
Recent Preclinical and Clinical Progress in Liposomal Doxorubicin


Abstract
:1. Introduction
2. Limitations of the Approved Liposomal Doxorubicin Formulations
- The poor release of DOX from the liposome [26]
- The entrapment of the liposome in the cellular lysosomes limits its delivery to the nucleus and reduces its bioavailable concentrations [27].
- These liposomes rely entirely on the enhanced permeability and retention (EPR) effect of the tumor. The liposomes should extravasate passively through the leaky tumor vasculature into the tumor interstitium (Figure 1A). This extravasation can be limited by
- The elevated interstitial fluid pressure (IFP) in most solid tumors [31].
- Interaction with extracellular matrix (ECM) components can hinder the distribution of the liposome to distant parts of the tumor [32].
- The lymphatic drainage in the metastatic microenvironment is not impaired as in the primary tumor; hence, the liposome is not entrapped in metastatic lesions. This fact explains the unsatisfactory effects of PLD and non-PLD in metastatic cancer [33].
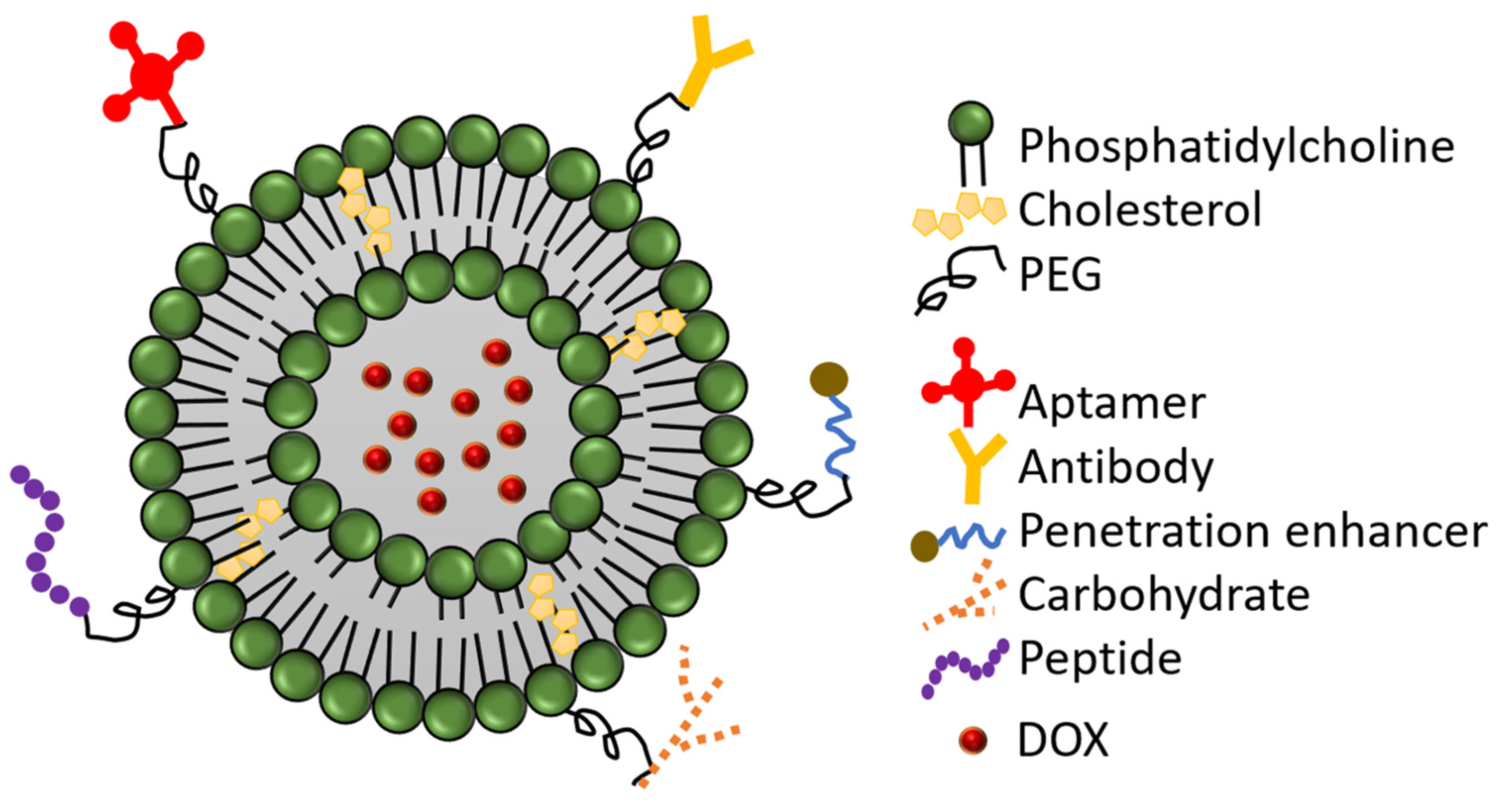
3. Modified-PEGylated Liposomal Doxorubicin:
3.1. Antibody-Conjugated PLD
3.2. Peptide-Conjugated PLD
3.3. Aptamer-Conjugated PLD
3.4. Carbohydrates-Conjugated PLD
3.5. CPE-Conjugated PLD
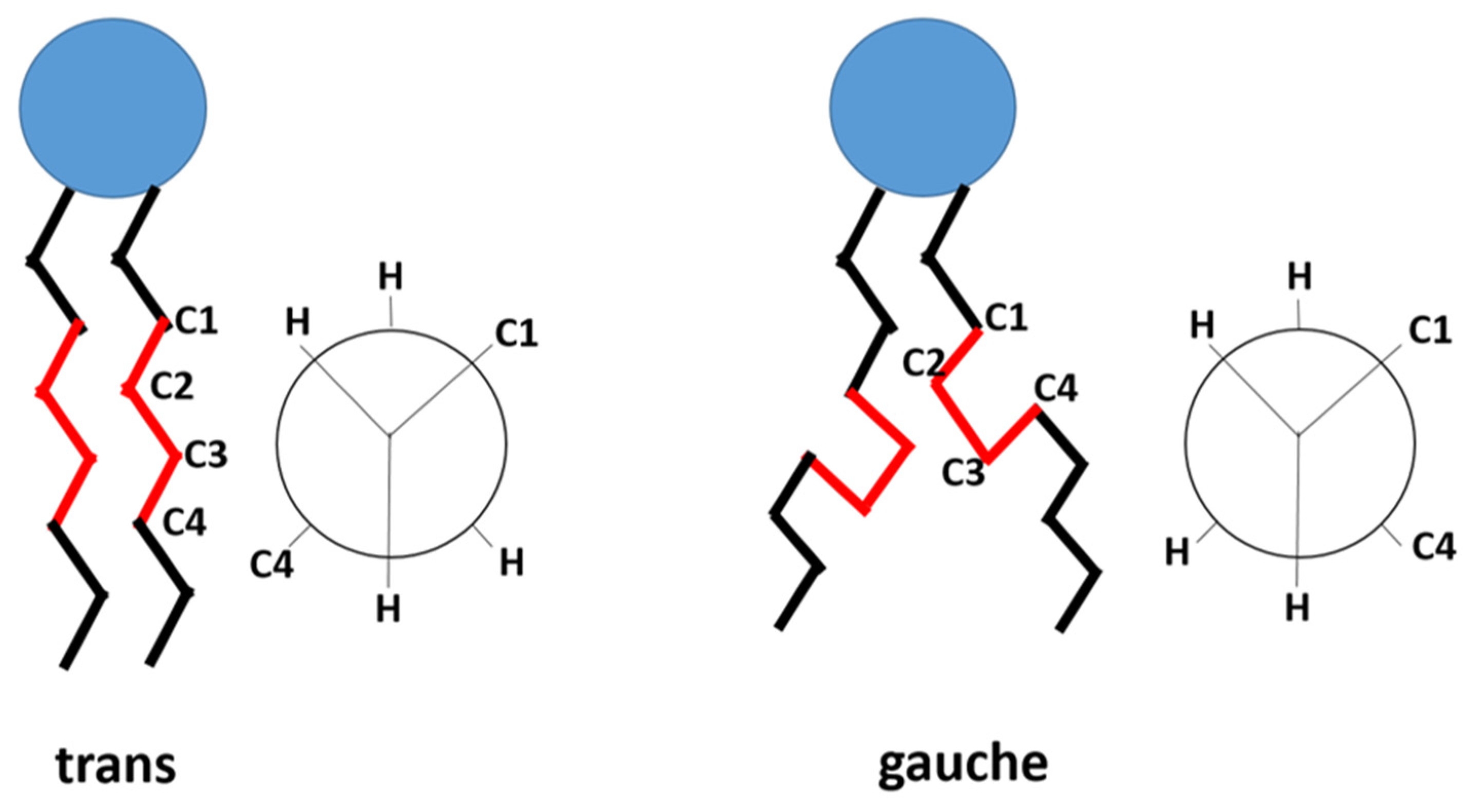
4. Thermosensitive Liposomal Doxorubicin
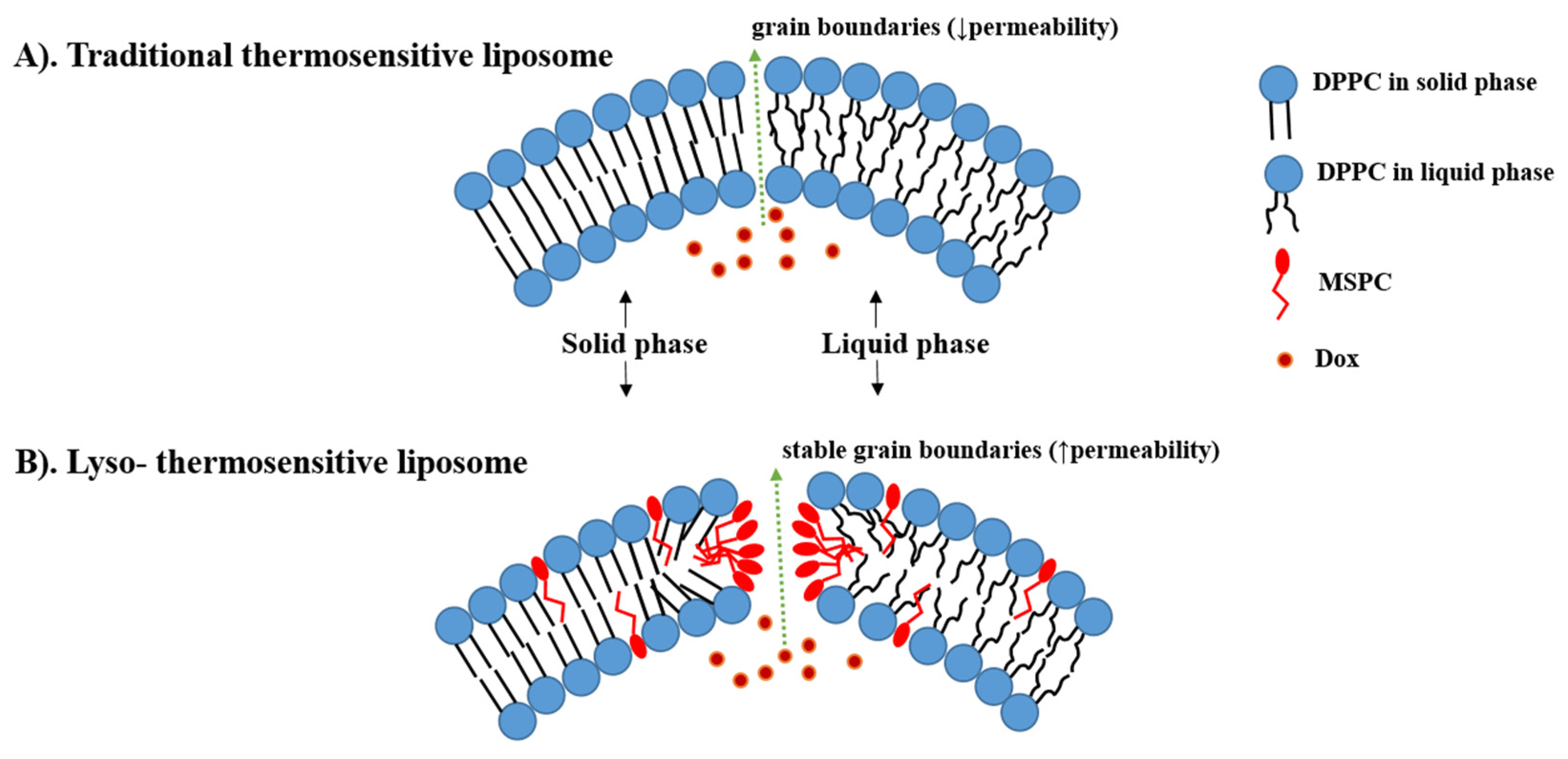
4.1. Traditional Thermosensitive Liposome (TTSL)
4.2. Lyso-Thermosensitive Liposome (LTSL)
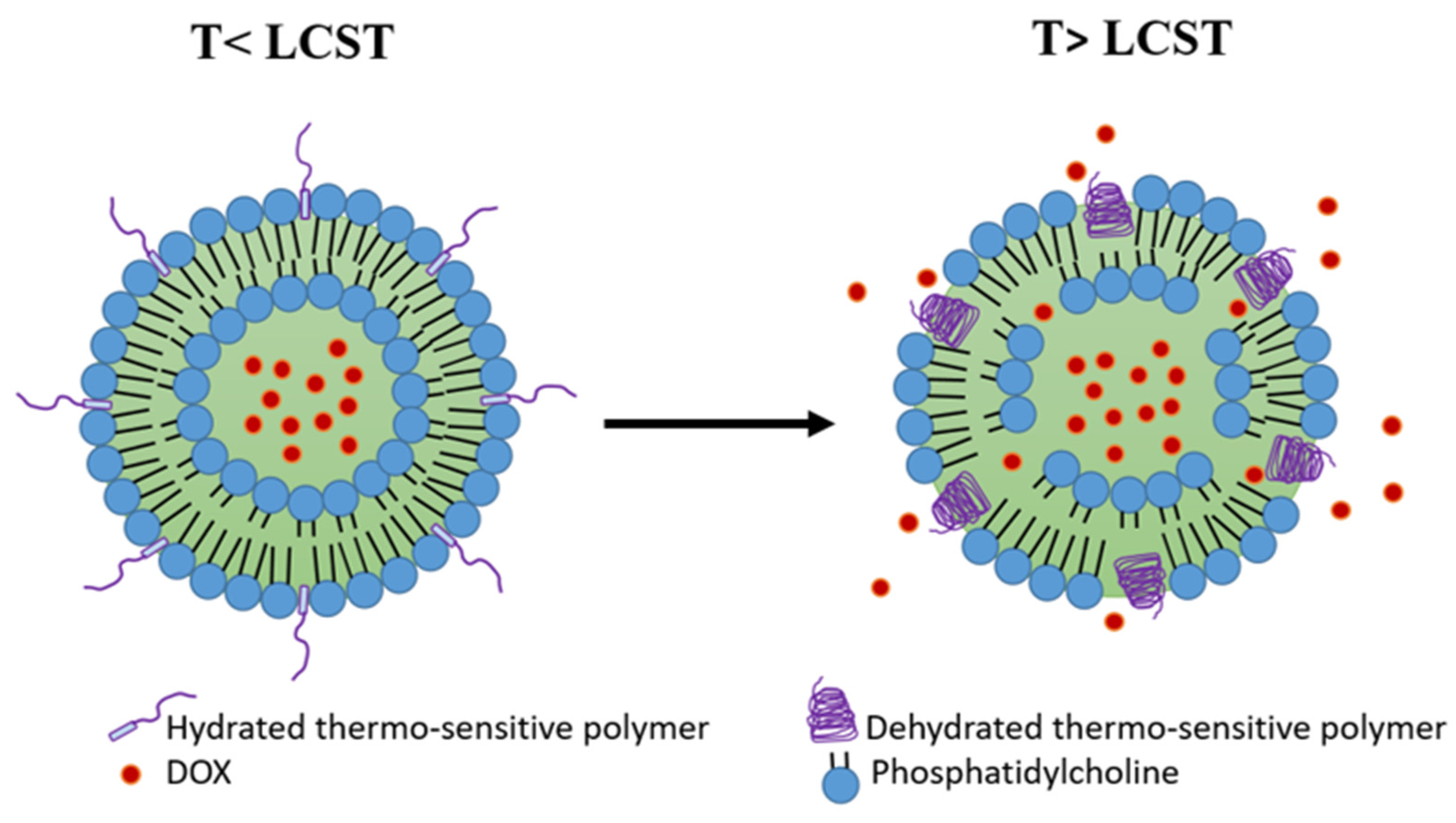
4.3. Polymer-Modified Thermosensitive Liposome (pTSL)
4.4. Phosphatidylglycerol-Based Thermosensitive Liposome (DPPG-TSL)
4.5. Multifunctional-Thermosensitive Liposome
5. pH-Sensitive Liposome (PSL)
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110, reprinted in Biotechnol. Bioeng. 2000, 67, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Prados, J.; Melguizo, C.; Ortiz, R.; Vélez, C.; Alvarez, P.J.; Arias, J.L.; Ruíz, M.A.; Gallardo, V.; Aranega, A. Doxorubicin-loaded nanoparticles: New advances in breast cancer therapy. Anticancer Agents Med. Chem. 2012, 12, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Che, S.; Hui, B.; Yang, Y.; Wang, X.; Zhang, X.; Qiang, Y.; Ma, H. Lung cancer therapy using doxorubicin and curcumin combination: Targeted prodrug based, pH sensitive nanomedicine. Biomed. Pharmacother. 2019, 112, 108614. [Google Scholar] [CrossRef] [PubMed]
- Sehouli, J.; Camara, O.; Schmidt, M.; Mahner, S.; Seipelt, G.; Otremba, B.; Schmalfeldt, B.; Tesch, H.; Lorenz-Schlüter, C.; Oskay-Ozcelik, G. Pegylated liposomal doxorubicin (CAELYX) in patients with advanced ovarian cancer: Results of a German multicenter observational study. Cancer Chemother. Pharmacol. 2009, 64, 585–591. [Google Scholar] [CrossRef]
- Cross, R.J.; Glashan, R.W.; Humphrey, C.S.; Robinson, R.G.; Smith, P.H.; Williams, R.E. Treatment of advanced bladder cancer with adriamycin and 5-fluorouracil. Br. J. Urol. 1976, 48, 609–615. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Bhinge, K.N.; Gupta, V.; Hosain, S.B.; Satyanarayanajois, S.D.; Meyer, S.A.; Blaylock, B.; Zhang, Q.J.; Liu, Y.Y. The opposite effects of doxorubicin on bone marrow stem cells versus breast cancer stem cells depend on glucosylceramide synthase. Int. J. Biochem. Cell Biol. 2012, 44, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.M.; Yusuf, S.W.; Ewer, M.S. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomed. 2007, 2, 567–583. [Google Scholar]
- Hequet, O.; Le, Q.H.; Moullet, I.; Pauli, E.; Salles, G.; Espinouse, D.; Dumontet, C.; Thieblemont, C.; Arnaud, P.; Antal, D.; et al. Subclinical late cardiomyopathy after doxorubicin therapy for lymphoma in adults. J. Clin. Oncol. 2004, 22, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, D.S.; dos Santos Goldenberg, R.C. Doxorubicin-Induced Cardiotoxicity: From Mechanisms to Development of Efficient Therapy. In Cardiotoxicity; Wenyong, T., Ed.; IntechOpen: Rijeka, Croatia, 2018; Chapter 1. [Google Scholar]
- Kinoshita, T.; Yuzawa, H.; Natori, K.; Wada, R.; Yao, S.; Yano, K.; Akitsu, K.; Koike, H.; Shinohara, M.; Fujino, T.; et al. Early electrocardiographic indices for predicting chronic doxorubicin-induced cardiotoxicity. J. Cardiol. 2021, 77, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Yuan, D.; Wu, Y.; Cao, Y. Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs. Pharmaceutics 2019, 11, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, M.E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.F.; et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [CrossRef]
- Hunault-Berger, M.; Leguay, T.; Thomas, X.; Legrand, O.; Huguet, F.; Bonmati, C.; Escoffre-Barbe, M.; Legros, L.; Turlure, P.; Chevallier, P.; et al. A randomized study of pegylated liposomal doxorubicin versus continuous-infusion doxorubicin in elderly patients with acute lymphoblastic leukemia: The GRAALL-SA1 study. Haematologica 2011, 96, 245–252. [Google Scholar] [CrossRef]
