

Self-Assembled Lecithin-Chitosan Nanoparticles Improved Rotigotine Nose-to-Brain Delivery and Brain Targeting Efficiency
 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Rotigotine Nanoparticles
2.3. Size and Zeta Potential Measurements
2.4. Entrapment Efficiency and Drug Loading
2.5. Differential Scanning Calorimetry
2.6. Field Emission Scanning Electron Microscopy
2.7. Transmission Electron Microscopy
2.8. Storage Stability of Nanoparticles
2.9. In Vitro Drug Release
2.10. Ex Vivo Nasal Drug Permeation
2.11. In Vivo studies in Wistar Rats
2.11.1. Administration of Intranasal (i.n.) Formulation to Rats
2.11.2. Mucociliary Transport Time RTG-LCNP
2.11.3. Brain and Plasma PK Analysis
2.12. Histopathology of Brain
3. Results and Discussion
3.1. Effect of Drug: Lecithin Ratio on Nanoparticle Size
3.2. Effect of Lecithin:CS Ratio on the Particle Size and PDI
3.3. Effect of the Amount of Poloxamer 407 on the Particle Size, PDI and %EE
3.4. Effect of pH of CS Solution on the Particle Size and %DL
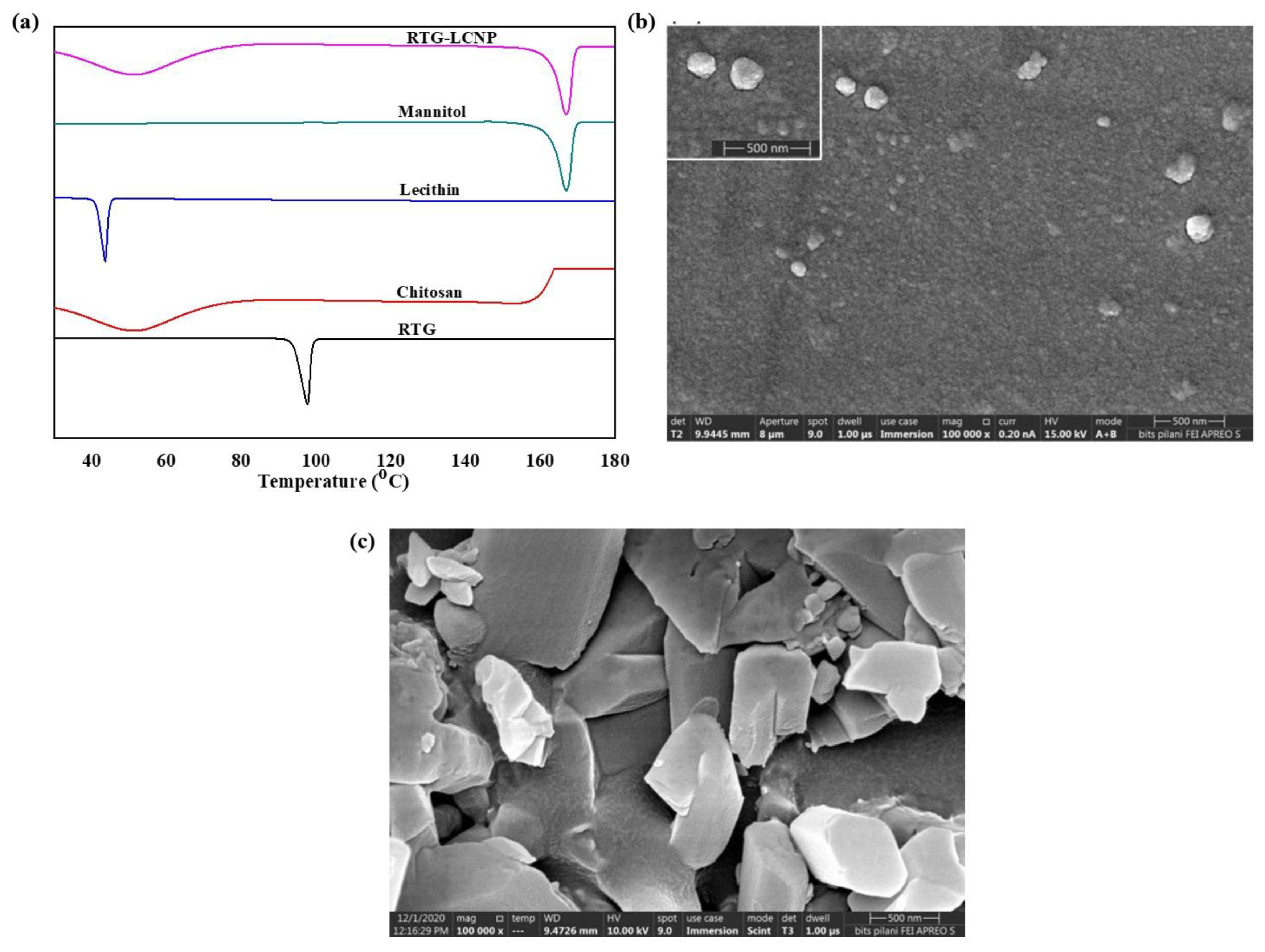
3.5. Differential Scanning Calorimetry
3.6. Field Emission Scanning Electron Microscopy
3.7. Tranmission Electron Microscopy
3.8. Stability Study of RTG-LCNP
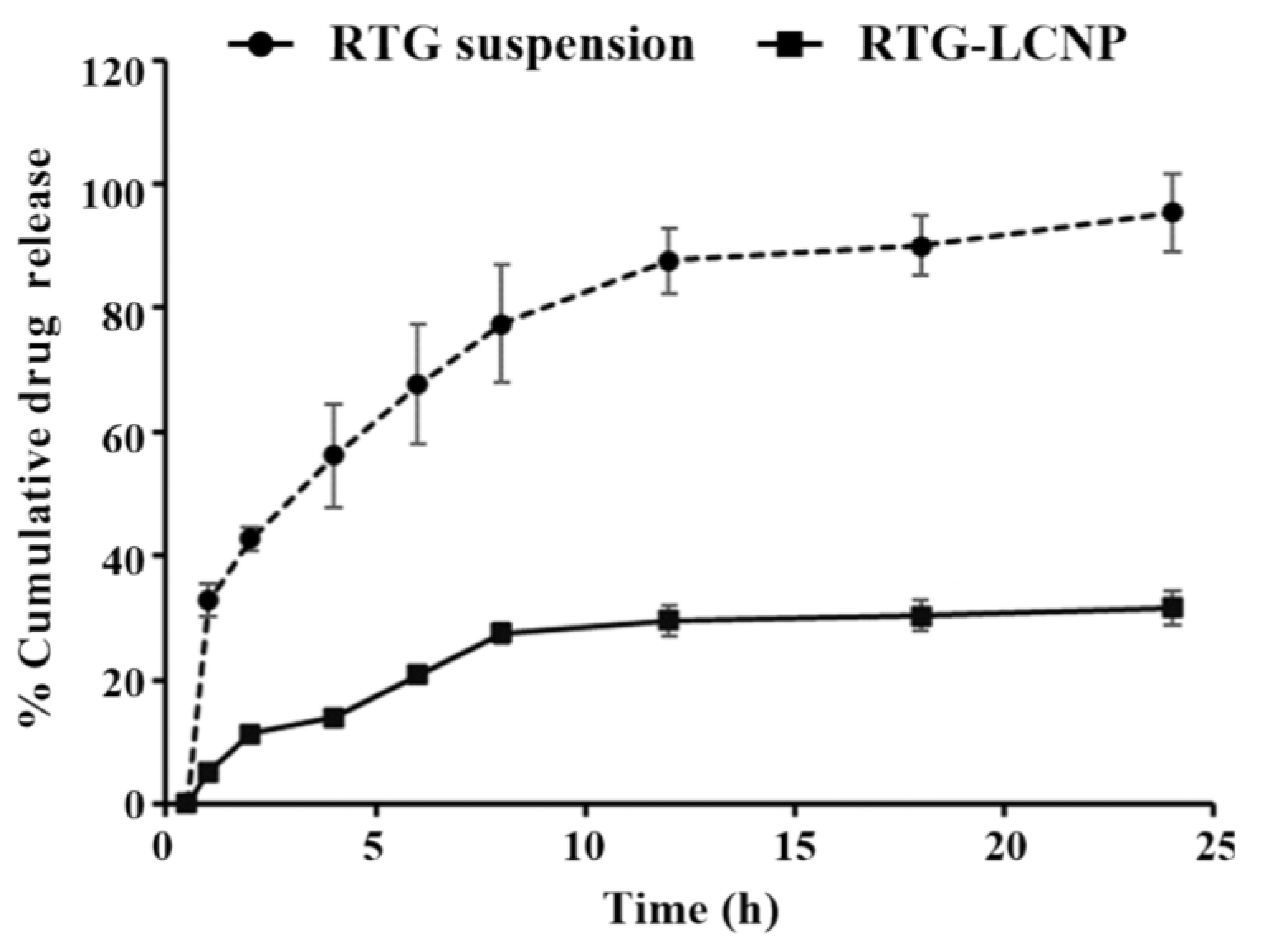
3.9. In Vitro Drug Release
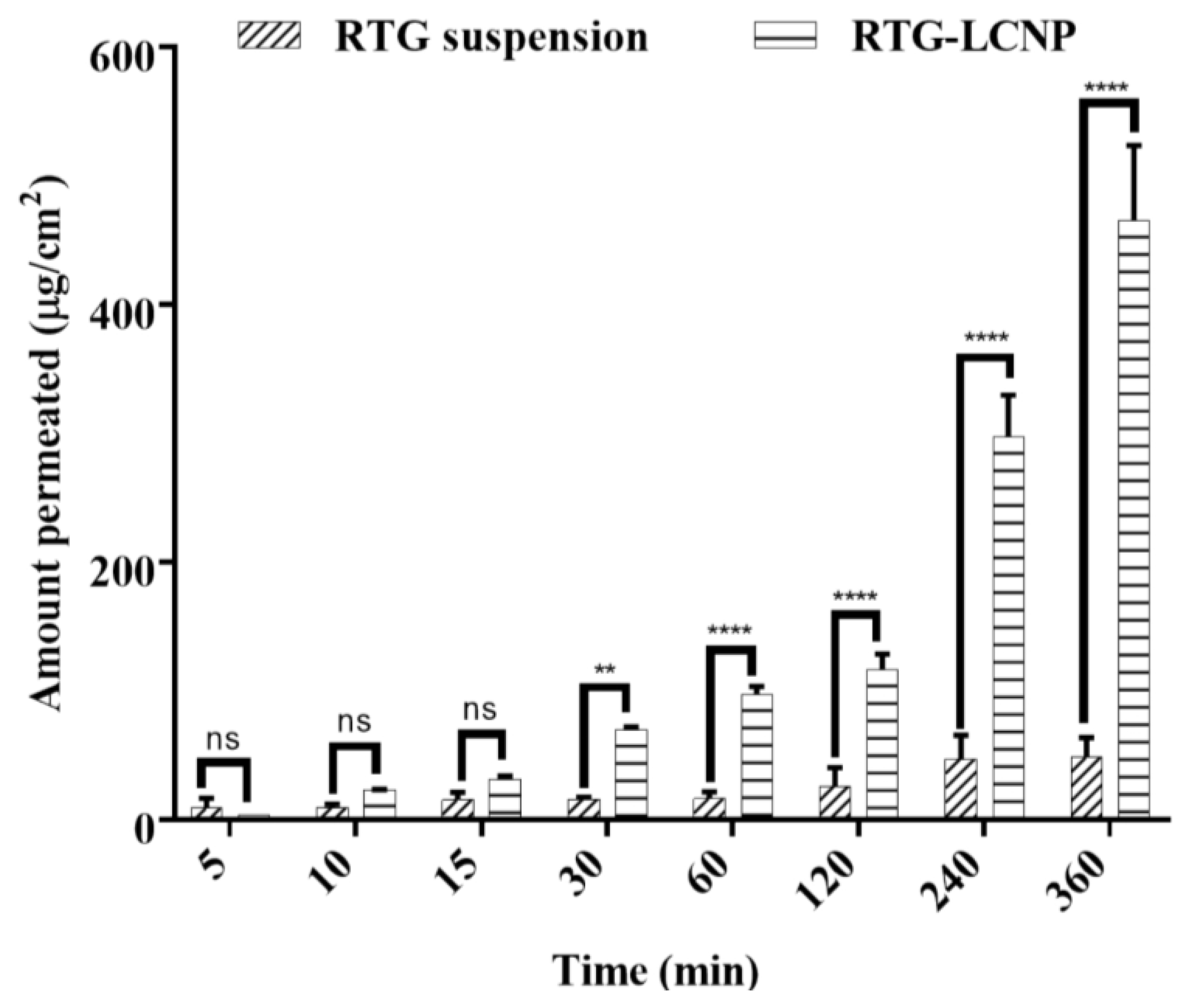
3.10. Ex Vivo Nasal Permeation
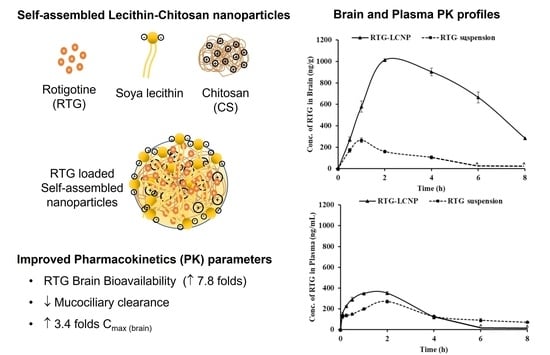
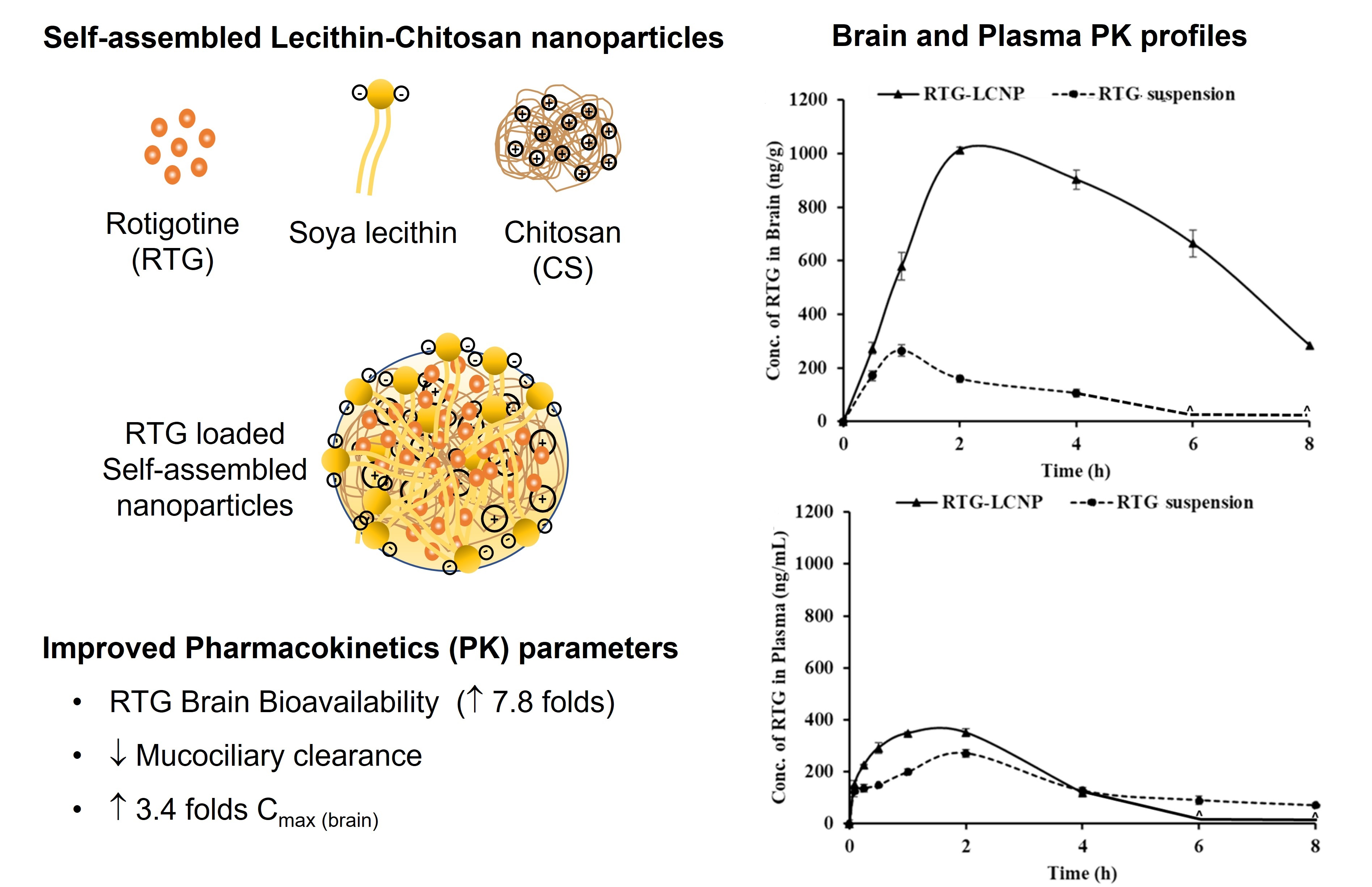
3.11. In Vivo Studies
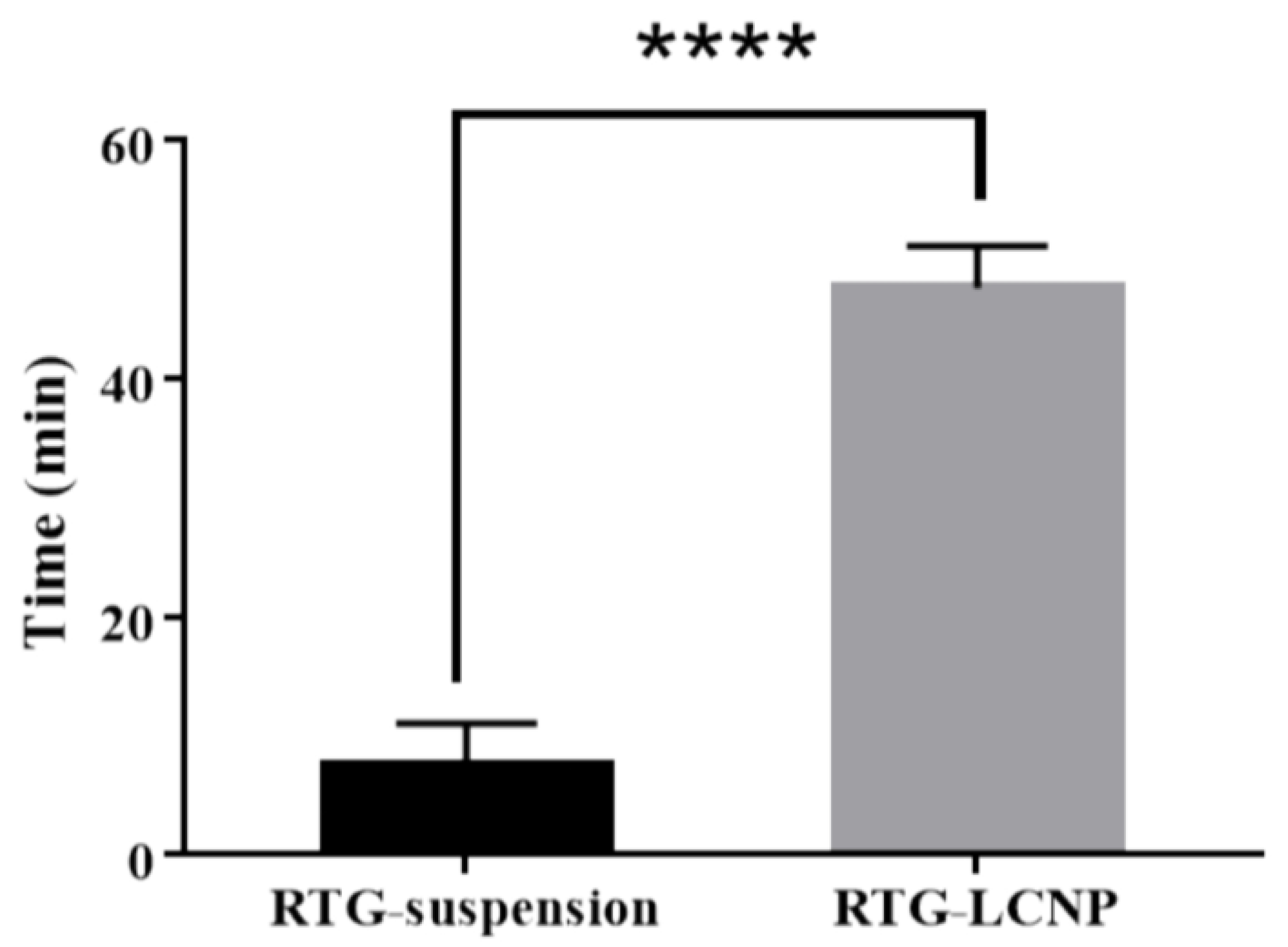
3.11.1. Mucociliary Transport Time of Nanoparticles
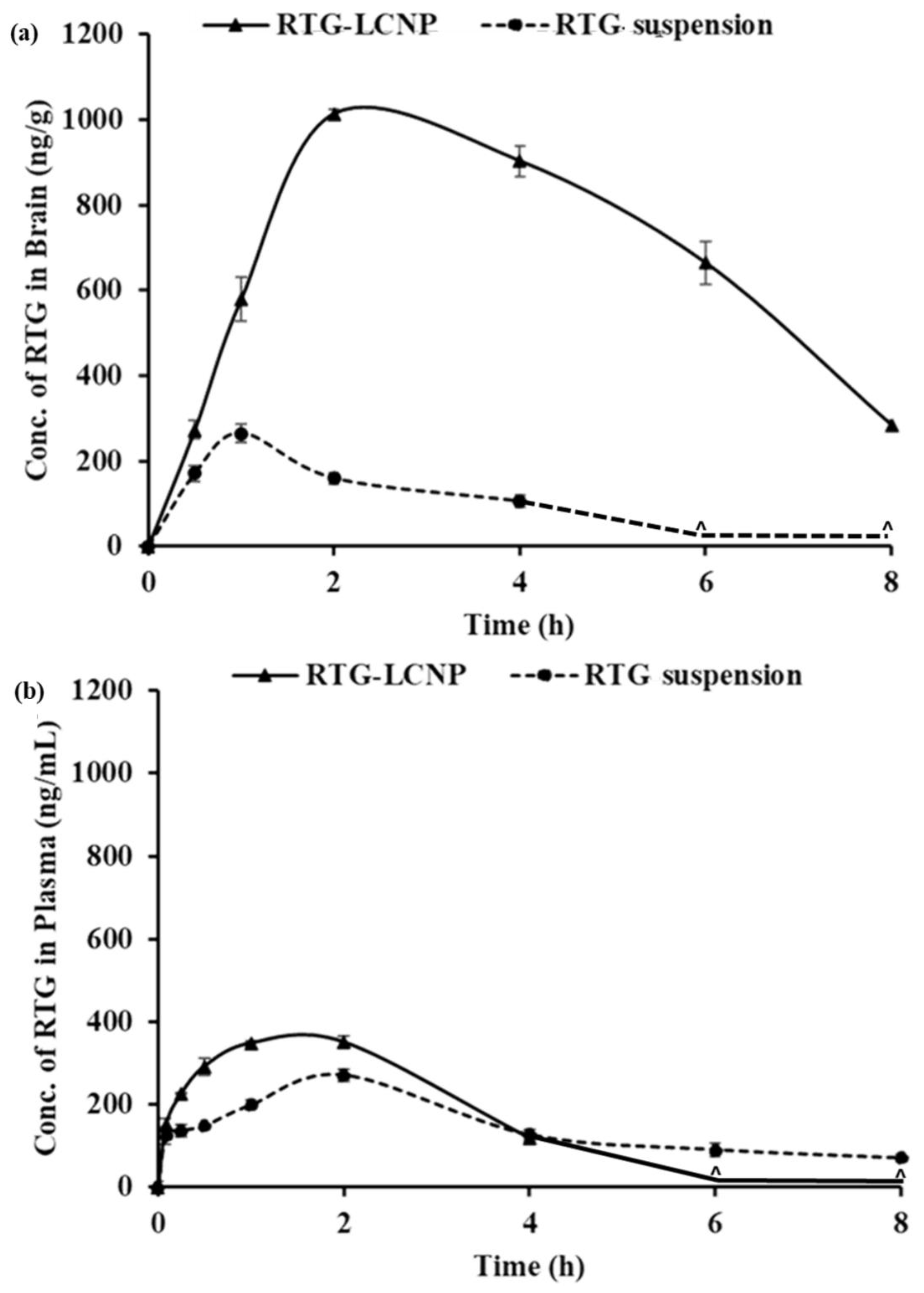
3.11.2. Plasma and Brain PK Analysis
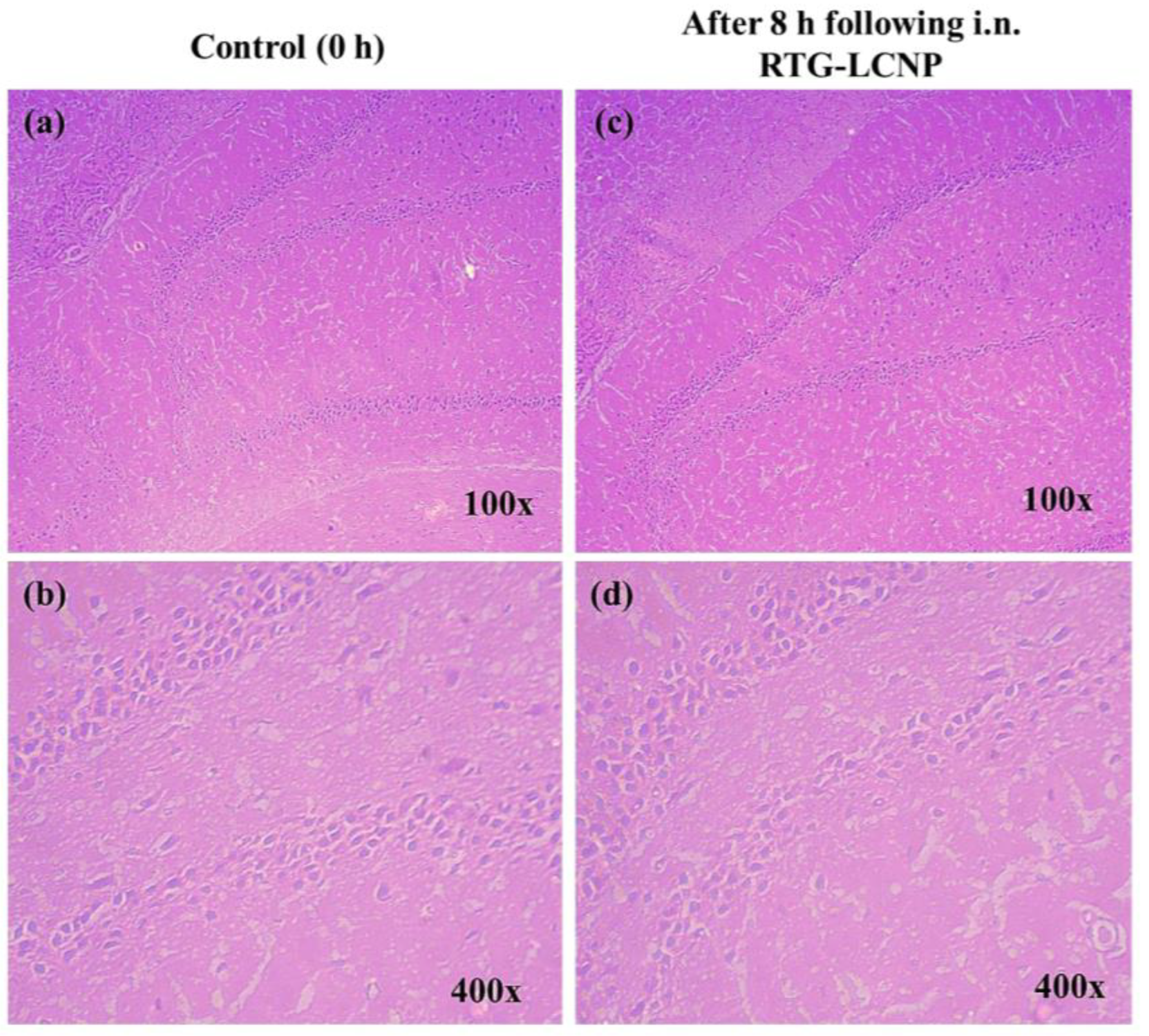
