1. Introduction
Colorectal cancer (CRC) is a malignant tumor occurring in the colon or rectum characterized by high invasiveness, strong metastasis, easy recurrence and poor prognosis [
1]. CRC is the third most common cancer and the second leading cause of cancer-related deaths in the world with an incidence of 10.2% and a mortality rate of 9.2% of all cancers [
2]. So far, standard conventional treatments for CRC are chemoradiotherapy and surgical resection [
3]. However, the former is accompanied by severe toxic and side effects and the latter has a high risk of anal loss [
3]. Despite the emergence of total neoadjuvant therapy (TNT), limited progress has been made in improving the survival outcomes in patients with CRC [
4]. Therefore, the prognosis for patients with metastatic CRC remains poor with a median 5-year survival of only 18.5–27.7% [
4]. The global burden of CRC is 1.93 million new cases and 0.93 million deaths in 2020 [
1]. What is worse, this burden is projected to increase to 3.2 million new cases and 1.6 million deaths by 2040 [
5]. The increasing incidence and deaths make CRC a serious threat to human health and survival. It is urgent to search for safe and effective therapies to address the CRC crisis.
Programmed death-1 (PD-1) is a cell surface receptor that functions as a T cell checkpoint and plays a central role in regulating T cell exhaustion [
6]. A variety of tumor cells, such as CRC, lung cancer and leukemia, can avoid the recognition and killing of T cells by overexpressing the programmed death ligand 1 (PD-L1), resulting in T cell exhaustion in the tumor microenvironment (TME) and realizing immune escape [
7]. The immune checkpoint blockade (ICB) therapy, with PD-L1 as the representative therapeutic target, has achieved great success in the field of cancer immunotherapy due to its unprecedented and durable clinical response in various cancers [
8,
9]. However, with the emergence of drug resistance, low therapeutic response rate and cytotoxicity caused by cytokine storm, the wide application of ICBs in clinical application is limited [
10]. Accordingly, developing other therapeutic methodologies to target PD-L1 for cancer treatment is urgent. Interference with PD-L1 function by various experimental methods in tumor therapy has proven that immune checkpoint silencing may be a better strategy for strengthening therapeutic efficacy than immune checkpoint blocking [
11,
12,
13,
14,
15,
16,
17]. Blocking PD-L1 by silencing can effectively inhibit tumor growth and invasion of CRC [
14,
15,
18,
19]. RNA interference (RNAi) technology can specifically silence the expression of target genes [
11,
12]. siRNA-mediated RNAi, one of the most widely studied techniques in gene therapy, is considered a promising approach to treating refractory cancer due to its high specificity, rapid and robust inhibition and low toxicity [
20,
21,
22]. Therefore, the siRNA-based RNAi-targeted silencing of PD-L1 for CRC treatment has also aroused great research enthusiasm and shown broad potential [
7,
19,
23].
Moreover, many studies have shown that cytosine-phosphate-guanine oligodeoxynucleotides (CpG ODNs) are the most effective immune adjuvants. They can be recognized as pathogen-related molecular patterns (PAMPs) by toll-like receptor 9 (TLR9) to activate innate and acquired immune responses [
24]. Thus, CpG ODNs have been used alone or in combination with other drugs for preventing or treating cancer [
25,
26,
27]. Furthermore, CpG ODNs are excellent candidates for combination with PD-L1 blockades [
24]. Immunoregulation using CpG ODNs at the tumor site could promote the maturation and antigen presentation of dendritic cells (DCs) and activate the rapid proliferation and infiltration of cytotoxic T lymphocytes (CTLs) in TME, thereby restoring resistance to the PD-L1 blockade and inhibiting the growth, metastasis and recurrence of CT26 or MC38 cell-driven CRC [
27,
28]. Based on the above evidence, we suggest that CpG ODNs may have great potential as immune adjuvants to improve the efficacy of siPD-L1-mediated immune checkpoint silencing in CRC.
The therapeutic effects of either siPD-L1 or CpG ODNs largely depend on the payload and delivery efficiency of the carrier. Finding a safe and efficient co-delivery carrier of siPD-L1 and CpG ODNs is the premise of realizing their joint application. Mesoporous SiO
2 (or Mesoporous silica, MS) nanomaterials are promising gene or drug carriers for improving cancer therapy due to their attractive properties of a high porosity and loading capacity, efficient delivery efficiency, good biocompatibility, facile surface modification and self-adjuvanticity [
29,
30]. Gold nanomaterials, with elegant thermal, optical or chemical properties due to quantum size effects, can be used as photothermal therapy (PTT) reagents, carriers and biosensors for therapeutic or diagnostic applications [
31,
32,
33,
34,
35,
36]. The nanocomposites (NPs) formed by gold nanorods (AuNRs) coated with MS possess the above advantages while functioning as carriers. MS-coated AuNR NPs could be used in siPD-L1-mediated immune checkpoint silencing for bladder cancer treatment [
37] or chemotherapeutic/photothermal synergistic therapy for CRC treatment [
38]. Such nanocomposites could also co-deliver doxorubicin (DOX) and Bcl-2-targeted siRNA to mediate the three-fold synergic therapy of PTT, chemotherapy and gene therapy for the potential treatment of breast cancer [
39]. However, the co-delivery of CpG ODNs and siPD-L1 by MS-coated AuNR NPs for the immunological and gene synergistic therapy of CRC is still extremely rare. In addition, PTT can be divided into traditional photothermal therapy (>45 °C) and mild photothermal therapy (MPTT) (42–45 °C) [
40]. MPTT could not only avoid the damage of normal tissues around the tumor caused by hyperthermia, but also increase the cell membrane permeability and improve cellular uptake and gene transfection efficiency [
40,
41]. Importantly, MPTT was beneficial to the survival and release of tumor-associated antigens (TAAs), it could effectively activate the antigen presentation of DCs, it could promote the proliferation and differentiation of CTLs and finally, it could effectively enhance the anti-tumor immune efficacy [
40,
42]. Due to the adjustability of laser power and radiation time, MS-coated AuNRs NPs have the potential to realize mild photothermal heating for promoting delivery efficiency and tumor immunotherapy efficacy.