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K.; et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [CrossRef]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal Doxorubicin: Review of animal and human studies. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Lorusso, D.; Di Stefano, A.; Carone, V.; Fagotti, A.; Pisconti, S.; Scambia, G. Pegylated liposomal doxorubicin-related palmar-plantar erythrodysesthesia (‘hand-foot’ syndrome). Ann. Oncol. 2007, 18, 1159–1164. [Google Scholar] [CrossRef]
- Hama, S.; Itakura, S.; Nakai, M.; Nakayama, K.; Morimoto, S.; Suzuki, S.; Kogure, K. Overcoming the polyethylene glycol dilemma via pathological environment-sensitive change of the surface property of nanoparticles for cellular entry. J. Control. Release 2015, 206, 67–74. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Harashima, H. The polyethyleneglycol dilemma: Advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiyath, S.M.; Rasul, M.; Lee, B.; Wei, G.; Lamba, G.; Liu, D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: A meta-analysis. Exp. Hematol. Oncol. 2012, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brucker, J.; Mayer, C.; Gebauer, G.; Mallmann, P.; Belau, A.K.; Schneeweiss, A.; Sohn, C.; Eichbaum, M. Non-pegylated liposomal doxorubicin for patients with recurrent ovarian cancer: A multicentric phase II trial. Oncol. Lett. 2016, 12, 1211–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Alakhova, D.Y.; Kim, J.O.; Bronich, T.K.; Kabanov, A.V. A simple way to enhance Doxil® therapy: Drug release from liposomes at the tumor site by amphiphilic block copolymer. J. Control. Release 2013, 168, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seynhaeve, A.L.B.; Dicheva, B.M.; Hoving, S.; Koning, G.A.; Ten Hagen, T.L.M. Intact Doxil is taken up intracellularly and released doxorubicin sequesters in the lysosome: Evaluated by in vitro/in vivo live cell imaging. J. Control. Release 2013, 172, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.Y.; Kano, M.R. Stromal barriers to nanomedicine penetration in the pancreatic tumor microenvironment. Cancer Sci. 2018, 109, 2085–2092. [Google Scholar] [CrossRef]
- Vinhas, R.; Mendes, R.; Fernandes, A.R.; Baptista, P.V. Nanoparticles-Emerging Potential for Managing Leukemia and Lymphoma. Front. Bioeng. Biotechnol. 2017, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Ali, E.S.; Sharker, S.M.; Islam, M.T.; Khan, I.N.; Shaw, S.; Rahman, M.A.; Uddin, S.J.; Shill, M.C.; Rehman, S.; Das, N.; et al. Targeting cancer cells with nanotherapeutics and nanodiagnostics: Current status and future perspectives. Semin. Cancer Biol. 2021, 69, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.; Merrigan, S.; Turpin, J.; Lavoie, C.; Papavasiliou, V.; Geretti, E.; Espelin, C.W.; Luus, L.; Kamoun, W.S.; Ghasemi, O.; et al. Nanoliposome targeting in breast cancer is influenced by the tumor microenvironment. Nanomedicine 2019, 17, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: State of the art review. Int. J. Nanomed. 2014, 9, 4387–4398. [Google Scholar]
- Moosavian, S.A.; Abnous, K.; Badiee, A.; Jaafari, M.R. Improvement in the drug delivery and anti-tumor efficacy of PEGylated liposomal doxorubicin by targeting RNA aptamers in mice bearing breast tumor model. Colloids Surf. B Biointerfaces 2016, 139, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Aldughaim, M.S.; Muthana, M.; Alsaffar, F.; Barker, M.D. Specific Targeting of PEGylated Liposomal Doxorubicin (Doxil(®)) to Tumour Cells Using a Novel TIMP3 Peptide. Molecules 2020, 26, 100. [Google Scholar] [CrossRef]
- Ara, M.N.; Matsuda, T.; Hyodo, M.; Sakurai, Y.; Hatakeyama, H.; Ohga, N.; Hida, K.; Harashima, H. An aptamer ligand based liposomal nanocarrier system that targets tumor endothelial cells. Biomaterials 2014, 35, 7110–7120. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Allemailem, K.S.; Almatroodi, S.A.; Almatroudi, A.; Rahmani, A.H. Recent strategies towards the surface modification of liposomes: An innovative approach for different clinical applications. 3 Biotech 2020, 10, 163. [Google Scholar] [CrossRef]
- Cardoso, M.M.; Peça, I.N.; Roque, A.C. Antibody-conjugated nanoparticles for therapeutic applications. Curr. Med. Chem. 2012, 19, 3103–3127. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, Y.; Ho, R.J.; Zhao, Y.; Zhang, T.; Guo, C. Antibody-modified liposomes for tumor-targeting delivery of timosaponin AIII. Int. J. Nanomed. 2018, 13, 1927–1944. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Cao, H.; He, W.; Gao, F.; Liu, Y.; Yin, L. Novel drug delivery liposomes targeted with a fully human anti-VEGF165 monoclonal antibody show superior antitumor efficacy in vivo. Biomed. Pharmacother. 2015, 73, 48–57. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Ishida, E.; Hashimoto, K.; Kobayashi, H.; Aoki, T.; Yasuda, J.; Obata, K.; Kikuchi, H.; Ishida, T.; et al. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int. J. Pharm. 2007, 342, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Krop, I.E.; LoRusso, P.; Ma, C.; Siegel, B.A.; Shields, A.F.; Molnár, I.; Wickham, T.J.; Reynolds, J.; Campbell, K.; et al. Safety and pharmacokinetics of MM-302, a HER2-targeted antibody–liposomal doxorubicin conjugate, in patients with advanced HER2-positive breast cancer: A phase 1 dose-escalation study. Br. J. Cancer 2018, 119, 1086–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamot, C.; Ritschard, R.; Wicki, A.; Stehle, G.; Dieterle, T.; Bubendorf, L.; Hilker, C.; Deuster, S.; Herrmann, R.; Rochlitz, C. Tolerability, safety, pharmacokinetics, and efficacy of doxorubicin-loaded anti-EGFR immunoliposomes in advanced solid tumours: A phase 1 dose-escalation study. Lancet Oncol. 2012, 13, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.G.; Geretti, E.; Hendriks, B.S.; Lee, H.; Leonard, S.C.; Klinz, S.G.; Noble, C.O.; Lücker, P.B.; Zandstra, P.W.; Drummond, D.C.; et al. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol. Appl. Pharmacol. 2012, 262, 1–10. [Google Scholar] [CrossRef]
