
Drug Combination of Ciprofloxacin and Polymyxin B for the Treatment of Multidrug–Resistant Acinetobacter baumannii Infections: A Drug Pair Limiting the Development of Resistance

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Strains
2.3. Wet Milling
2.4. The Spray–Drying Process
2.5. Morphology
2.6. Particle Size
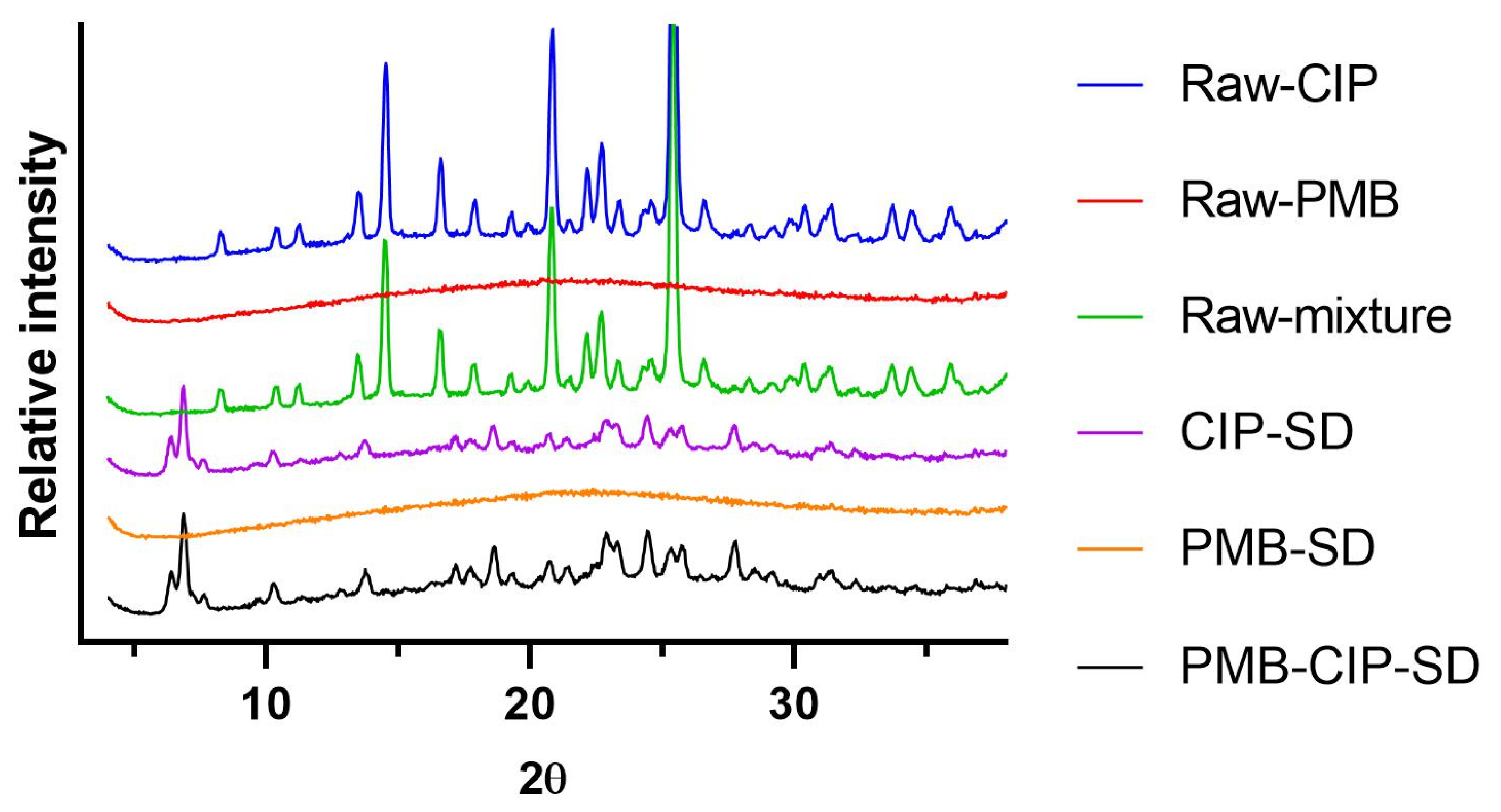
2.7. X-ray Powder Diffraction (XRPD)
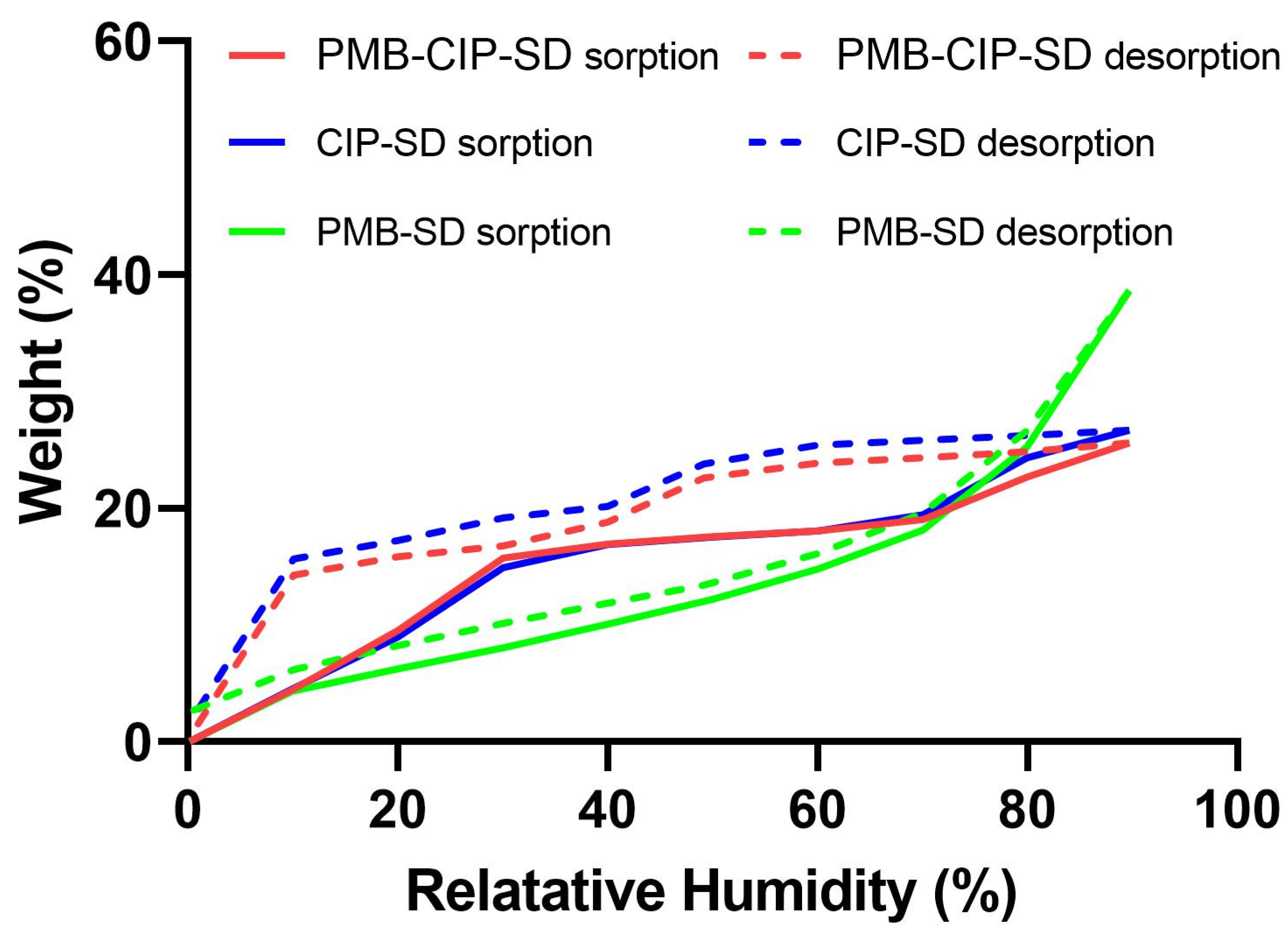