In this context, we successfully constructed a novel multifunctional mesoporous silica-coated gold nanorod to mediate mild photothermal heating-enhanced gene/immunotherapy for CRC. As illustrated in
Scheme 1, AuNRs@MS (AS) NPs were synthesized by coating the mesoporous silica (MS) shells with gold nanorods (AuNRs) as the cores. Then, the co-delivery vector AuNRs@MS/CpG ODN@PEG-bPEI NPs (ASCP) were prepared by two-step surface modification of CpG ODNs-loading and polyethylene glycol-branched polyethyleneimine (PEG-bPEI)-coating. Exhibiting excellent biosafety in vitro, ASCP-based NPs promoted the maturity and antigen presentation ability of DCs by delivering CpG ODNs. Next, ASCP-based NPs not only mediated mild photothermal heating to kill tumor cells, but also facilitated the release of tumor-related antigens (TTAs) from tumor lysates to promote the DCs maturity and antigen presentation further. Furthermore, the transfection and endo-lysosomal escape of payloads were strengthened by mild photothermal heating mediated by ASCP-based NPs, resulting in an enhanced PD-L1 gene silencing effect to relieve T cell exhaustion and inhibit tumor immune escape. The enhanced DCs maturity and PD-L1 gene silencing effectively promoted the proliferation and infiltration of effector T lymphocytes and natural killer cells (NKs) in TME, which could further effectively kill tumor cells following the tumor ablation effect of PTT, and ultimately lead to the strong inhibition of CRC. The findings in this study provide new insights into the design of gene/immune/mild photothermal synergies for tumor therapy and may contribute to translational nanomedicine for CRC treatment.
2. Materials and Methods
2.1. Materials
Cetyltrimethylammonium bromide (CTAB, No. H9151), polyethylene glycol-branched polyetherimide (PEG-bPEI, No. 900926), ribonuclease A (RNase A, No. 10109169001) and 4’,6-diamidino-2-phenylindole (DAPI, MBD0015) were purchased from Sigma-Aldrich Co., Ltd. (St. Louis, MO, USA). Gold chloride solution (HAuCl4, No. g109456), NaBH4 (No. s108355), silver nitrate solution (AgNO3, No. s116264), sodium hydroxide solution (NaOH, No. S817971), formaldehyde solution (37%, No. F809702), ethyl acetate (EA, No. E809178), L-ascorbic acid (No. A800296) and tetraethyl orthosilicate (TEOS, No. T819507) were obtained from Macklin Co., Ltd. (Shanghai, China). Phosphate buffer solution (PBS, No. P1010), tris-acetate-EDTA buffer (TAE, No. T1060) and 0.25% trypsin-EDTA solution (No. T1300) were purchased from Solarbio Biotech (Shanghai, China). Hoechst 33342 (0.01 mg/mL; No. 62249), Roswell Park Memorial Institute 1640 medium (RPMI 1640, No. 11875119), penicillin-streptomycin solution (PS, No. 15070063), fetal bovine serum (FBS, NO.10100147) and Lipofectamine™ 3000 (Lipo3000, No. L3000001) were purchased from Gibco Biotech (ThermoFisher Scientific, Waltham, MA, USA). Lyso-tracker red (DND 99, No. C1046), calcein-AM (No. C2012) and red blood cell lysate (No. C3702) were purchased from Beyotime Biotech (Shanghai, China). Cell Counting Kit-8 (CCK-8, No. BS350A), granulocyte-macrophage colony-stimulating factor (GM-CSF, No. BSEM-026), interleukin-4 (IL-4, No. BSEM-004) and lipopolysaccharide (LPS, No. BS904) were from Biosharp Co., Ltd. (Beijing, China). TRIcom reagent (No. TR251) was from TIANMO BIOTECH Co., Ltd. (Beijing, China). QuantiTect Reverse Transcription Kit (No. 205311) was purchased from QIAGEN Biology Co., Ltd. (Frankfurt, Germany). Bestar SybrGreen qPCR Master Mix Kit (No. DBI-2043) was from DBI Bioscience Co., Ltd. (Ludwigshafen, Germany). 5-(and 6-)carboxyfluorescein diacetate succinimidyl ester (CFSE or CFDA-SE, No. abs9106) and propyl iodide solution (PI, No. abs9358) were purchased from Absin Biology (Shanghai, China). All the flow cytometry antibodies of Ms CD45 PerCP 30-F11 (No. 557235), Ms CD3e APC 145-2C11 (No. 553066), Ms CD4 PE-Cy7 RM4-5 (No. 552775), Ms CD8a FITC 53-6.7 (No. 553030), Ms CD49b PE HM Alp2 (No. 558759), Ms CD11c APC HL3 (No. 550261), Ms CD86 PE GL1 (No. 561963) and Ms I-A I-E Alexa 488 M5/114.15.2 (No. 562352) were purchased from BD Pharmingen (Franklin Lake, NJ, USA).
2.2. Synthesis of siRNAs and Primers
All the custom synthesized siRNAs were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China) and all the custom synthesized primers and CpG ODN 2395 (abbreviated as CpG) were purchased from Sangon Biotech (Shanghai, China).