3.12. Histopathology of Brain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raza, C.; Anjum, R.; Shakeel, N.u.A. Parkinson’s Disease: Mechanisms, Translational Models and Management Strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Saha, P.; Chitkara, D.; Pandey, M.M. (Re)Formulating Rotigotine: A Potential Molecule with Unmet Needs. Ther. Deliv. 2023, 13, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Elshoff, J.P.; Cawello, W.; Andreas, J.O.; Mathy, F.X.; Braun, M. An Update on Pharmacological, Pharmacokinetic Properties and Drug-Drug Interactions of Rotigotine Transdermal System in Parkinson’s Disease and Restless Legs Syndrome. Drugs 2015, 75, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Elshoff, J.P.; Braun, M.; Andreas, J.O.; Middle, M.; Cawello, W. Steady-State Plasma Concentration Profile of Transdermal Rotigotine: An Integrated Analysis of Three, Open-Label, Randomized, Phase I Multiple Dose Studies. Clin. Ther. 2012, 34, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Nordling-David, M.M.; Yaffe, R.; Guez, D.; Meirow, H.; Last, D.; Grad, E.; Salomon, S.; Sharabi, S.; Levi-Kalisman, Y.; Golomb, G.; et al. Liposomal Temozolomide Drug Delivery Using Convection Enhanced Delivery. J. Control. Release 2017, 261, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Asadi, S.; Roohbakhsh, A.; Shamsizadeh, A.; Fereidoni, M.; Kordijaz, E.; Moghimi, A. The Effect of Intracerebroventricular Administration of Orexin Receptor Type 2 Antagonist on Pentylenetetrazol-Induced Kindled Seizures and Anxiety in Rats. BMC Neurosci. 2018, 19, 49. [Google Scholar] [CrossRef] [Green Version]
- Dalvi, A.V.; Ravi, P.R.; Uppuluri, C.T.; Mahajan, R.R.; Katke, S.V.; Deshpande, V.S. Thermosensitive Nasal in Situ Gelling Systems of Rufinamide Formulated Using Modified Tamarind Seed Xyloglucan for Direct Nose-to-Brain Delivery: Design, Physical Characterization, and in Vivo Evaluation. J. Pharm. Investig. 2021, 51, 199–211. [Google Scholar] [CrossRef]
