part 5: Tests for in Vitro Cytotoxicity. ISO: Geneva, Switzerland, 2009.
- Geissmann, Q. OpenCFU, a New Free and Open-Source Software to Count Cell Colonies and Other Circular Objects. PLoS ONE 2013, 8, e54072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic Assay of Cells in Vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Kleiner, D.E. Drugs and Toxins, 7th, ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; ISBN 9780702066979. [Google Scholar]
- Lee, S.; Choi, M.C.; Al Adem, K.; Lukman, S.; Kim, T.Y. Aggregation and Cellular Toxicity of Pathogenic or Non-Pathogenic Proteins. Sci. Rep. 2020, 10, 5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurunathan, S.; Han, J.W.; Eppakayala, V.; Kim, J.H. Biocompatibility of Microbially Reduced Graphene Oxide in Primary Mouse Embryonic Fibroblast Cells. Colloids Surf. B Biointerfaces 2013, 105, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Sarkar, J.; Ghosh, M.; Bhattacharya, A.; Mukherjee, A.; Chattopadhyay, D.; Acharya, K. Green Conversion of Graphene Oxide to Graphene Nanosheets and Its Biosafety Study. PLoS ONE 2017, 12, e0171607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Nie, S.; Li, S.; Peng, H.; Yang, H.; Ma, L.; Sun, S.; Zhao, C. Biopolymer Functionalized Reduced Graphene Oxide with Enhanced Biocompatibility via Mussel Inspired Coatings/Anchors. J. Mater. Chem. B 2013, 1, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Kim, S.H.; Yang, J.Y.; Jung, K.; Jeong, J.; Oh, J.H.; Lee, J.H. Colony-Forming Efficiency Assay to Assess Nanotoxicity of Graphene Nanomaterials. Toxics 2022, 10, 236. [Google Scholar] [CrossRef]
- Yu, Z.; Li, Q.; Wang, J.; Yu, Y.; Wang, Y.; Zhou, Q.; Li, P. Reactive Oxygen Species-Related Nanoparticle Toxicity in the Biomedical Field. Nanoscale Res. Lett. 2020, 15, 115. [Google Scholar] [CrossRef]
- Gurunathan, S.; Han, J.W.; Eppakayala, V.; Kim, J.H. Green Synthesis of Graphene and Its Cytotoxic Effects in Human Breast Cancer Cells. Int. J. Nanomed. 2013, 8, 1015–1027. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, Y.; Fu, Y.; Wei, T.; Le Guyader, L.; Gao, G.; Liu, R.S.; Chang, Y.Z.; Chen, C. The Triggering of Apoptosis in Macrophages by Pristine Graphene through the MAPK and TGF-Beta Signaling Pathways. Biomaterials 2012, 33, 402–411. [Google Scholar] [CrossRef]
- Huang, Y.W.; Cambre, M.; Lee, H.J. The Toxicity of Nanoparticles Depends on Multiple Molecular and Physicochemical Mechanisms. Int. J. Mol. Sci. 2017, 18, 2702. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Guo, C.; Xia, Y.; Niu, P.; Jiang, L.; Duan, J.; Yu, Y.; Zhou, X.; Li, Y.; Sun, Z. Silica Nanoparticles Induce Oxidative Stress, Inflammation, and Endothelial Dysfunction in Vitro via Activation of the MAPK/Nrf2 Pathway and Nuclear Factor-ΚB Signaling. Int. J. Nanomed. 2015, 10, 1463–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-ΚB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tang, M. Research Progress of Nanoparticle Toxicity Signaling Pathway. Life Sci. 2020, 263, 118542. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papanikolaou, E.; Simos, Y.V.; Spyrou, K.; Patila, M.; Alatzoglou, C.; Tsamis, K.; Vezyraki, P.; Stamatis, H.; Gournis, D.P.; Peschos, D.; et al. Does Green Exfoliation of Graphene Produce More Biocompatible Structures? Pharmaceutics 2023, 15, 993. https://doi.org/10.3390/pharmaceutics15030993
Papanikolaou E, Simos YV, Spyrou K, Patila M, Alatzoglou C, Tsamis K, Vezyraki P, Stamatis H, Gournis DP, Peschos D, et al. Does Green Exfoliation of Graphene Produce More Biocompatible Structures? Pharmaceutics. 2023; 15(3):993. https://doi.org/10.3390/pharmaceutics15030993
Chicago/Turabian StylePapanikolaou, Eirini, Yannis V. Simos, Konstantinos Spyrou, Michaela Patila, Christina Alatzoglou, Konstantinos Tsamis, Patra Vezyraki, Haralambos Stamatis, Dimitrios P. Gournis, Dimitrios Peschos, and et al. 2023. "Does Green Exfoliation of Graphene Produce More Biocompatible Structures?" Pharmaceutics 15, no. 3: 993. https://doi.org/10.3390/pharmaceutics15030993