The siRNA sequences were listed as follows: sense of siPD-L1: 5′-GACUCAAGAUGGAACCUGAdTdT-3′, antisense of siPD-L1:5′-UCAGGUUCCAUCUUGAGUCdTdT-3′, sense of siNC: 5′-CGAAGUGUGUGUGUGUGGCdTdT-3′, antisense of siNC: 5′-GCCACACACACACACUUCGdTdT-3′. siNC: negative control siRNA. The sequence of CpG was 5′-TCGTCGTTTTCGGCGCGCGCCG-3′. The siRNA labeled with Cy5 (siCy5) or FAM (siFAM) in the 5’ tag and CpG labeled with Cy3 (CpGCy3) in the 5’ tag was used for fluorescence detection. The primer sequences for qPCR were listed as follows: Mouse-PD-L1-F: 5′-TGCTGCATAATCAGCTACGG-3′, Mouse-PD-L1-R: 5′-CCACGGAAATTCTCTGGTTG-3′. Mouse-GAPDH-F: 5′-GTGGACCTCATGGCCTACAT-3′, Mouse-GAPDH-R: 5′-TGTGAGGGAGATGCTCAGTG-3′.
2.3. Preparation of AS NPs
The silica-coated gold cores with a shell-core structure were prepared by a method previously reported in the literature [
39,
43,
44]. Typically, the gold seed solution was prepared by mixing aqueous HAuCl
4 (0.01 M, 0.25 mL) and cetyltrimethylammonium bromide (CTAB, 0.1 M, 10 mL) in a 15 mL plastic tube. A freshly prepared, ice-cold NaBH
4 solution (0.01 M, 0.6 mL) was then injected quickly into the mixture solution, followed by rapid inversion for 2 min. The seed solution was kept at room temperature (RT) for at least 2 h before use. To grow gold nanorods (AuNRs), HAuCl
4 (0.01 M, 2.0 mL) and AgNO
3 (0.01 M, 0.4 mL) were mixed with CTAB (0.1 M, 40 mL) in a 50 mL plastic tube. HCl (1.0 M, 0.8 mL) was then added to adjust the pH of the solution to 1–2, followed by the addition of ascorbic acid (0.1 M, 0.32 mL). Finally, the seed solution (0.096 mL) was injected into the growth solution. The solution was gently mixed for 10 s and left undisturbed at RT for at least 6 h before use. Notably, the amount of AuNRs before and after should be quantified. For further preparation of AuNRs@MS NPs (AuNRs NPs coated with mesoporous silica shells), the CTAB was subsequently removed by centrifugation, the pH was adjusted to 10 by a NaOH solution (0.1 M), TEOS/ethanol solution was added to the mixture in three portions and the reaction was gently stirred for 48 h at RT to obtain AuNRs@MS NPs (abbreviated as AS NPs) with a shell-core structure.
2.4. Modification from AS NPs to ASCP NPs