- Espelin, C.W.; Leonard, S.C.; Geretti, E.; Wickham, T.J.; Hendriks, B.S. Dual HER2 Targeting with Trastuzumab and Liposomal-Encapsulated Doxorubicin (MM-302) Demonstrates Synergistic Antitumor Activity in Breast and Gastric Cancer. Cancer Res. 2016, 76, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Manikhas, A.; Cortés, J.; Llombart, A.; Roman, L.; Semiglazov, V.F.; Byakhov, M.; Lokanatha, D.; Forenza, S.; Goldfarb, R.H.; et al. Phase III trial of nonpegylated liposomal doxorubicin in combination with trastuzumab and paclitaxel in HER2-positive metastatic breast cancer. Ann. Oncol. 2014, 25, 592–598. [Google Scholar] [CrossRef]
- Wicki, A.; Ritschard, R.; Loesch, U.; Deuster, S.; Rochlitz, C.; Mamot, C. Large-scale manufacturing of GMP-compliant anti-EGFR targeted nanocarriers: Production of doxorubicin-loaded anti-EGFR-immunoliposomes for a first-in-man clinical trial. Int. J. Pharm. 2015, 484, 8–15. [Google Scholar] [CrossRef]
- Mamot, C.; Drummond, D.C.; Noble, C.O.; Kallab, V.; Guo, Z.; Hong, K.; Kirpotin, D.B.; Park, J.W. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005, 65, 11631–11638. [Google Scholar] [CrossRef] [Green Version]
- Mamot, C.; Ritschard, R.; Wicki, A.; Küng, W.; Schuller, J.; Herrmann, R.; Rochlitz, C. Immunoliposomal delivery of doxorubicin can overcome multidrug resistance mechanisms in EGFR-overexpressing tumor cells. J. Drug Target. 2012, 20, 422–432. [Google Scholar] [CrossRef]
- Kasenda, B.; König, D.; Manni, M.; Ritschard, R.; Duthaler, U.; Bartoszek, E.; Bärenwaldt, A.; Deuster, S.; Hutter, G.; Cordier, D.; et al. Targeting immunoliposomes to EGFR-positive glioblastoma. ESMO Open 2022, 7, 100365. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Pan, G.; Ni, J.; Xie, F.; Jiang, B.; Wei, L.; Gao, J.; Zhou, W. Enhanced doxorubicin delivery to hepatocellular carcinoma cells via CD147 antibody-conjugated immunoliposomes. Nanomedicine 2018, 14, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Shahin, M.; Ahmed, S.; Kaur, K.; Lavasanifar, A. Decoration of polymeric micelles with cancer-specific peptide ligands for active targeting of paclitaxel. Biomaterials 2011, 32, 5123–5133. [Google Scholar] [CrossRef] [PubMed]
- Shadidi, M.; Sioud, M. Selective targeting of cancer cells using synthetic peptides. Drug Resist. Updates 2003, 6, 363–371. [Google Scholar] [CrossRef]
- Shahraki, N.; Mehrabian, A.; Amiri-Darban, S.; Moosavian, S.A.; Jaafari, M.R. Preparation and characterization of PEGylated liposomal Doxorubicin targeted with leptin-derived peptide and evaluation of their anti-tumor effects, in vitro and in vivo in mice bearing C26 colon carcinoma. Colloids Surf. B Biointerfaces 2021, 200, 111589. [Google Scholar] [CrossRef]
- Amiri Darban, S.; Nikoofal-Sahlabadi, S.; Amiri, N.; Kiamanesh, N.; Mehrabian, A.; Zendehbad, B.; Gholizadeh, Z.; Jaafari, M.R. Targeting the leptin receptor: To evaluate therapeutic efficacy and anti-tumor effects of Doxil, in vitro and in vivo in mice bearing C26 colon carcinoma tumor. Colloids Surf. B Biointerfaces 2018, 164, 107–115. [Google Scholar] [CrossRef]
- Zahmatkeshan, M.; Gheybi, F.; Rezayat, S.M.; Jaafari, M.R. Improved drug delivery and therapeutic efficacy of PEgylated liposomal doxorubicin by targeting anti-HER2 peptide in murine breast tumor model. Eur. J. Pharm. Sci. 2016, 86, 125–135. [Google Scholar] [CrossRef]
- Haftcheshmeh, S.M.; Jaafari, M.R.; Mashreghi, M.; Mehrabian, A.; Alavizadeh, S.H.; Zamani, P.; Zarqi, J.; Darvishi, M.H.; Gheybi, F. Liposomal doxorubicin targeting mitochondria: A novel formulation to enhance anti-tumor effects of Doxil® in vitro and in vivo. J. Drug Deliv. Sci. Technol. 2021, 62, 102351. [Google Scholar] [CrossRef]
- D’Avanzo, N.; Torrieri, G.; Figueiredo, P.; Celia, C.; Paolino, D.; Correia, A.; Moslova, K.; Teesalu, T.; Fresta, M.; Santos, H.A. LinTT1 peptide-functionalized liposomes for targeted breast cancer therapy. Int. J. Pharm. 2021, 597, 120346. [Google Scholar] [CrossRef]
- Kato, N.; Sato, T.; Fuchigami, Y.; Suga, T.; Geng, L.; Tsurumaru, M.; Hagimori, M.; Mukai, H.; Kawakami, S. Synthesis and evaluation of a novel adapter lipid derivative for preparation of cyclic peptide-modified PEGylated liposomes: Application of cyclic RGD peptide. Eur. J. Pharm. Sci. 2022, 176, 106239. [Google Scholar] [CrossRef]
- Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G.C.; Cheng, J.; Lu, Y. Reversible cell-specific drug delivery with aptamer-functionalized liposomes. Angew. Chem. Int. Ed. 2009, 48, 6494–6498. [Google Scholar] [CrossRef] [PubMed]
- Moosavian, S.A.; Abnous, K.; Akhtari, J.; Arabi, L.; Gholamzade Dewin, A.; Jafari, M. 5TR1 aptamer-PEGylated liposomal doxorubicin enhances cellular uptake and suppresses tumour growth by targeting MUC1 on the surface of cancer cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2054–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashreghi, M.; Zamani, P.; Moosavian, S.A.; Jaafari, M.R. Anti-Epcam Aptamer (Syl3c)-Functionalized Liposome for Targeted Delivery Of Doxorubicin: In Vitro And In Vivo Antitumor Studies in Mice Bearing C26 Colon Carcinoma. Nanoscale Res. Lett. 2020, 15, 101. [Google Scholar] [CrossRef] [PubMed]
- Mashreghi, M.; Zamani, P.; Karimi, M.; Mehrabian, A.; Arabsalmani, M.; Zarqi, J.; Moosavian, S.A.; Jaafari, M.R. Anti-epithelial cell adhesion molecule RNA aptamer-conjugated liposomal doxorubicin as an efficient targeted therapy in mice bearing colon carcinoma tumor model. Biotechnol. Prog. 2021, 37, e3116. [Google Scholar] [CrossRef]
- Dalle Vedove, E.; Costabile, G.; Merkel, O.M. Mannose and Mannose-6-Phosphate Receptor-Targeted Drug Delivery Systems and Their Application in Cancer Therapy. Adv. Healthc. Mater. 2018, 7, e1701398. [Google Scholar] [CrossRef]
- Li, C.; Lai, C.; Qiu, Q.; Luo, X.; Hu, L.; Zheng, H.; Lu, Y.; Liu, M.; Zhang, H.; Liu, X.; et al. Dual-Ligand Modification of PEGylated Liposomes Used for Targeted Doxorubicin Delivery to Enhance Anticancer Efficacy. AAPS PharmSciTech 2019, 20, 188. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Regberg, J.; Srimanee, A.; Langel, U. Applications of cell-penetrating peptides for tumor targeting and future cancer therapies. Pharmaceuticals 2012, 5, 991–1007. [Google Scholar] [CrossRef] [Green Version]
- Popilski, H.; Feinshtein, V.; Kleima, S.; Mattarei, A.; Garofalo, M.; Salmaso, S.; Stepensky, D. Doxorubicin liposomes cell penetration enhancement and its potential drawbacks for the tumor targeting efficiency. Int. J. Pharm. 2021, 592, 120012. [Google Scholar] [CrossRef]
- Teymouri, M.; Badiee, A.; Golmohammadzadeh, S.; Sadri, K.; Akhtari, J.; Mellat, M.; Nikpoor, A.R.v.; Jaafari, M.R. Tat peptide and hexadecylphosphocholine introduction into pegylated liposomal doxorubicin: An in vitro and in vivo study on drug cellular delivery, release, biodistribution and antitumor activity. Int. J. Pharm. 2016, 511, 236–244. [Google Scholar] [CrossRef]
- Zarazvand, F.; Karimi, M.; Moosavian, S.A.; Arabi, L.; Badiee, A.; Jaafari, M.R.; Mashreghi, M. Efficacy Comparison of TAT Peptide-Functionalized PEGylated Liposomal Doxorubicin in C26 and B16F0 Tumor Mice Models. Int. J. Pept. Res. Ther. 2021, 27, 2099–2109. [Google Scholar] [CrossRef]
- Deshpande, P.; Jhaveri, A.; Pattni, B.; Biswas, S.; Torchilin, V. Transferrin and octaarginine modified dual-functional liposomes with improved cancer cell targeting and enhanced intracellular delivery for the treatment of ovarian cancer. Drug Deliv. 2018, 25, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Mansourian, M.; Burgers, P.C.; Amin, B.; Jaafari, M.R.; Ten Hagen, T.L.M. Increased Targeting Area in Tumors by Dual-Ligand Modification of Liposomes with RGD and TAT Peptides. Pharmaceutics 2022, 14, 458. [Google Scholar] [CrossRef] [PubMed]
- Mashreghi, M.; Faal Maleki, M.; Karimi, M.; Kalalinia, F.; Badiee, A.; Jaafari, M.R. Improving anti-tumour efficacy of PEGylated liposomal doxorubicin by dual targeting of tumour cells and tumour endothelial cells using anti-p32 CGKRK peptide. J Drug Target 2021, 29, 617–630. [Google Scholar] [CrossRef]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int J Mol Sci 2018, 19, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirpotin, D.; Park, J.W.; Hong, K.; Zalipsky, S.; Li, W.L.; Carter, P.; Benz, C.C.; Papahadjopoulos, D. Sterically Stabilized Anti-HER2 Immunoliposomes: Design and Targeting to Human Breast Cancer Cells in Vitro. Biochemistry 1997, 36, 66–75. [Google Scholar] [CrossRef]
- Mamot, C.; Drummond, D.C.; Greiser, U.; Hong, K.; Kirpotin, D.B.; Marks, J.D.; Park, J.W. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res. 2003, 63, 3154–3161. [Google Scholar] [PubMed]
- Kim, K.Y. Nanotechnology platforms and physiological challenges for cancer therapeutics. Nanomedicine 2007, 3, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Leekumjorn, S. and A.K. Sum, Molecular studies of the gel to liquid-crystalline phase transition for fully hydrated DPPC and DPPE bilayers. Biochim. Biophys. Acta Biomembr. 2007, 1768, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Papahadjopoulos, D.; Jacobson, K.; Nir, S.; Isac, T. Phase transitions in phospholipid vesicles Fluorescence polarization and permeability measurements concerning the effect of temperature and cholesterol. Biochim. Biophys. Acta Biomembr. 1973, 311, 330–348. [Google Scholar] [CrossRef]