- Dhaliwal, H.K.; Fan, Y.; Kim, J.; Amiji, M.M. Intranasal Delivery and Transfection of MRNA Therapeutics in the Brain Using Cationic Liposomes. Mol. Pharm. 2020, 17, 1996–2005. [Google Scholar] [CrossRef]
- Saha, P.; Kathuria, H.; Pandey, M.M. Nose-to-Brain Delivery of Rotigotine Redispersible Nanosuspension: In Vitro and in Vivo Characterization. J. Drug Deliv. Sci. Technol. 2023, 79, 104049. [Google Scholar] [CrossRef]
- Bi, C.C.; Wang, A.P.; Chu, Y.C.; Liu, S.; Mu, H.J.; Liu, W.H.; Wu, Z.M.; Sun, K.X.; Li, Y.X. Intranasal Delivery of Rotigotine to the Brain with Lactoferrin-Modified PEG-PLGA Nanoparticles for Parkinson’s Disease Treatment. Int. J. Nanomedicine 2016, 11, 6547–6559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Yang, Z.; Liu, M.; Tao, Y.; Li, Z.; Wu, Z.; Gui, S. Facile Nose-to-Brain Delivery of Rotigotine-Loaded Polymer Micelles Thermosensitive Hydrogels: In Vitro Characterization and in Vivo Behavior Study. Int. J. Pharm. 2020, 577, 119046. [Google Scholar] [CrossRef] [PubMed]
- Bhattamisra, S.K.; Shak, A.T.; Xi, L.W.; Safian, N.H.; Choudhury, H.; Lim, W.M.; Shahzad, N.; Alhakamy, N.A.; Anwer, M.K.; Radhakrishnan, A.K.; et al. Nose to Brain Delivery of Rotigotine Loaded Chitosan Nanoparticles in Human SH-SY5Y Neuroblastoma Cells and Animal Model of Parkinson’s Disease. Int. J. Pharm. 2020, 579, 119148. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, C.; Xia, X.; Liu, Y. Self-Assembled Lecithin/Chitosan Nanoparticles for Oral Insulin Delivery: Preparation and Functional Evaluation. Int. J. Nanomedicine 2016, 11, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.; Ye, J.; Wang, W.; Yang, Y.; Wang, H.; Sun, T.; Gao, L.; Liu, Y. Self-Assembled Lecithin/Chitosan Nanoparticles Based on Phospholipid Complex: A Feasible Strategy to Improve Entrapment Efficiency and Transdermal Delivery of Poorly Lipophilic Drug. Int. J. Nanomedicine 2020, 15, 5629. [Google Scholar] [CrossRef]