The AS NPs solution and CpG solution were mixed at the AS:CpG mass ratio of 5:1. Next, the mixture was stirred with a magnetic mixer at RT, 100 rpm for 6 h. The excess CpG was removed three times by repeated operations of 5 min-centrifugation (10,000 rpm, 25 °C) and pure water washing. In order to get the siRNA-load capacity, PEG-bPEI was modified in the outer layer of the ASC NPs. Briefly, 1 mg of AS NPs was dissolved in 10 mL of ultra-pure water, then mixed with 0.05 mL of PEG-bPEI solution (100 mg/mL). Then, the mixture was magnetically stirred overnight at RT, 300 rpm. After the three repeated operations of 10 min-centrifugation (10,000 rpm, 25 °C) and pure water washing, the excess PEG-bPEI was removed. Finally, the CpG ODN/siRNA co-delivery vectors of ASCP were successfully obtained.
2.5. Preparation of ASCP/siRNA NPs
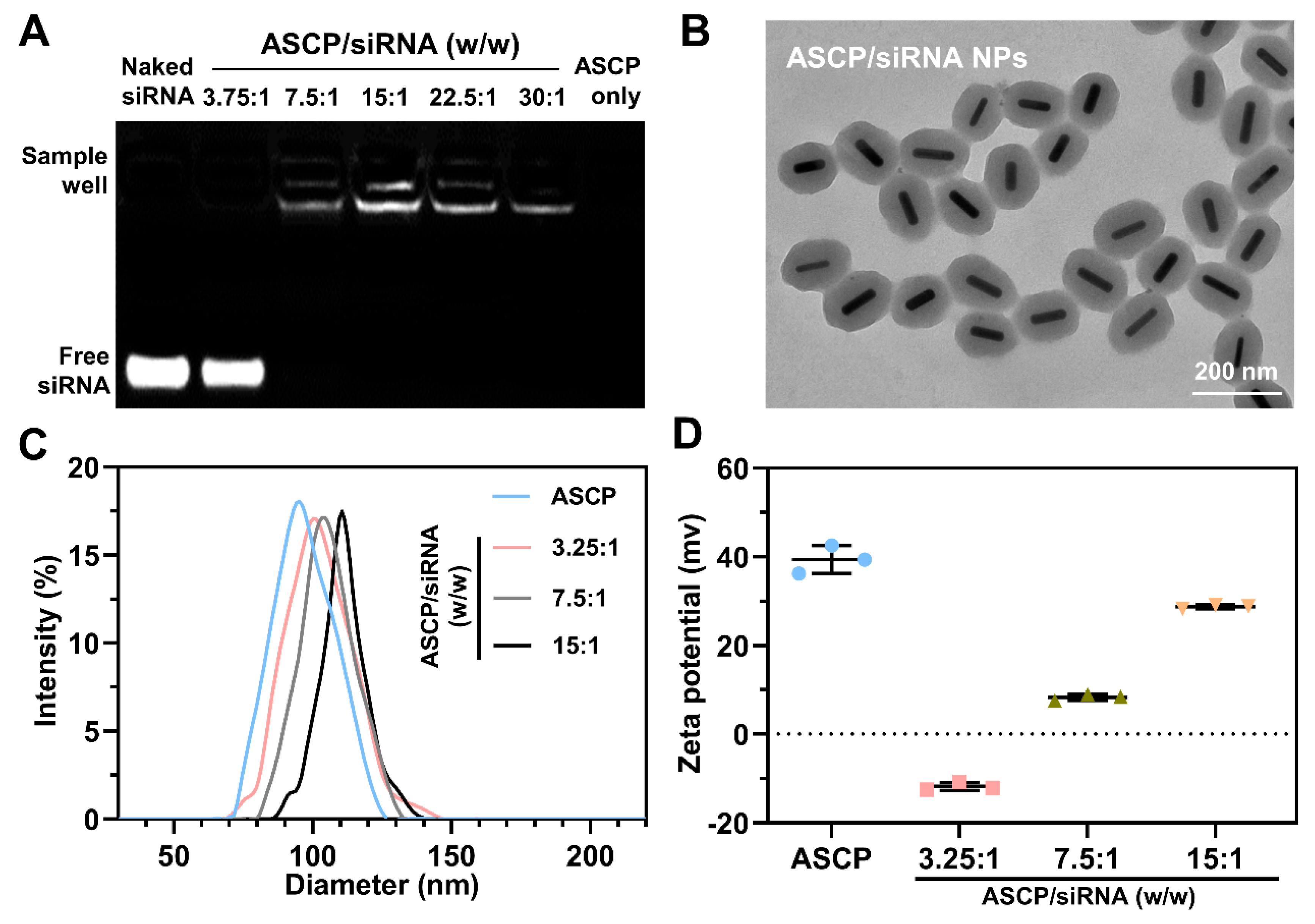
ASCP NPs and siRNA were dissolved in diethylpyrocarbonate (DEPC) water. ASCP/siRNA NPs were prepared by gently mixing the ASCP solution with the siRNA solution at different mass ratios (3.75:1, 7.5:1, 15:1, 22.5:1, 30:1) and incubated at RT for at least 40 min. All the weights of ASCP-based NPs were calculated by gold nanorod cores. The dose of siRNA was fixed at 0.133 μg (10 pmol) in each sample well. Then the siRNA binding capacity of ASCP NPs was evaluated by the agarose gel electrophoresis (110 V, 10 min) in a tris-acetate-EDTA (TAE) running buffer. The gel was imaged under UV transillumination (Fluor Chem E, Protein Simple, San Jose, CA, USA) and the gray values of the gel images were analyzed by Image J software v.1.53 (National Institutes of Health, NIH, Bethesda, MD, USA).
2.6. Characterization
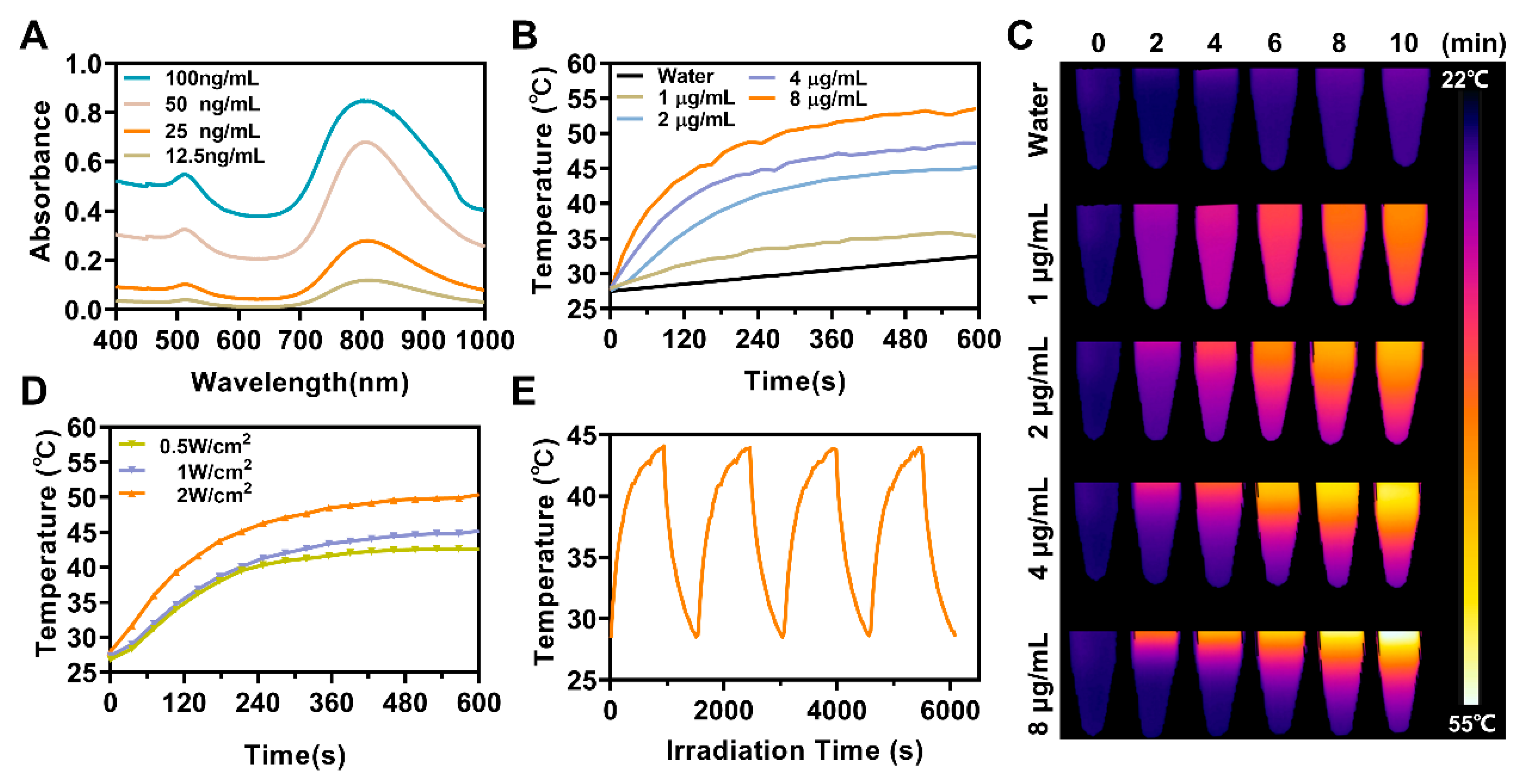
The zeta potential (surface potential) values and hydrodynamic diameters of these NPs with different degrees of modification (AS NPs, ASC NPs, ASCP NPs and ASCP/siRNA NPs) were measured by dynamic light scattering (DLS, Nano-ZS90, Malvern, UK) and their morphological properties were detected by transmission electron microscopy (TEM, HT7700, Hitach Ltd., Ibaraki, Japan). The absorbance intensity of the ASC aqueous solution was measured at 275 nm by a UV-Vis-NIR spectrophotometer (TP-720, Tuopu Instrument, Tianjin, China). The plasmonic absorptions of AuNRs before and after coating with MS shells were also measured using the UV-Vis-NIR spectrophotometer. The stability of ASCP NPs (2 μg/mL) before and after irradiation (808 nm laser, 1 W/cm2) was further investigated by TEM.