- Kuć, M.; Cieślik-Boczula, K.; Rospenk, M. Anesthetic-dependent changes in the chain-melting phase transition of DPPG liposomes studied using near-infrared spectroscopy supported by PCA. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 186, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.; Porter, T.M. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J. Control. Release 2013, 169, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Yatvin, M.B.; Weinstein, J.N.; Dennis, W.H.; Blumenthal, R. Design of Liposomes for Enhanced Local Release of Drugs by Hyperthermia. Science 1978, 202, 1290–1293. [Google Scholar] [CrossRef]
- Gaber, M.H.; Hong, K.; Huang, S.K.; Papahadjopoulos, D. Thermosensitive Sterically Stabilized Liposomes: Formulation and in Vitro Studies on Mechanism of Doxorubicin Release by Bovine Serum and Human Plasma. Pharm. Res. 1995, 12, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Arouri, A.; Mouritsen, O.G. Membrane-perturbing effect of fatty acids and lysolipids. Prog. Lipid Res. 2013, 52, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Landon, C.D.; Park, J.Y.; Needham, D.; Dewhirst, M.W. Nanoscale Drug Delivery and Hyperthermia: The Materials Design and Preclinical and Clinical Testing of Low Temperature-Sensitive Liposomes Used in Combination with Mild Hyperthermia in the Treatment of Local Cancer. Open Nanomed. J. 2011, 3, 38–64. [Google Scholar] [CrossRef]
- Mills, J.K.; Needham, D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim. Biophys. Acta Biomembr. 2005, 1716, 77–96. [Google Scholar] [CrossRef] [Green Version]
- De Matos, M.B.C.; Beztsinna, N.; Heyder, C.; Fens, M.H.A.M.; Mastrobattista, E.; Schiffelers, R.M.; Leneweit, G.; Kok, R.J. Thermosensitive liposomes for triggered release of cytotoxic proteins. Eur. J. Pharm. Biopharm. 2018, 132, 211–221. [Google Scholar] [CrossRef]
- Anyarambhatla, G.R.; Needham, D. Enhancement of the Phase Transition Permeability of DPPC Liposomes by Incorporation of MPPC: A New Temperature-Sensitive Liposome for use with Mild Hyperthermia. J. Liposome Res. 1999, 9, 491–506. [Google Scholar] [CrossRef]
- Needham, D.; Anyarambhatla, G.; Kong, G.; Dewhirst, M.W. A New Temperature-sensitive Liposome for Use with Mild Hyperthermia: Characterization and Testing in a Human Tumor Xenograft Model1. Cancer Res. 2000, 60, 1197–1201. [Google Scholar] [PubMed]
- Kong, G.; Anyarambhatla, G.; Petros, W.P.; Braun, R.D.; Colvin, O.M.; Needham, D.; Dewhirst, M.W. Efficacy of Liposomes and Hyperthermia in a Human Tumor Xenograft Model: Importance of Triggered Drug Release1. Cancer Res. 2000, 60, 6950–6957. [Google Scholar] [PubMed]
- Manzoor, A.A.; Lindner, L.H.; Landon, C.D.; Park, J.Y.; Simnick, A.J.; Dreher, M.R.; Das, S.; Hanna, G.; Park, W.; Chilkoti, A. Overcoming limitations in nanoparticle drug delivery: Triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012, 72, 5566–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; ten Hagen, T.L.; Haeri, A.; Soullié, T.; Scholten, C.; Seynhaeve, A.L.; Eggermont, A.M.; Koning, G.A. A novel two-step mild hyperthermia for advanced liposomal chemotherapy. J. Control. Release 2014, 174, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, W.T.; Kostarelos, K. Mild hyperthermia accelerates doxorubicin clearance from tumour-extravasated temperature-sensitive liposomes. Nanotheranostics 2022, 6, 230–242. [Google Scholar] [CrossRef]
- Banno, B.; Ickenstein, L.M.; Chiu, G.N.; Bally, M.B.; Thewalt, J.; Brief, E.; Wasan, E.K. The functional roles of poly(ethylene glycol)-lipid and lysolipid in the drug retention and release from lysolipid-containing thermosensitive liposomes in vitro and in vivo. J. Pharm. Sci. 2010, 99, 2295–2308. [Google Scholar] [CrossRef]
- Sadeghi, N.; Deckers, R.; Ozbakir, B.; Akthar, S.; Kok, R.J.; Lammers, T.; Storm, G. Influence of cholesterol inclusion on the doxorubicin release characteristics of lysolipid-based thermosensitive liposomes. Int. J. Pharm. 2018, 548, 778–782. [Google Scholar] [CrossRef]
- Needham, D.; Park, J.Y.; Wright, A.M.; Tong, J. Materials characterization of the low temperature sensitive liposome (LTSL): Effects of the lipid composition (lysolipid and DSPE-PEG2000) on the thermal transition and release of doxorubicin. Faraday Discuss. 2013, 161, 515–534; discussion 563–589. [Google Scholar] [CrossRef]
- Besse, H.C.; Barten-van Rijbroek, A.D.; van der Wurff-Jacobs, K.M.G.; Bos, C.; Moonen, C.T.W.; Deckers, R. Tumor Drug Distribution after Local Drug Delivery by Hyperthermia, In Vivo. Cancers 2019, 11, 1512. [Google Scholar] [CrossRef] [Green Version]