2.8. Dynamic Vapor Sorption (DVS)
2.9. In Vitro Dissolution
2.10. In Vitro Aerosol Performance
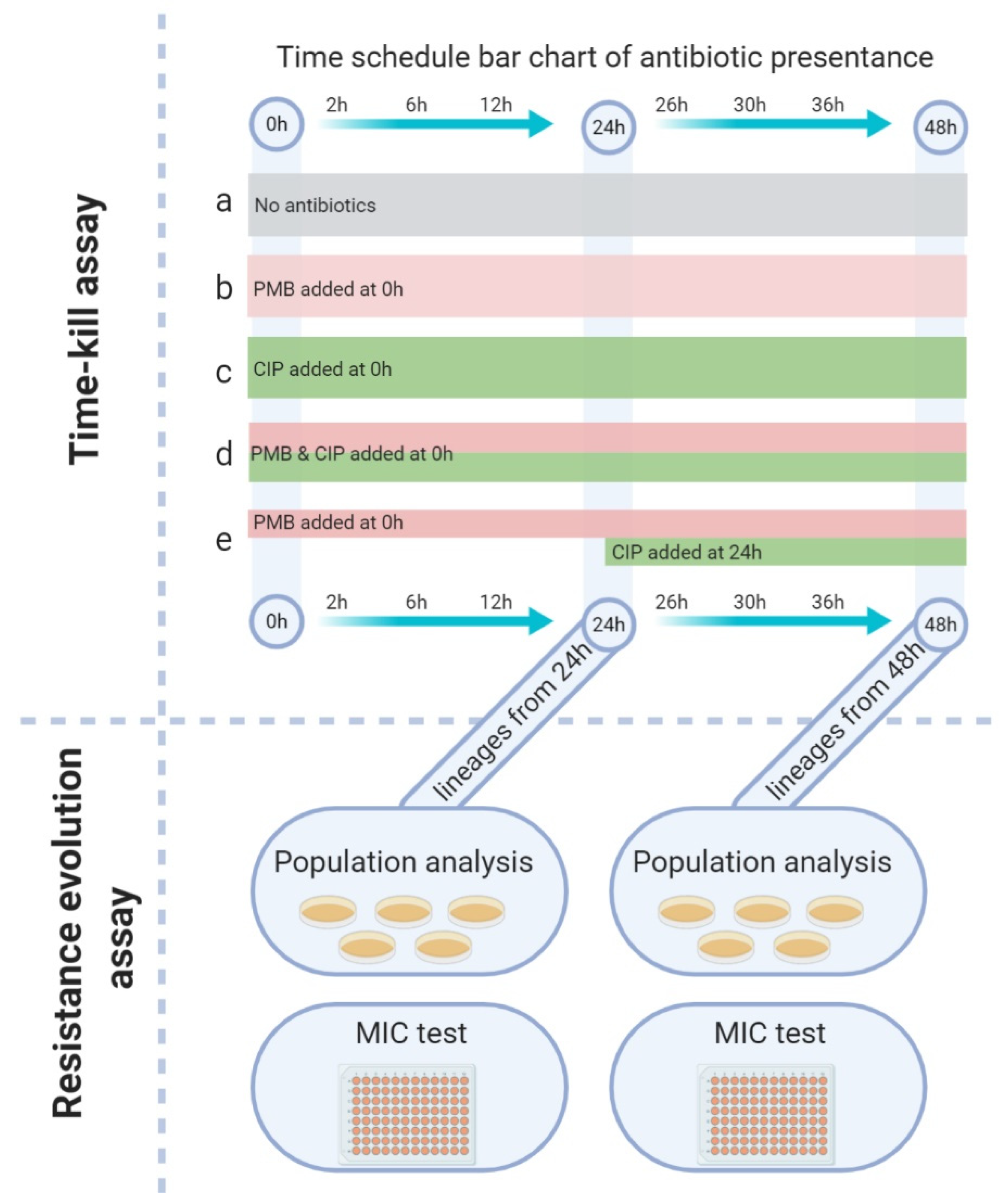
2.11. Time–Kill Assay
2.12. Population Analysis Profiling (PAP)
2.13. MIC Testing