2.7. In Vitro Photothermal Properties Measurement
For the concentration-dependent evaluation of photothermal properties in vitro, 1 mL aqueous dispersion of ASCP NPs with different concentrations (1, 2, 4, 8 μg/mL) was irradiated by an 808 nm laser (1 W/cm2) for 10 min. For power density-dependent evaluation, a 1 mL aqueous dispersion of ASCP (2 μg/mL) was irradiation by an 808 nm laser at various laser densities for 10 min. For the photothermal stability measurement, a 1 mL aqueous dispersion of ASCP (2 μg/mL) was irradiated in four consecutive laser-on/off cycles with a power density of 0.5 W/cm2. During the observation of irradiation, the real-time temperature images and temperature alteration with an accuracy of 0.1 °C were recorded by a FLIR A300 infrared thermal imaging camera.
2.8. Cells and Culture
The mouse colorectal cancer cell line MC38 and mouse breast cancer cell line 4T1 were purchased from the National Infrastructure of Cell Line Resource (Beijing, China). After the mice were sacrificed with an overdose of isoflurane, the spleen was isolated, cut up, gently ground and filtered through a 200-mesh screen to obtain the single-cell suspension of the spleen cell. Meanwhile, the tibia, femur and ilium of the lower extremities in mice were isolated. Then a 1 mL syringe was used for flushing out bone marrow cells (BMCs) from the marrow cavity of these bones with RPMI 1640 complete medium. After 2 min mild treatment with red blood cell lysate and washing with PBS, experimental splenocytes and BMCs were obtained, respectively. All the cells were cultured in RPMI 1640 complete medium containing 10% FBS and 1% PS, and maintained in a 37 °C incubator with 5% CO2.
2.9. Mice and Feeding
Six–eight weeks old female C57BL/6J mice were purchased from the Medical Laboratory Animal Center of Guangdong Province, China. The mice were kept in an individually ventilated cages (IVCs) system in a specific pathogen freedom (SPF) animal house. The SPF environment conditions were set to a temperature of 22.5 ± 2.5 °C, humidity of 55 ± 15%, noise of less than 60 decibels, accompanied by a natural 12-h cycle of alternating light and dark. All the animal experiments were approved by the Animal Ethical and Welfare Committee of Shenzhen University (AEWC) and were performed strictly according to animal care guidelines.
2.10. Hemolysis Assay
The hemolysis assay was performed to evaluate the biocompatibility of ASCP NPs on red blood cells (RBCs). An amount of 1 mL of blood from healthy C57BL/6J mice was diluted in a 9 mL phosphate-buffered solution (PBS, pH 7.4). Then, the blood diluent was centrifuged at 4 °C, 5000 rpm for 10 min and the supernatant was discarded. After repeating the above operation three times, the precipitation of RBCs was diluted in 1 mL PBS. Finally, the RBCs working solution for the hemolysis assay consisted of 5% (v/v) of RBCs in PBS. To evaluate the hemolytic effects, 200 μL of RBCs working solution were incubated with 800 μL of ASCP NPs with different concentrations for 4 h in a 37 °C incubator. An equal volume of incubated H2O or PBS was used as a positive control or negative control (NC), respectively. After incubation, the samples were centrifugated at 5000 rpm for 5 min at RT. In addition, 100 μL of supernatants were extracted to quantify hemoglobin by recording the absorbance at 540 nm (A540) with a microplate reader (EPOCH-2, Bio-Tek Instruments, Winooski, VT, USA). The percentage of the hemolysis rate was calculated as follows: hemolysis rate (%) = (A540 of sample − A540 of NC) / (A540 of PC − A540 of NC) × 100%.
2.11. In Vitro Cell Cytotoxicity
Cell viability was measured by utilizing the Cell Counting Kit-8 (CCK-8) assay. Different cells (MC38, 4T1 and splenocytes) were seeded into 96-well plates at a density of 1 × 104 cells/well and incubated overnight, respectively. Subsequently, cells were incubated in 100 μL RPMI 1640 complete medium with different concentrations of ASCP NPs (0, 0.25, 0.5, 1, 2, 4, 8, 16 μg/mL) for 24 h. ASCP NPs-free treated cells were used as the control group (Blk group). After adding 10 μL CCK-8 in each well, cells were incubated for 2 h at 37 °C. Then, the absorbance was measured with a microplate reader (EPOCH-2, Bio-Tek Instruments, Winooski, VT, USA) at a wavelength of 450 nm. The cell viability was calculated according to the following formula: cell viability (%) = A450 of the test well/A450 of the control well (Blk group) × 100%.