- Ponce, A.M.; Viglianti, B.L.; Daohai, Y.u.; Yarmolenko, P.S.; Michelich, C.R.; Woo, J.; Bally, M.B.; Dewhirst, M.W. Magnetic Resonance Imaging of Temperature-Sensitive Liposome Release: Drug Dose Painting and Antitumor Effects. J. Natl. Cancer Inst. 2007, 99, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Hauck, M.L.; LaRue, S.M.; Petros, W.P.; Poulson, J.M.; Yu, D.; Spasojevic, I.; Pruitt, A.F.; Klein, A.; Case, B.; Thrall, D.E.; et al. Phase I trial of doxorubicin-containing low temperature sensitive liposomes in spontaneous canine tumors. Clin. Cancer Res. 2006, 12, 4004–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhail, A.S.; Negussie, A.H.; Pritchard, W.F.; Haemmerich, D.; Woods, D.; Bakhutashvili, I.; Esparza-Trujillo, J.; Brancato, S.J.; Karanian, J.; Agarwal, P.K.; et al. Lyso-thermosensitive liposomal doxorubicin for treatment of bladder cancer. Int. J. Hyperth. 2017, 33, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Poon, R.T.P.; Borys, N. Lyso-thermosensitive liposomal doxorubicin: A novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin. Pharmacother. 2009, 10, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; Cioni, D. RFA plus lyso-thermosensitive liposomal doxorubicin: In search of the optimal approach to cure intermediate-size hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Tak, W.Y.; Lin, S.M.; Wang, Y.; Zheng, J.; Vecchione, A.; Park, S.Y.; Chen, M.H.; Wong, S.; Xu, R.; Peng, C.Y.; et al. Phase III HEAT Study Adding Lyso-Thermosensitive Liposomal Doxorubicin to Radiofrequency Ablation in Patients with Unresectable Hepatocellular Carcinoma Lesions. Clin. Cancer Res. 2018, 24, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regenold, M.; Bannigan, P.; Evans, J.C.; Waspe, A.; Temple, M.J.; Allen, C. Turning down the heat: The case for mild hyperthermia and thermosensitive liposomes. Nanomed. Nanotechnol. Biol. Med. 2022, 40, 102484. [Google Scholar] [CrossRef]
- Dou, Y.; Hynynen, K.; Allen, C. To heat or not to heat: Challenges with clinical translation of thermosensitive liposomes. J. Control. Release 2017, 249, 63–73. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Kuo, S.J.; Tsai, H.D.; Chou, M.C.; Yeh, G.P. Clinical Application of High-intensity Focused Ultrasound in Cancer Therapy. J. Cancer 2016, 7, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Partanen, A.; Talcott, M.R.; Gach, H.M.; Greco, S.C.; Henke, L.E.; Contreras, J.A.; Zoberi, I.; Hallahan, D.E.; Chen, H.; et al. Feasibility and safety assessment of magnetic resonance-guided high-intensity focused ultrasound (MRgHIFU)-mediated mild hyperthermia in pelvic targets evaluated using an in vivo porcine model. Int. J. Hyperth. 2019, 36, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Schvarcz, C.A.; Danics, L.; Krenács, T.; Viana, P.; Béres, R.; Vancsik, T.; Nagy, Á.; Gyenesei, A.; Kun, J.; Fonović, M.; et al. Modulated Electro-Hyperthermia Induces a Prominent Local Stress Response and Growth Inhibition in Mouse Breast Cancer Isografts. Cancers 2021, 13, 1744. [Google Scholar] [CrossRef]
- Danics, L.; Schvarcz, C.A.; Viana, P.; Vancsik, T.; Krenács, T.; Benyó, Z.; Kaucsár, T.; Hamar, P. Exhaustion of Protective Heat Shock Response Induces Significant Tumor Damage by Apoptosis after Modulated Electro-Hyperthermia Treatment of Triple Negative Breast Cancer Isografts in Mice. Cancers 2020, 12, 2581. [Google Scholar] [CrossRef] [PubMed]
- Zagar, T.M.; Vujaskovic, Z.; Formenti, S.; Rugo, H.; Muggia, F.; O’Connor, B.; Myerson, R.; Stauffer, P.; Hsu, I.C.; Diederich, C.; et al. Two phase I dose-escalation/pharmacokinetics studies of low temperature liposomal doxorubicin (LTLD) and mild local hyperthermia in heavily pretreated patients with local regionally recurrent breast cancer. Int. J. Hyperth. 2014, 30, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Swenson, C.E.; Haemmerich, D.; Maul, D.H.; Knox, B.; Ehrhart, N.; Reed, R.A. Increased Duration of Heating Boosts Local Drug Deposition during Radiofrequency Ablation in Combination with Thermally Sensitive Liposomes (ThermoDox) in a Porcine Model. PLoS ONE 2015, 10, e0139752. [Google Scholar] [CrossRef] [PubMed]
- Yarmolenko, P.S.; Zhao, Y.; Landon, C.; Spasojevic, I.; Yuan, F.; Needham, D.; Viglianti, B.L.; Dewhirst, M.W. Comparative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumours. Int. J. Hyperth. 2010, 26, 485–498. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Zhong, T.; Zhao, Y.; Zhang, W.Q.; Ren, W.; Huang, D.; Zhang, S.; Guo, Y.; Yao, X.; et al. The antitumor activity of tumor-homing peptide-modified thermosensitive liposomes containing doxorubicin on MCF-7/ADR: In vitro and in vivo. Int. J. Nanomed. 2015, 10, 2229–2248. [Google Scholar]
- Yin, X.; Hoffman, A.S.; Stayton, P.S. Poly(N-isopropylacrylamide-co-propylacrylic acid) Copolymers That Respond Sharply to Temperature and pH. Biomacromolecules 2006, 7, 1381–1385. [Google Scholar] [CrossRef]
- Kono, K.; Nakai, R.; Morimoto, K.; Takagishi, T. Thermosensitive polymer-modified liposomes that release contents around physiological temperature. Biochim. Biophys. Acta Biomembr. 1999, 1416, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Pennadam, S.S.; Firman, K.; Alexander, C.; Górecki, D.C. Protein-polymer nano-machines. Towards synthetic control of biological processes. J. Nanobiotechnology 2004, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Du, H.; Chen, B.; Liu, D.; Yin, Q.; Yan, Y.; Wang, Z.; Wan, F.; Qi, T.; Wang, Y.; et al. Quick-Responsive Polymer-Based Thermosensitive Liposomes for Controlled Doxorubicin Release and Chemotherapy. ACS Biomater. Sci. Eng. 2019, 5, 2316–2329. [Google Scholar] [CrossRef]