2.14. Genome Sequencing and Analysis
2.15. Statistical Analysis
3. Results
3.1. Preparation and Characterization of Spray–Dried Powders
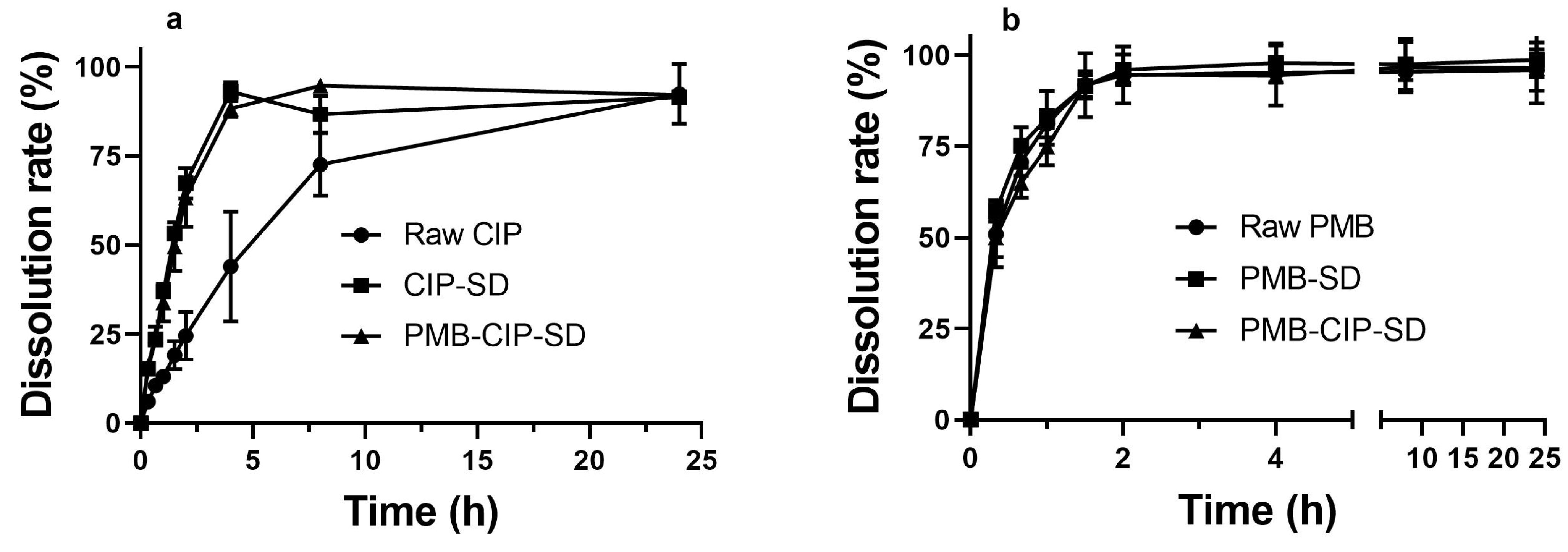
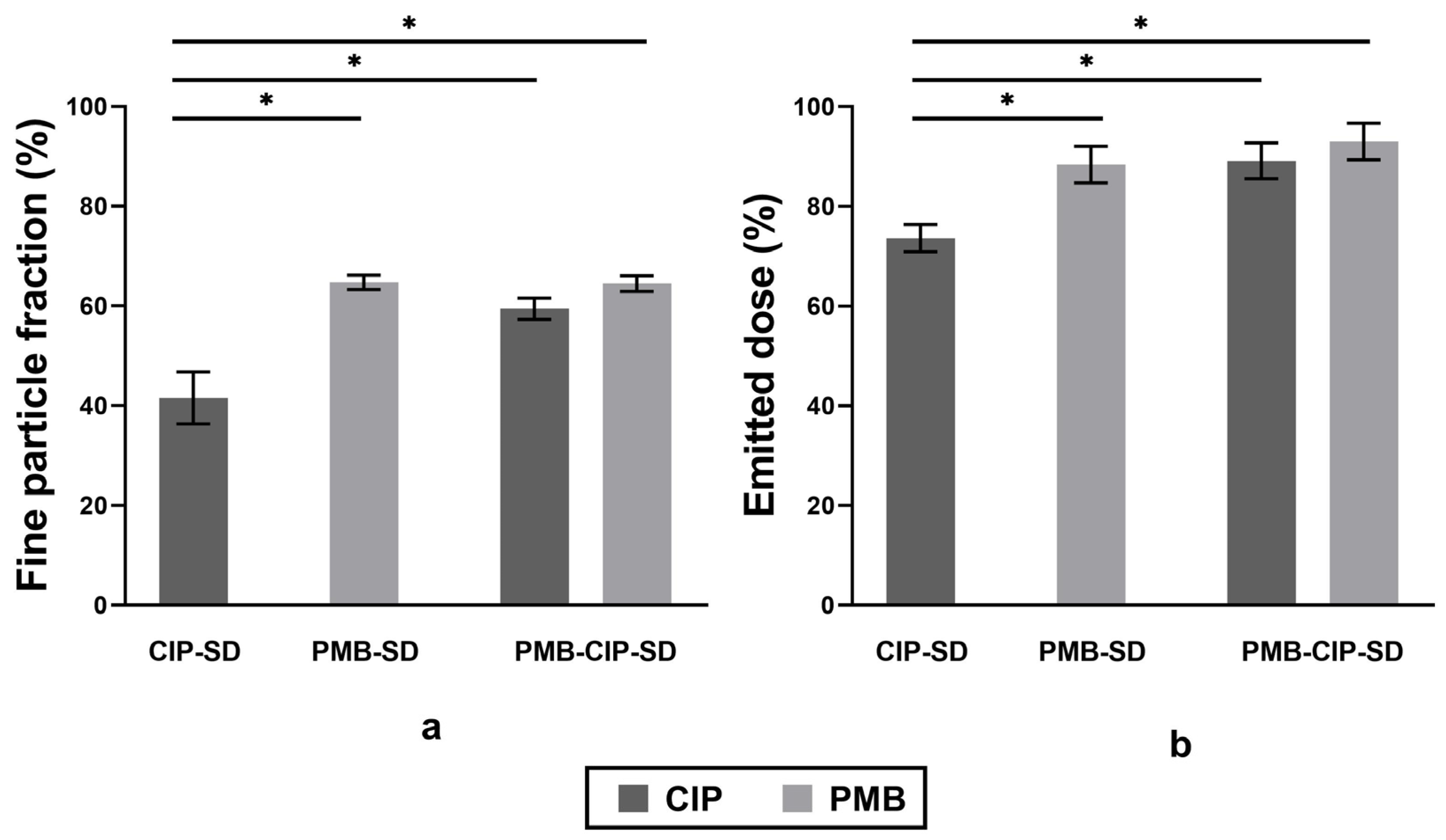