2.12. Preparation of BMDCs
Mouse bone marrow cells (BMCs) were isolated and co-induced to prepare bone marrow-derived dendritic cells (BMDCs) using interleukin-4 (IL-4) and granulocyte macrophage colony-stimulating factor (GM-CSF) according to the method reported by Tang et al. [
45]. In brief, mouse BMCs were seeded in a 24-well plate at a density of 1 × 10
5 cells per well and cultured in 1 mL of RPMI 1640 complete medium. GM-CSF (20 ng/mL) and IL-4 (10 ng/mL) were added into the cultures on day 1 and day 3, respectively, to promote the differentiation of BMCs into BMDCs. After observing most cells sticking to the wall on day 8, the treated BMCs were digested by trypsin, washed with PBS and then stained with the antibody of Ms CD11c APC HL3. Finally, the expression of CD11c
+ in the cells was detected by flow cytometry (FCM; FACS Aria II, BD medical device, Franklin Lake, NJ, USA) to confirm whether the preparation of BMDCs was successful.
2.13. Transfection Efficiency of ASCP/siRNA NPs
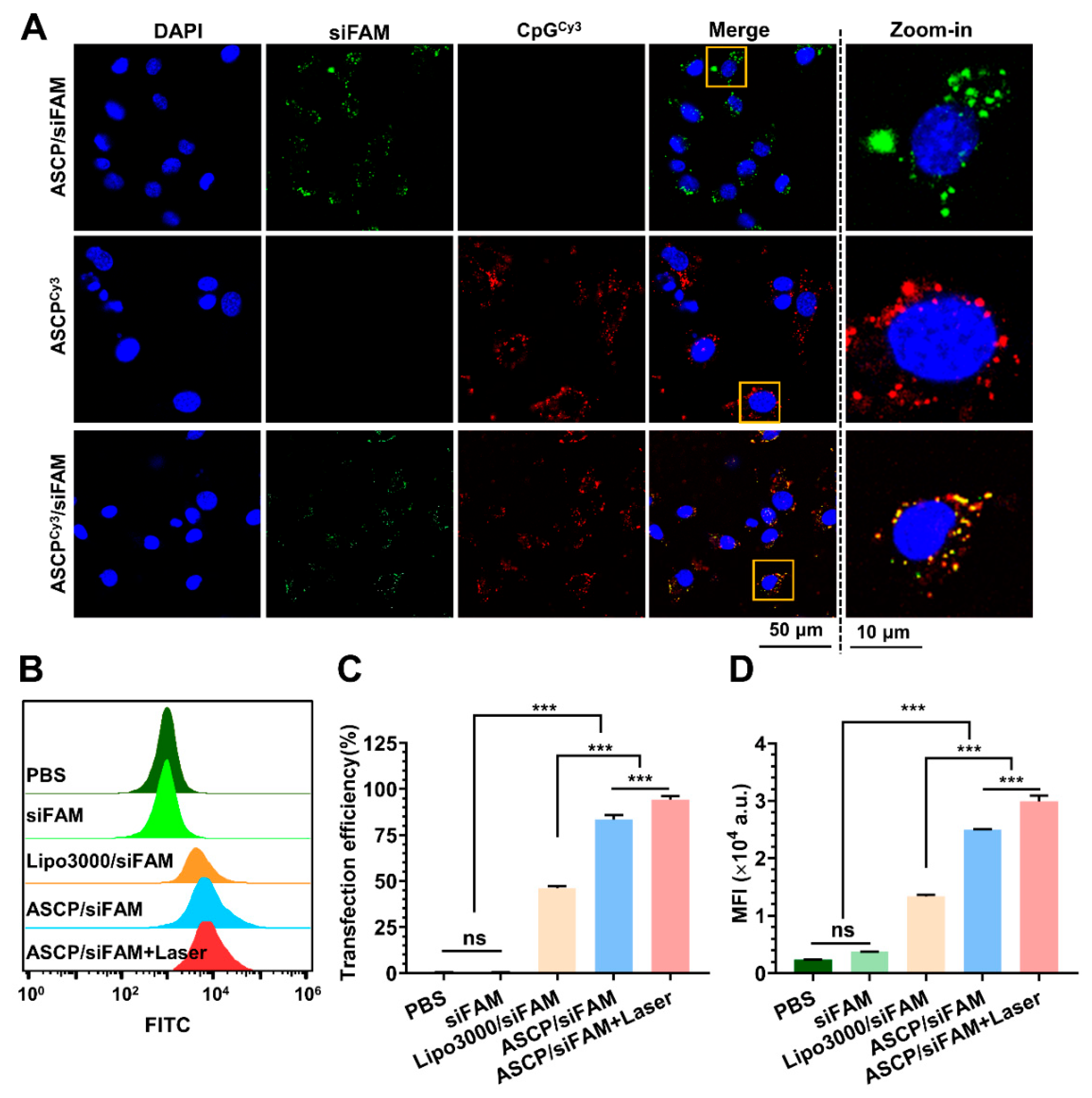
MC38 cells were seeded in a 24-well plate at a density of 1 × 105 cells per well and cultured in 1 mL of RPMI 1640 complete medium overnight for adherent. After removing the complete medium, cells were washed twice with 1 mL of PBS and 0.5 mL of serum-free RPMI 1640 medium was added to each well. Meanwhile, the ASCP NPs and siRNA labeled with FAM (siFAM) were mixed and incubated at the weight ratio of 7.5:1 at RT for at least 40 min to form ASCP/siFAM NPs for transfection. The final transfected concentration of ASCP NPs was 2 μg/mL and the dose of siRNA was 0.3253 μg (25 pmol). The Lipo3000 was used as the positive control and PBS was used as the negative control. Afterward, cells were transfected with the ASCP/siRNA NPs for 4 h in a 37 °C incubator with 5% CO2. Then, cells were treated with or without a 10 min-irradiation inspired by an 808 nm laser at the power density of 0.5 W/cm2 and cultured for another 4 h. After transfection, the old medium was discarded, the cells were washed twice with 1 mL PBS and 1 mL of RPMI 1640 complete medium was added for further culturing. Next, the 8 h post-transfection cells were washed three times with PBS and collected after trypsinization with a 0.25% Trypsin-EDTA solution. Finally, flow cytometry (FCM; CytoFLEX, Beckman, CA USA) was used to detect the intracellular transfection efficiency of FAM-labeled-ASCP/siRNA NPs with the channel set as FAM. Meanwhile, the mean fluorescence intensity (MFI) of the FAM channel recorded in FCM detection was also quantified.