- Ta, T.; Convertine, A.J.; Reyes, C.R.; Stayton, P.S.; Porter, T.M. Thermosensitive liposomes modified with poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers for triggered release of doxorubicin. Biomacromolecules 2010, 11, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Ta, T.; Bartolak-Suki, E.; Park, E.J.; Karrobi, K.; McDannold, N.J.; Porter, T.M. Localized delivery of doxorubicin in vivo from polymer-modified thermosensitive liposomes with MR-guided focused ultrasound-mediated heating. J. Control. Release 2014, 194, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Kono, K.; Takagishi, T. Temperature-controlled release property of phospholipid vesicles bearing a thermo-sensitive polymer. Biochim. Biophys. Acta Biomembr. 1996, 1280, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, K.; Murakami, T.; Yoshida, T.; Haba, Y.; Kanaoka, S.; Takagishi, T.; Aoshima, S. Temperature sensitization of liposomes by use of thermosensitive block copolymers synthesized by living cationic polymerization: Effect of copolymer chain length. Bioconjugate Chem. 2005, 16, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Ozawa, T.; Yoshida, T.; Ozaki, F.; Ishizaka, Y.; Maruyama, K.; Kojima, C.; Harada, A.; Aoshima, S. Highly temperature-sensitive liposomes based on a thermosensitive block copolymer for tumor-specific chemotherapy. Biomaterials 2010, 31, 7096–7105. [Google Scholar] [CrossRef] [PubMed]
- van Elk, M.; van den Dikkenberg, J.B.; Storm, G.; Hennink, W.E.; Vermonden, T.; Heger, M. Preclinical evaluation of thermosensitive poly(N-(2-hydroxypropyl) methacrylamide mono/dilactate)-grafted liposomes for cancer thermochemotherapy. Int. J. Pharm. 2018, 550, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Van Elk, M.; Deckers, R.; Oerlemans, C.; Shi, Y.; Storm, G.; Vermonden, T.; Hennink, W.E. Triggered release of doxorubicin from temperature-sensitive poly(N-(2-hydroxypropyl)-methacrylamide mono/dilactate) grafted liposomes. Biomacromolecules 2014, 15, 1002–1009. [Google Scholar] [CrossRef]
- Lindner, L.H.; Eichhorn, M.E.; Eibl, H.; Teichert, N.; Schmitt-Sody, M.; Issels, R.D.; Dellian, M. Novel Temperature-Sensitive Liposomes with Prolonged Circulation Time. Clin. Cancer Res. 2004, 10, 2168–2178. [Google Scholar] [CrossRef] [Green Version]
- Hossann, M.; Wiggenhorn, M.; Schwerdt, A.; Wachholz, K.; Teichert, N.; Eibl, H.; Issels, R.D.; Lindner, L.H. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2491–2499. [Google Scholar] [CrossRef] [Green Version]
- Hossann, M.; Hirschberger, J.; Schmidt, R.; Baumgartner, C.; Zimmermann, K.; Baer, S.; Ratzlaff, C.; Peller, M.; Troedson, K.; Limmer, S.; et al. A Heat-Activated Drug-Delivery Platform Based on Phosphatidyl-(oligo)-glycerol Nanocarrier for Effective Cancer Treatment. Adv. NanoBiomed Res. 2021, 1, 2000089. [Google Scholar] [CrossRef]
- Zimmermann, K.; Hossann, M.; Hirschberger, J.; Troedson, K.; Peller, M.; Schneider, M.; Brühschwein, A.; Meyer-Lindenberg, A.; Wess, G.; Wergin, M.; et al. A pilot trial of doxorubicin containing phosphatidyldiglycerol based thermosensitive liposomes in spontaneous feline soft tissue sarcoma. Int. J. Hyperth. 2017, 33, 178–190. [Google Scholar] [CrossRef]
- Van Valenberg, F.J.P.; Brummelhuis, I.S.G.; Lindner, L.H.; Kuhnle, F.; Wedmann, B.; Schweizer, P.; Hossann, M.; Witjes, J.A.; Oosterwijk, E. DPPG(2)-Based Thermosensitive Liposomes with Encapsulated Doxorubicin Combined with Hyperthermia Lead to Higher Doxorubicin Concentrations in the Bladder Compared to Conventional Application in Pigs: A Rationale for the Treatment of Muscle-Invasive Bladder Cancer. Int. J. Nanomed. 2021, 16, 75–88. [Google Scholar]
- Brummelhuis, I.S.G.; Simons, M.; Lindner, L.H.; Kort, S.; de Jong, S.; Hossann, M.; Witjes, J.A.; Oosterwijk, E. DPPG(2)-based thermosensitive liposomes as drug delivery system for effective muscle-invasive bladder cancer treatment in vivo. Int. J. Hyperth. 2021, 38, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Sebeke, L.C.; Castillo Gómez, J.D.; Heijman, E.; Rademann, P.; Simon, A.C.; Ekdawi, S.; Vlachakis, S.; Toker, D.; Mink, B.L.; Schubert-Quecke, C.; et al. Hyperthermia-induced doxorubicin delivery from thermosensitive liposomes via MR-HIFU in a pig model. J. Control. Release 2022, 343, 798–812. [Google Scholar] [CrossRef]
- Deng, Z.; Xiao, Y.; Pan, M.; Li, F.; Duan, W.; Meng, L.; Liu, X.; Yan, F.; Zheng, H. Hyperthermia-triggered drug delivery from iRGD-modified temperature-sensitive liposomes enhances the anti-tumor efficacy using high intensity focused ultrasound. J. Control. Release 2016, 243, 333–341. [Google Scholar] [CrossRef]
- Lin, W.; Xie, X.; Yang, Y.; Fu, X.; Liu, H.; Yang, Y.; Deng, J. Thermosensitive magnetic liposomes with doxorubicin cell-penetrating peptides conjugate for enhanced and targeted cancer therapy. Drug Deliv. 2016, 23, 3436–3443. [Google Scholar] [CrossRef]
- Dorjsuren, B.; Chaurasiya, B.; Ye, Z.; Liu, Y.; Li, W.; Wang, C.; Shi, D.; Evans, C.E.; Webster, T.J.; Shen, Y. Cetuximab-Coated Thermo-Sensitive Liposomes Loaded with Magnetic Nanoparticles and Doxorubicin for Targeted EGFR-Expressing Breast Cancer Combined Therapy. Int. J. Nanomed. 2020, 15, 8201–8215. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Takashima, M.; Yuba, E.; Harada, A.; Hiramatsu, Y.; Kitagawa, H.; Otani, T.; Maruyama, K.; Aoshima, S. Multifunctional liposomes having target specificity, temperature-triggered release, and near-infrared fluorescence imaging for tumor-specific chemotherapy. J. Control. Release 2015, 216, 69–77. [Google Scholar] [CrossRef]