- Hafner, A.; Lovrić, J.; Pepić, I.; Filipović-Grčić, J. Lecithin/Chitosan Nanoparticles for Transdermal Delivery of Melatonin. J. Microencapsul. 2011, 28, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Uppuluri, C.T.; Ravi, P.R.; Dalvi, A.V. Design and Evaluation of Thermo-Responsive Nasal in Situ Gelling System Dispersed with Piribedil Loaded Lecithin-Chitosan Hybrid Nanoparticles for Improved Brain Availability. Neuropharmacology 2021, 201, 108832. [Google Scholar] [CrossRef]
- Murthy, A.; Ravi, P.R.; Kathuria, H.; Vats, R. Self-Assembled Lecithin-Chitosan Nanoparticles Improve the Oral Bioavailability and Alter the Pharmacokinetics of Raloxifene. Int. J. Pharm. 2020, 588, 119731. [Google Scholar] [CrossRef]
- Dalvi, A.; Ravi, P.R.; Uppuluri, C.T. Rufinamide-Loaded Chitosan Nanoparticles in Xyloglucan-Based Thermoresponsive In Situ Gel for Direct Nose to Brain Delivery. Front. Pharmacol. 2021, 12, 1274. [Google Scholar] [CrossRef]
- Mistry, A.; Glud, S.Z.; Kjems, J.; Randel, J.; Howard, K.A.; Stolnik, S.; Illum, L. Effect of Physicochemical Properties on Intranasal Nanoparticle Transit into Murine Olfactory Epithelium. J. Drug Target. 2009, 17, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Brooking, J.; Davis, S.S.; Illum, L. Transport of Nanoparticles Across the Rat Nasal Mucosa. J. Drug Target. 2001, 9, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Hinge, N.S.; Kathuria, H.; Pandey, M.M. Engineering of Structural and Functional Properties of Nanotherapeutics and Nanodiagnostics for Intranasal Brain Targeting in Alzheimer’s. Appl. Mater. Today 2022, 26, 101303. [Google Scholar] [CrossRef]