2.14. Intracellular Uptake
To explore the co-delivery ability of ASCP to CpG and siRNA in MC38 cells, siRNA labeled with FAM (siFAM) and CpG labeled with or without Cy3 were used in the preparation process of ASCP-based NPs from the previous method. Firstly, the ASCP NPs or ASCPCy3 NPs and siFAM were mixed and incubated at a mass ratio of 7.5:1 at RT for 40 min to form ASCP/siFAM NPs and ASCPCy3/siFAM NPs. Subsequently, MC38 cells seeded in confocal dishes (5 × 105 cells/dish) were transfected with the NPs of ASCPCy3, ASCP/siFAM and ASCPCy3/siFAM. The final transfected concentration of ASCP NPs was 2 μg/mL and the dose of siRNA was 50 pmol. After 6 h-transfection, cells were washed three times with 1 mL PBS and the nuclei were stained with 500 μL of Hoechst 33342 (0.01 mg/mL) for 15 min at 37 °C. Next, cells were washed with 1 mL PBS three times after removing the staining buffer. Finally, cells were soaked in 1 mL PBS for fluorescence imaging by using a confocal laser scanning microscope (CLSM; ZEISS LSM880 AiryScan, Jena, Germany) with the filters set for DAPI, FAM and Cy3.
For the intracellular uptake test of NPs by BMDCs, the prepared BMDCs were seeded in confocal dishes (5 × 105 cells/dish) and cultured overnight for adherence. Then, BMDCs were transfected with ASCPCy3 NPs and ASCPCy3/siFAM NPs, same as the above methods. After the 6 h transfection, BMDCs were treated with three repeated PBS washings, followed by 30-min-staining of DAPI (0.01 mg/mL). The BMDCs were then washed with PBS 3 times and 1 mL of PBS was added for confocal fluorescence detection. Images were taken by the CLSM with the filters set for DAPI, FAM and Cy3 to assess the cellular uptake of ASCP-based NPs by BMDCs.
2.15. Endo-Lysosome Escape Observation
MC38 cells seeded in confocal dishes (5 × 105 cells/dish) were transfected with ASCP/siFAM NPs prepared at a weight ratio of 7.5:1 for 4 h. The final transfected concentration of ASCP NPs was 2 μg/mL and the dose of siRNA was 50 pmol. Next, cells were irradiated with or without an 808 nm laser (0.5 W/cm2) for 10 min and cultured for another 4 h. After a total 8-h transfection, cells were washed three times with PBS. Then, the cells were stained with 1 mL PBS containing 50 nM of Lysotracker Red and 1 μg/mL Hoechst 33342 for 30 min. Next, the staining buffer was removed and the cells were washed three times with PBS. Finally, the cells were re-immersed in 1 mL PBS for endo-lysosome escape observation under the above CLSM with the filters set for DAPI, FAM and Cy3.
2.16. In Vitro Immune Activation Effect Induced by ASCP-Based NPs Only
The prepared BMDCs and splenocytes were seeded in a 24-well plate at a density of 1 × 105 cells per well and cultured overnight. Then PBS, CpG (1 mg/mL), lipopolysaccharides (LPS, 1 mg/mL), ASCP NPs (2 μg/mL) and ASCP/siPD-L1 NPs (2 μg/mL by ASCP NPs mass) were added to different culture wells of cells for 24-h-incubation. Then, cells were digested by trypsin and washed with PBS for three times. For the activation effect test of ASCP-based NPs on BMDCs, the treated BMDCs were stained with the antibodies of Ms CD45 PerCP 30-F11, Ms CD11c APC HL3, Ms CD86 PE GL1 and Ms I-A I-E Alexa 488 M5/114.15.2 (dye for MHC-II+ cells). Finally, the maturity of BMDCs gated in CD45+CD11c+ cells was measured by flow cytometry to assess the in vitro immune activation effect induced by co-incubation on BMDCs. The CD86+MHC-II+ cells were mature BMDCs. For the activation effect test of ASCP-based NPs on splenic lymphocytes, the treated splenocytes were stained with the antibodies of Ms CD45 PerCP 30-F11, Ms CD3e APC 145-2C11, Ms CD4 PE-Cy7 RM4-5, Ms CD8a FITC 53-6.7 and Ms CD49b PE HM Alp2. Finally, the activation effect of different formulations of NPs on the splenic lymphocyte differentiation was detected by flow cytometry gated in CD45+ cells. CD3 was the marker of T lymphocytes, CD4 and CD8 were the markers of effector T lymphocytes and CD49b was the marker of natural killer cells (NKs).
2.17. Fluorescence Images of Live–Dead Staining
MC38 cells were seeded into a 24-well plate (5 × 104 cells/well) and incubated overnight for adherence. After removing the complete medium, cells were washed twice with 1 mL PBS and transfected with 100 μL of serum-free RPMI 1640 medium containing ASCP NPs (final concentrations: 2 or 4 μg/mL) for 4 h. Then, cells were treated with or without irradiation inspired by an 808 nm laser (0.5 W/cm2) for 10 min. After being cultured for 24 h, cells were treated with twice PBS washing and stained with calcein-AM (for live cells: green fluorescence) and PI (for dead cells: red fluorescence). Fluorescence images of live–dead staining of MC38 cells were obtained with a fluorescent microscope (Leica M205 FCA, Wetzlar, Germany) with filters set for FITC and PI.