3.2. In Vitro Dissolution and Aerosol Performance of Spray–Dried Powders
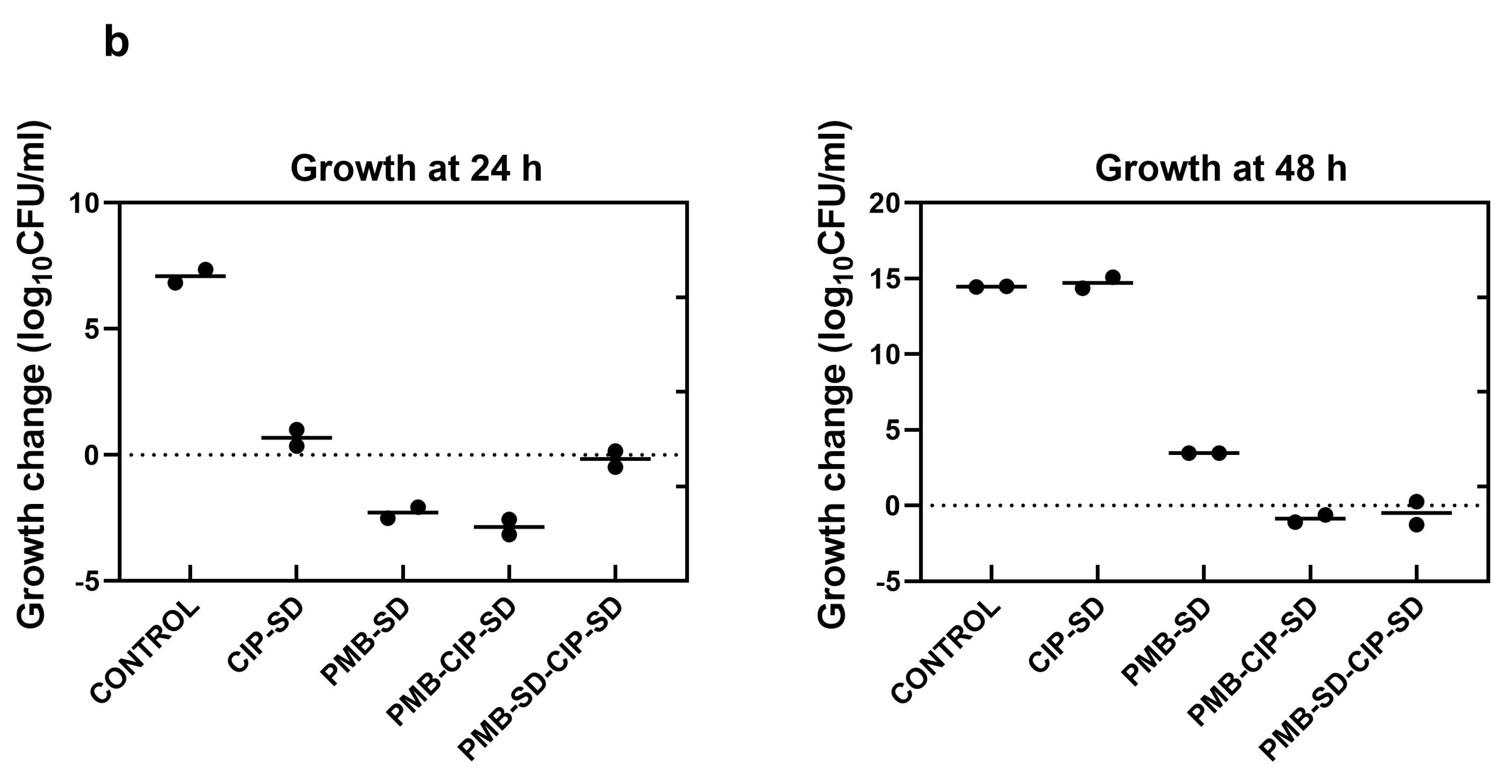
3.3. Time–Kill Assay
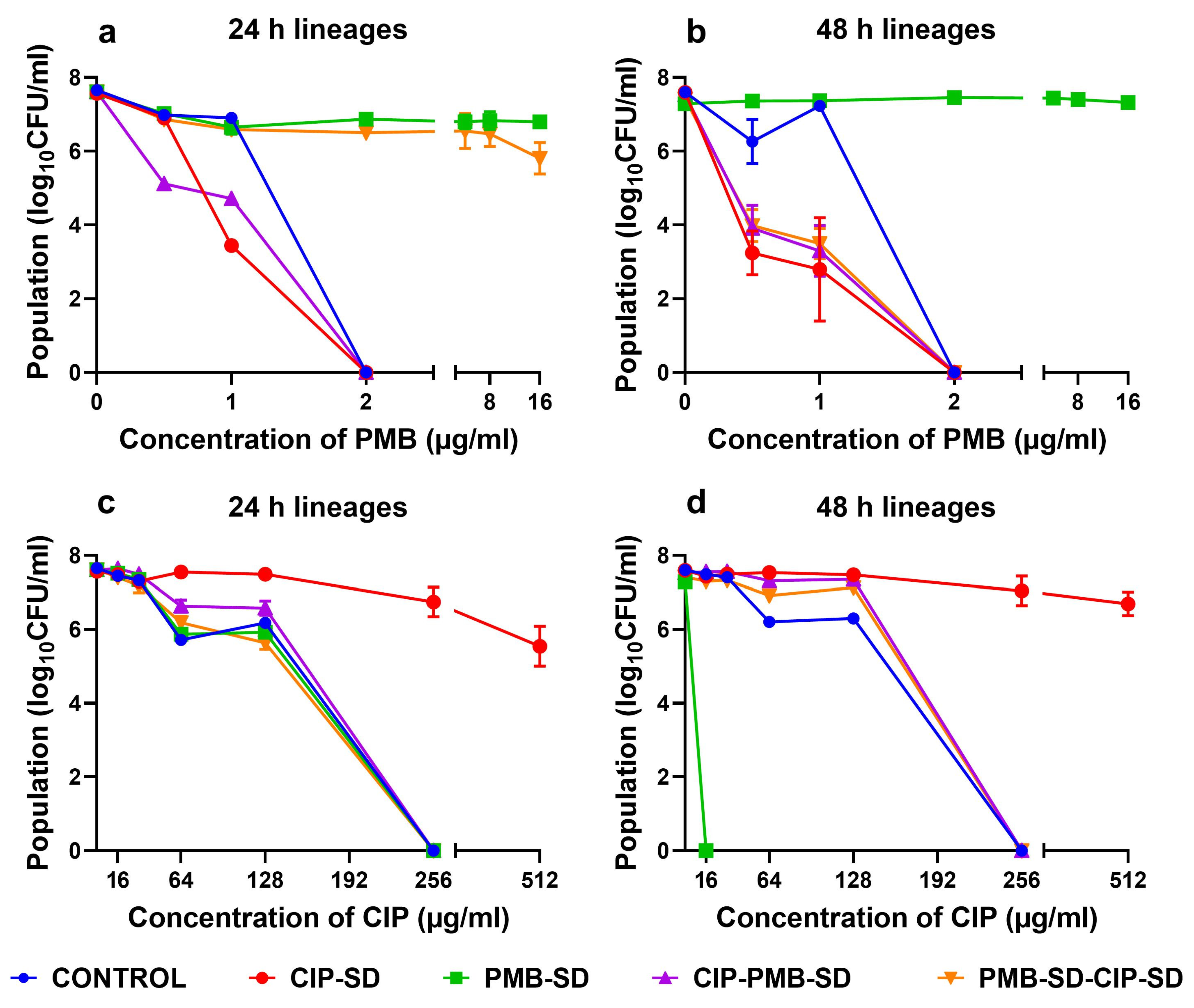
3.4. Population Analysis Profile
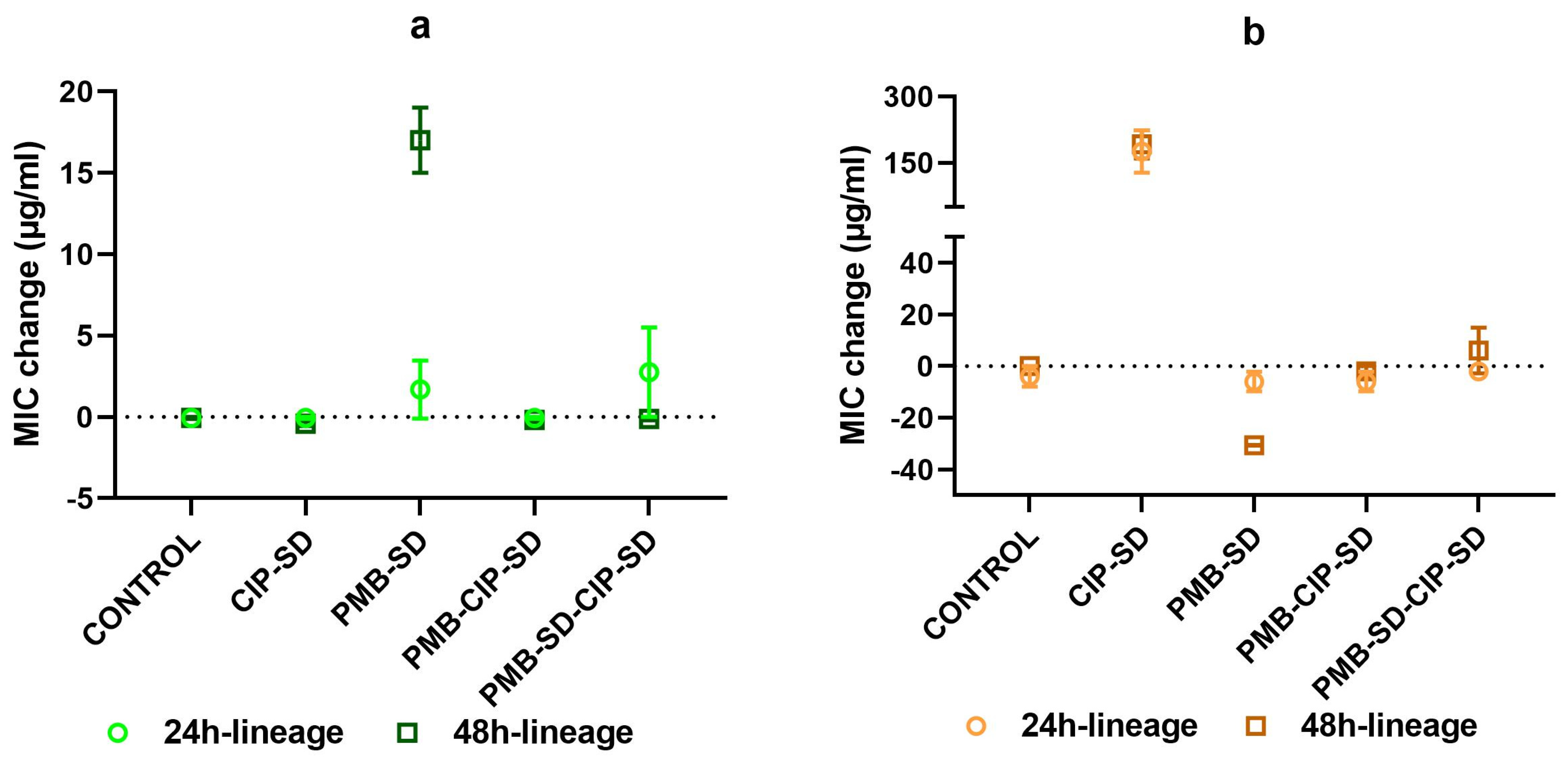
3.5. Changes in MIC of Lineages
3.6. Genomic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. The Top 10 Causes of Death. 2018. Available online: http://www.who.int (accessed on 10 February 2020).