- Saha, P.; Pandey, M.M. DoE-Based Validation of a HPLC–UV Method for Quantification of Rotigotine Nanocrystals: Application to in Vitro Dissolution and Ex Vivo Nasal Permeation Studies. Electrophoresis 2022, 43, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Lujan, H.; Griffin, W.C.; Taube, J.H.; Sayes, C.M. Synthesis and Characterization of Nanometer-Sized Liposomes for Encapsulation and MicroRNA Transfer to Breast Cancer Cells. Int. J. Nanomedicine 2019, 14, 5159–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saka, R.; Chella, N.; Khan, W. Development of Imatinib Mesylate-Loaded Liposomes for Nose to Brain Delivery: In Vitro and in Vivo Evaluation. AAPS PharmSciTech 2021, 22, 192. [Google Scholar] [CrossRef]
- Roy, J.C.; Salaün, F.; Giraud, S.; Ferri, A.; Roy, J.C.; Salaün, F.; Giraud, S.; Ferri, A. Solubility of Chitin: Solvents, Solution Behaviors and Their Related Mechanisms. In Solubility of Polysaccharides; IntechOpen: London, UK, 2017; pp. 20–60. ISBN 978-953-51-3650-7. [Google Scholar]
- Wolff, H.-M.; Quere, L.; Riedner, J. Polymorphic Form of Rotigotine. 2008. Available online: https://patents.google.com/patent/EP2215072B1/en (accessed on 12 December 2022).
- Paul, A.; Shi, L.; Bielawski, C.W. A Eutectic Mixture of Galactitol and Mannitol as a Phase Change Material for Latent Heat Storage. Energy Convers. Manag. 2015, 103, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Pandey, M.M. Spectrochimica Acta Part A Molecular and Biomolecular Spectroscopy A New Fluorescence-Based Method for Rapid and Specific Quantification of Rotigotine in Chitosan Nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 267, 120555. [Google Scholar] [CrossRef] [PubMed]