2.18. Proliferation Inhibition Analysis with or without Irradiation
A CCK-8 assay was employed to carry out the proliferation inhibition analysis. MC38 cells were seeded into 96-well plates at a density of 1 × 104 cells/well and incubated overnight for adherence. Subsequently, cells were treated with 100 μL RPMI 1640 complete medium with different concentrations of ASCP NPs (0, 0.25, 0.5, 1, 2, 4, 8, 16 μg/mL). Cells treated with an equal volume of PBS were used as the control group. After a 4-h treatment with NPs, MC38 cells were irradiated with or without an 808 nm laser (0.5 W/cm2) for 10 min. After continuing the culture for another 20 h, the cells were added with 10 μL CCK-8 in each well and then incubated for 2 h at 37 °C. Finally, the cell viability was evaluated with a CCK-8 kit by measuring the absorbance of 450 nm (A450) and calculated as follows: cell viability (%) = A450 of test well/A450 of PBS well without irradiation × 100%.
2.19. Gene Expression Assay
After being cultured overnight, MC38 cells seeded in 6-well plates (5 × 10
5 cells per well) were transfected with PBS, siPD-L1 only, ASCP/siNC NPs, ASCP/siPD-L1 NPs and Lipo3000/siPD-L1 NPs for 4 h following the methods described in the above siRNA transfection. The final transfected concentration of ASCP NPs was 2 μg/mL and the dose of siRNA was 50 pmol. Next, the cells were irradiated with or without the 808 nm laser (0.5 W/cm
2) for 10 min and cultured for another 20 h. Then, the transcription level of the PD-L1 gene of the 24 h post-transfection MC38 cells was investigated using quantitative real-time polymerase chain reaction (qPCR) according to previous experience [
46]. Briefly, the total RNA was extracted from MC38 cells using a TRIzol reagent and quantified using a micro-spectrophotometer (EPOCH-2, Bio-Tek Instruments, Winooski, VT, USA). Then, the total RNA (1 μg) was reversely transcribed to complementary deoxyribonucleic acid (cDNA) by using the QuantiTect Reverse Transcription Kit in accordance with the manufacturer’s instructions. Finally, the mRNA level of the PD-L1 gene was measured by a qPCR instrument (QuantStudio-1, Appliedbio systems, Waltham, MA, USA) by using the Bestar SybrGreen qPCR Master Mix, followed by normalizing to the expression of GAPDH.
2.20. In Vitro Killing Effect of Co-Incubation on MC38 Cells
In order to simulate the T-cell-mediated anti-cancer immune response in vitro, the killing effects of co-incubation of BMDCs and splenocytes on ASCP-based NPs-treated MC38 cells were detected with or without laser irradiation. Briefly, MC38 cells in the logarithmic phase were trypsinized, collected and stained with CFSE (5 μM) at 37 °C for 10 min under gentle shaking. After washing them twice with an equal volume of PBS, the CFSE labeled cells were resuspended in the RPMI 1640 complete medium and seeded in 12-well plates at 1 mL volume with 1 × 105 cells. After being cultured overnight for adherent, the MC38 cells were transfected with PBS, CpG (1 mg/mL), ASCP (1 μg/mL), ASCP (2 μg/mL), ASCP/siPD-L1 (1 μg/mL by ASCP mass) and L1 ASCP/siPD-L1 (2 μg/mL by ASCP mass) for 4 h. Then, the MC38 cells treated with or without 10-min mild photothermal heating inspired by an 808 nm laser (0.5 W/cm2) and cultured for another 4 h. Next, BMDCs and splenocytes were added in each well at the number ratio of effector cells to target cells of 50:1. After being co-incubated with BMDCs and splenocytes for 48 h, the killing effects on MC38 cells in different treatment groups were analyzed by flow cytometry with a PI staining buffer by gating in PI+CFSE+ cells.
2.21. In Vitro Immune Activation Effect Induced by Co-Incubation
A similar experimental condition as described above was used to test the in vitro immune activation effect induced by co-incubation. In brief, after being treated with different formulations of NPs with or without 10-min mild photothermal heating, MC38 cells were co-incubated with BMDCs and splenocytes for 48 h. Then, the co-incubated cells in each group were divided into 2 tubes. For the activation effect detection on BMDCs, the co-incubated cells were stained with the antibodies of Ms CD45 PerCP 30-F11, Ms CD11c APC HL3, Ms CD86 PE GL1 and Ms I-A I-E Alexa 488 M5/114.15.2. Finally, the maturity of BMDCs (CD86+MHC-II+%) gated in CD45+CD11c+ cells was measured by flow cytometry to assess the in vitro activation effect of ASCP NPs and ASCP/siPD-L1 NPs on BMDCs. For the activation effect detection on splenic lymphocytes, the co-incubated cells were stained with the antibodies of Ms CD45 PerCP 30-F11, Ms CD3e APC 145-2C11, Ms CD4 PE-Cy7 RM4-5, Ms CD8a FITC 53-6.7 and Ms CD49b PE HM Alp2. Finally, the in vitro immune activation effect induced by co-incubation on splenic lymphocyte differentiation was detected by flow cytometry gated in CD45+ cells. The CD3+CD4+ cells and CD3+CD8+ cells were effector T lymphocytes, and the CD49b+ cells were NKs.
2.22. Statistical Analysis Methods
The number of biological samples in experiments was not less than three (n ≥ 3). Statistical analysis and graphs collection were performed using one-way analysis of variance (ANOVA) with Tukey multi-comparisons by GraphPad Prism v.8.0 (GraphPad Software Inc., San Diego, CA, USA). All data were shown as means ± SD. A p-value less than 0.05 was considered statistically significant, and * p < 0.05, ** p < 0.01, *** p < 0.001.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