- Trinh, T.D.; Zasowski, E.J.; Claeys, K.C.; Lagnf, A.M.; Kidambi, S.; Davis, S.L.; Rybak, M.J. Multidrug-resistant Pseudomonas aeruginosa lower respiratory tract infections in the intensive care unit: Prevalence and risk factors. Diagn. Microbiol. Infect. Dis. 2017, 89, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Righi, E.; Carnelutti, A.; Graziano, E.; Russo, A. Multidrug-resistant Klebsiella pneumoniae: Challenges for treatment, prevention and infection control. Expert Rev. Anti-Infect. Ther. 2018, 16, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Al-Saryi, N.; Al-Kadmy, I.M.S.; Aziz, S.N. Multidrug-resistant Acinetobacter baumannii as an emerging concern in hospitals. Mol. Biol. Rep. 2021, 48, 6987–6998. [Google Scholar] [CrossRef]
- Ikuta, K.S.; Swetschinski, L.R.; Aguilar, G.R.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Weaver, N.D.; Wool, E.; Han, C.; Hayoon, A.G.; et al. Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef] [PubMed]
- Ayoub Moubareck, C.; Hammoudi Halat, D. Insights into Acinetobacter baumannii: A Review of Microbiological, Virulence, and Resistance Traits in a Threatening Nosocomial Pathogen. Antibiotics 2020, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Pogue, J.M.; Li, Z.; Nation, R.L.; Kaye, K.S.; Li, J. Agents of Last Resort: An Update on Polymyxin Resistance. Infect. Dis. Clin. 2020, 34, 723–750. [Google Scholar] [CrossRef]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Cheah, S.-E.; Johnson, M.D.; Zhu, Y.; Tsuji, B.T.; Forrest, A.; Bulitta, J.B.; Boyce, J.D.; Nation, R.L.; Li, J. Polymyxin Resistance in Acinetobacter baumannii: Genetic Mutations and Transcriptomic Changes in Response to Clinically Relevant Dosage Regimens. Sci. Rep. 2016, 6, 26233. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, L.A.; Herrera, C.M.; Fernandez, L.; Hankins, J.V.; Trent, M.S.; Hancock, R.E.W. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob. Agents Chemother. 2011, 55, 3743–3751. [Google Scholar] [CrossRef] [Green Version]
- Lima, W.G.; de Brito, J.C.M.; Cardoso, B.G.; Cardoso, V.N.; Paiva, M.; De Lima, M.E.; Fernandes, S.O.A. Rate of polymyxin resistance among Acinetobacter baumannii recovered from hospitalized patients: A systematic review and meta-analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1427–1438. [Google Scholar] [CrossRef]
- Moo, C.-L.; Yang, S.-K.; Yusoff, K.; Ajat, M.; Thomas, W.; Abushelaibi, A.; Lim, S.-H.-E.; Lai, K.-S. Mechanisms of Antimicrobial Resistance (AMR) and Alternative Approaches to Overcome AMR. Curr. Drug Discov. Technol. 2020, 17, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef] [PubMed]
- Wences, M.; Wolf, E.R.; Li, C.; Singh, N.; Bah, N.; Tan, X.; Huang, Y.; Bulman, Z.P. Combatting Planktonic and Biofilm Populations of Carbapenem-Resistant Acinetobacter baumannii with Polymyxin-Based Combinations. Antibiotics 2022, 11, 959. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Teo, J.; Heng, D.; Ng, W.K.; Zhao, Y.; Tan, R.B. Tailored Antibiotic Combination Powders for Inhaled Rotational Antibiotic Therapy. J. Pharm. Sci. 2016, 105, 1501–1512. [Google Scholar] [CrossRef]
- Almangour, T.A.; Garcia, E.; Zhou, Q.; Forrest, A.; Kaye, K.S.; Li, J.; Velkov, T.; Rao, G.G. Polymyxins for the treatment of lower respiratory tract infections: Lessons learned from the integration of clinical pharmacokinetic studies and clinical outcomes. Int. J. Antimicrob. Agents 2021, 57, 106328. [Google Scholar] [CrossRef]
- Sobieszczyk, M.E.; Furuya, E.Y.; Hay, C.M.; Pancholi, P.; Della-Latta, P.; Hammer, S.M.; Kubin, C.J. Combination therapy with polymyxin B for the treatment of multidrug-resistant Gram-negative respiratory tract infections. J. Antimicrob. Chemother. 2004, 54, 566–569. [Google Scholar] [CrossRef]