- Lisik, A. Witold Musial Conductomeric Evaluation of the Release Kinetics of Active Substances from Pharmaceutical Preparations Containing Iron Ions. Materials 2019, 12, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alomrani, A.; Badran, M.; Harisa, G.I.; ALshehry, M.; Alshamsan, A.; Alkholief, M. The Use of Chitosan-Coated Flexible Liposomes as a Remarkable Carrier to Enhance the Antitumor Efficacy of 5-Fluorouracil against Colorectal Cancer. Saudi Pharm. J. 2019, 27, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Ilk, S.; Saglam, N.; Özgen, M. Kaempferol Loaded Lecithin/Chitosan Nanoparticles: Preparation, Characterization, and Their Potential Applications as a Sustainable Antifungal Agent. Artif. Cells Nanomedicine Biotechnol. 2016, 45, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Şenyiğit, T.; Sonvico, F.; Rossi, A.; Tekmen, I.; Santi, P.; Colombo, P.; Nicoli, S.; Özer, Ö. In Vivo Assessment of Clobetasol Propionate-Loaded Lecithin-Chitosan Nanoparticles for Skin Delivery. Int. J. Mol. Sci. 2016, 18, 32. [Google Scholar] [CrossRef] [Green Version]
- Şenyiǧit, T.; Sonvico, F.; Barbieri, S.; Özer, Ö.; Santi, P.; Colombo, P. Lecithin/Chitosan Nanoparticles of Clobetasol-17-Propionate Capable of Accumulation in Pig Skin. J. Control. Release 2010, 142, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Jafarieh, O.; Md, S.; Ali, M.; Baboota, S.; Sahni, J.K.; Kumari, B.; Bhatnagar, A.; Ali, J. Design, Characterization, and Evaluation of Intranasal Delivery of Ropinirole-Loaded Mucoadhesive Nanoparticles for Brain Targeting. Drug Dev. Ind. Pharm. 2015, 41, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Shadab, M.D.; Khan, R.A.; Mustafa, G.; Chuttani, K.; Baboota, S.; Sahni, J.K.; Ali, J. Bromocriptine Loaded Chitosan Nanoparticles Intended for Direct Nose to Brain Delivery: Pharmacodynamic, Pharmacokinetic and Scintigraphy Study in Mice Model. Eur. J. Pharm. Sci. 2013, 48, 393–405. [Google Scholar] [CrossRef]