- Alawak, M.; Abu Dayyih, A.; Mahmoud, G.; Tariq, I.; Duse, L.; Goergen, N.; Engelhardt, K.; Reddy Pinnapireddy, S.; Jedelská, J.; Awak, M.; et al. ADAM 8 as a novel target for doxorubicin delivery to TNBC cells using magnetic thermosensitive liposomes. Eur. J. Pharm. Biopharm. 2021, 158, 390–400. [Google Scholar] [CrossRef]
- Feng, L.; Dong, Z.; Tao, D.; Zhang, Y.; Liu, Z. The acidic tumor microenvironment: A target for smart cancer nano-theranostics. Natl. Sci. Rev. 2018, 5, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Fang, M.; Dong, J.; Xu, C.; Liao, Z.; Ning, P.; Zeng, Q. pH sensitive liposomes delivering tariquidar and doxorubicin to overcome multidrug resistance of resistant ovarian cancer cells. Colloids Surf. B Biointerfaces 2018, 170, 514–520. [Google Scholar] [CrossRef]
- De Oliveira Silva, J.; Fernandes, R.S.; Ramos Oda, C.M.; Ferreira, T.H.; Machado Botelho, A.F.; Martins Melo, M.; de Miranda, M.C.; Assis Gomes, D.; Dantas Cassali, G.; Townsend, D.M.; et al. Folate-coated, long-circulating and pH-sensitive liposomes enhance doxorubicin antitumor effect in a breast cancer animal model. Biomed. Pharmacother. 2019, 118, 109323. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.O.; Fernandes, R.S.; Lopes, S.C.; Cardoso, V.N.; Leite, E.A.; Cassali, G.D.; Marzola, M.C.; Rubello, D.; Oliveira, M.C.; de Barros, A.L. pH-Sensitive, Long-Circulating Liposomes as an Alternative Tool to Deliver Doxorubicin into Tumors: A Feasibility Animal Study. Mol. Imaging Biol. 2016, 18, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, S.B.; de Oliveira Silva, J.; Garcia-Fossa, F.; Leite, E.A.; Malachias, A.; Pound-Lana, G.; Mosqueira, V.C.F.; Oliveira, M.C.; de Barros, A.L.B.; de Jesus, M.B. Mechanistic insights into the intracellular release of doxorubicin from pH-sensitive liposomes. Biomed. Pharmacother. 2021, 134, 110952. [Google Scholar] [CrossRef] [PubMed]
- Sonju, J.J.; Dahal, A.; Singh, S.S.; Gu, X.; Johnson, W.D.; Muthumula, C.M.R.; Meyer, S.A.; Jois, S.D. A pH-sensitive liposome formulation of a peptidomimetic-Dox conjugate for targeting HER2 + cancer. Int. J. Pharm. 2022, 612, 121364. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lee, D.; Lee, Y.; Jeong, E.; Kim, Z.; Koo, H. Double hit strategy using pH-sensitive liposomes containing doxorubicin and pheophorbide-a for combination tumor therapy. Colloid Interface Sci. Commun. 2022, 46, 100565. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Amin, M.; Haemmerich, D.; Van Rhoon, G.C.; Ten Hagen, T.L.M. Hyperthermia and smart drug delivery systems for solid tumor therapy. Adv. Drug Deliv. Rev. 2020, 163–164, 125–144. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugated PLD | Conjugate Type | Preclinical Results (Compared to PLD) | Year | Clinical Results/Trial Status/Possibility to Advance to Clinical Trial |
|---|---|---|---|---|
| MM-302 | antibody | 1997 [77] |
| |
| C225-ILS-DOX | antibody |
| 2003 [78] |
|
| ||||
| ||||
| anti-CD147 ILs-DOX | antibody |
| 2018 |
|
| ||||
| LP16-PLD | peptide |
| 2018 |
|
| LP31-PLD | peptide |
| 2021 |
|
| AHNP-PLD | peptide |
| 2016 |
|
| SS-02-PLD | peptide |
| 2021 |
|
| LinTT1-PLD | peptide |
| 2021 |
|
| PLD-P700 | peptide |
| 2020 |
|
| cRGD-PLD | peptide |
| 2022 |
|
| 5TR1-PLD | DNA aptamer |
| 2018 |
|
| Syl3c-PLD | DNA aptamer |
| 2020 |
|
| TSA14-PLD | RNA aptamer |
| 2016 |
|
| ER-lip | RNA aptamer |
| 2021 |
|
| DOX-MFPL | Carbohydrate |
| 2019 |
|
| TAT-PLD | CPE (linked to PEG2000) |
| 2016 |
|
| TAT-PLD | CPE (linked to PEG1000) |
| 2021 |
|
| DAG-Arg4-PLD | CPE |
| 2021 |
|
| Dual DOX-L | CPE + transferrin |
| 2018 |
|
| D31-PLD | peptide + CPE |
| 2022 |
|
| PLD-100 | CPE |
| 2020 |
|
| Trial ID/Name | Status | Phase | Disease | Intervention |
|---|---|---|---|---|
| NCT03749850/i-GO | recruiting | I | breast cancer | LTLD + Cyclophosphamide + MR-HIFU |
| NCT02536183 | recruiting | I | relapsed/refractory solid tumors | LTLD + MR-HIFU |
| NCT02112656/OPTIMA | completed | III | HCC | LTLD + RFA vs. RFA ( ≥ 45 min) |
| NCT00617981/HEAT | completed | III | HCC | LTLD + RFA vs. RFA (12–60 min based on tumor size) |
| NCT02181075/TARDOX | completed | I | Liver tumors | LTLD + Focused Ultrasound |
| NCT00826085/DIGNITY | completed | I/II | breast cancer | LTLD + Microwave HT (60 min) |
| NCT00441376 | completed | I | HCC | LTLD + RFA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aloss, K.; Hamar, P. Recent Preclinical and Clinical Progress in Liposomal Doxorubicin. Pharmaceutics 2023, 15, 893. https://doi.org/10.3390/pharmaceutics15030893
Aloss K, Hamar P. Recent Preclinical and Clinical Progress in Liposomal Doxorubicin. Pharmaceutics. 2023; 15(3):893. https://doi.org/10.3390/pharmaceutics15030893
Chicago/Turabian StyleAloss, Kenan, and Peter Hamar. 2023. "Recent Preclinical and Clinical Progress in Liposomal Doxorubicin" Pharmaceutics 15, no. 3: 893. https://doi.org/10.3390/pharmaceutics15030893