- Md, S.; Haque, S.; Fazil, M.; Kumar, M.; Baboota, S.; Sahni, J.K.; Ali, J. Optimised Nanoformulation of Bromocriptine for Direct Nose-to-Brain Delivery: Biodistribution, Pharmacokinetic and Dopamine Estimation by Ultra-HPLC/Mass Spectrometry Method. Expert Opin. Drug Deliv. 2014, 11, 827–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sita, V.G.; Jadhav, D.; Vavia, P. Niosomes for Nose-to-Brain Delivery of Bromocriptine: Formulation Development, Efficacy Evaluation and Toxicity Profiling. J. Drug Deliv. Sci. Technol. 2020, 58, 101791. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code a | Drug:Lecithin Ratio (w/w) | Particle Size (nm) | PDI |
|---|---|---|---|
| LCNP 1 | 1:1 | 220 ± 1.33 | 0.451 ± 0.011 |
| LCNP 2 | 1:2 | 182 ± 2.34 | 0.412 ± 0.014 |
| LCNP 3 | 1:3 | 123 ± 2.12 | 0.292 ± 0.002 |
| LCNP 4 | 1:4 | 263 ± 1.22 | 0.409 ± 0.009 |
| LCNP 5 | 1:5 | 294 ± 1.56 | 0.495 ± 0.003 |
| LCNP 6 | 1:6 | 322 ± 2.86 | 0.309 ± 0.001 |
| Formulation Code a | Lecithin:CS Ratio | Particle Size (nm) | PDI |
|---|---|---|---|
| LCNP 7 | 10 | 203.6 ± 1.22 | 0.430 ± 0.001 |
| LCNP 8 | 20 | 171.0 ± 2.31 | 0.394 ± 0.002 |
| LCNP 9 | 30 | 102.0 ± 1.22 | 0.312 ± 0.006 |
| Formulation Code a | Amount of Poloxamer 407 (mg) | Particle Size (nm) | PDI | %EE |
|---|---|---|---|---|
| LCNP 10 | 2.5 | 259.8 ± 5.17 | 0.309 ± 0.009 | 93.1 ± 3.61 |
| LCNP 11 | 5 | 110.3 ± 1.09 **** | 0.348 ± 0.012 | 87.6 ± 2.93 |
| LCNP 12 | 10 | 193.7 ± 4.05 **** | 0.421 ± 0.018 | 83.2 ± 1.90 |
| Formulation Code a | pH | Particle Size (nm) | %DL |
|---|---|---|---|
| LCNP 13 | 5.0 | 102.0 ± 0.0 | 6.33 ± 3.35 |
| LCNP 14 | 5.5 | 107.8 ± 2.0 | 10.72 ± 4.03 |
| LCNP 15 | 6.0 | 108.0 ± 4.0 | 14.43 ± 2.77 |
| Parameters | 0 Day | 7 Day | 30 Day | 60 Day |
|---|---|---|---|---|
| Particle size (d.nm) | 108.2 ± 4.40 | 105.1 ± 4.38 | 103.3 ± 1.56 | 119.8 ± 11.10 |
| PDI | 0.312 ± 0.001 | 0.310 ± 0.002 | 0.297 ± 0.022 | 0.371 ± 0.325 |
| Zeta potential (mV) | 14.9 ± 0.5 | 14.1 ± 0.3 | 13.8 ± 0.3 | 16.2 ± 0.3 |
| %DL | 14.43 ± 2.77 | 14.75 ± 0.12 | 15.01 ± 2.39 | 12.85 ± 4.03 |
| PK Parameters | Brain | Plasma | ||
|---|---|---|---|---|
| RTG-LCNP | RTG Suspension | RTG-LCNP | RTG Suspension | |
| AUC0→tlast (ng*h/g) b, (ng*h/mL) p | 5507.57± 23.91 | 628.11 ± 12.21 | 1060.44 ± 29.95 | 779.01 ± 14.11 |
| Cmax (ng/g) b, (ng/mL)p | 1013.47 ± 11.28 | 264.71 ± 21.12 | 230.87 ± 8.19 | 270.12 ± 18.50 |
| Tmax (h) | 2 ± 0.03 | 1 ± 0.01 | 1 ± 0.02 | 2 ± 0.01 |
| MRT (h) | 3.81 ± 0.38 | 1.82 ± 0.15 | 1.58 ± 0.05 | 3.15 ± 0.81 |
| Clearance (g/h) b, (mL/h) p | 78.57 ± 12.19 | - | - | 312.65 ± 15.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, P.; Singh, P.; Kathuria, H.; Chitkara, D.; Pandey, M.M. Self-Assembled Lecithin-Chitosan Nanoparticles Improved Rotigotine Nose-to-Brain Delivery and Brain Targeting Efficiency. Pharmaceutics 2023, 15, 851. https://doi.org/10.3390/pharmaceutics15030851
Saha P, Singh P, Kathuria H, Chitkara D, Pandey MM. Self-Assembled Lecithin-Chitosan Nanoparticles Improved Rotigotine Nose-to-Brain Delivery and Brain Targeting Efficiency. Pharmaceutics. 2023; 15(3):851. https://doi.org/10.3390/pharmaceutics15030851
Chicago/Turabian StyleSaha, Paramita, Prabhjeet Singh, Himanshu Kathuria, Deepak Chitkara, and Murali Monohar Pandey. 2023. "Self-Assembled Lecithin-Chitosan Nanoparticles Improved Rotigotine Nose-to-Brain Delivery and Brain Targeting Efficiency" Pharmaceutics 15, no. 3: 851. https://doi.org/10.3390/pharmaceutics15030851