- Michel, J.-B.; Yeh, P.J.; Chait, R.; Moellering, R.C.; Kishony, R. Drug interactions modulate the potential for evolution of resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 14918–14923. [Google Scholar] [CrossRef] [Green Version]
- Torella, J.P.; Chait, R.; Kishony, R. Optimal Drug Synergy in Antimicrobial Treatments. PLOS Comput. Biol. 2010, 6, e1000796. [Google Scholar] [CrossRef]
- Roemhild, R.; Andersson, D.I. Mechanisms and therapeutic potential of collateral sensitivity to antibiotics. PLOS Pathog. 2021, 17, e1009172. [Google Scholar] [CrossRef]
- Aulin, L.B.S.; Liakopoulos, A.; van der Graaf, P.H.; Rozen, D.E.; van Hasselt, J.G.C. Design principles of collateral sensitivity-based dosing strategies. Nat. Commun. 2021, 12, 5691. [Google Scholar] [CrossRef]
- Szybalski, W.; Bryson, V. Genetic studies on microbial cross resistance to toxic agents I: Cross resistance of Escherichia coli to fifteen antibiotics1, 2. J. Bacteriol. 1952, 64, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gefen, O.; Ronin, I.; Bar-Meir, M.; Balaban, N.Q. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 2020, 367, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-W.; Han, M.-L.; Zhao, J.; Zhu, Y.; Rao, G.; Forrest, A.; Song, J.; Kaye, K.S.; Hertzog, P.; Purcell, A.; et al. Synergistic Combination of Polymyxin B and Enrofloxacin Induced Metabolic Perturbations in Extensive Drug-resistant Pseudomonas aeruginosa. Front. Pharmacol. 2019, 10, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buyck, J.M.; Tulkens, P.M.; Van Bambeke, F. Activities of antibiotic combinations against resistant strains of Pseudomonas aeruginosa in a model of infected THP-1 monocytes. Antimicrob. Agents Chemother. 2015, 59, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 8.1. 2018. Available online: http://www.eucast.org (accessed on 20 November 2019).
- Wang, J.; Grégoire, N.; Marchand, S.; Kutter, J.P.; Mu, H.; Moodley, A.; Couet, W.; Yang, M. Improved antibacterial efficiency of inhaled thiamphenicol dry powders: Mathematical modelling of in vitro dissolution kinetic and in vitro antibacterial efficacy. Eur. J. Pharm. Sci. 2020, 152, 105435. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, S.; Kim, S.-M.; Cha, S.H.; Lim, S.-K.; Kim, J. Complete Genome Sequence of Multidrug-Resistant Acinetobacter baumannii Strain 1656-2, Which Forms Sturdy Biofilm. J. Bacteriol. 2011, 193, 6393–6394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahl, J.W.; Lemmer, D.; Travis, J.; Schupp, J.M.; Gillece, J.D.; Aziz, M.; Driebe, E.M.; Drees, K.P.; Hicks, N.D.; Williamson, C.H.D.; et al. NASP: An accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb. Genom. 2016, 2, e000074. [Google Scholar] [CrossRef]
- Durbin, L.R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Ziaee, A.; Albadarin, A.B.; Padrela, L.; Femmer, T.; O’Reilly, E.; Walker, G. Spray drying of pharmaceuticals and biopharmaceuticals: Critical parameters and experimental process optimization approaches. Eur. J. Pharm. Sci. 2019, 127, 300–318. [Google Scholar] [CrossRef]
- Weers, J.G.; Miller, D.P.; Tarara, T.E. Spray-Dried PulmoSphere™ Formulations for Inhalation Comprising Crystalline Drug Particles. AAPS PharmSciTech 2019, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Leng, D.; Kissi, E.O.; Löbmann, K.; Thanki, K.; Fattal, E.; Rades, T.; Foged, C.; Yang, M. Design of Inhalable Solid Dosage Forms of Budesonide and Theophylline for Pulmonary Combination Therapy. AAPS PharmSciTech 2019, 20, 137. [Google Scholar] [CrossRef] [PubMed]
- Sheokand, S.; Modi, S.R.; Bansal, A.K. Dynamic Vapor Sorption as a Tool for Characterization and Quantification of Amorphous Content in Predominantly Crystalline Materials. J. Pharm. Sci. 2014, 103, 3364–3376. [Google Scholar] [CrossRef]
- Mafra, L.; Santos, S.M.; Siegel, R.; Alves, I.; Paz, F.A.A.; Dudenko, D.; Spiess, H.W. Packing Interactions in Hydrated and Anhydrous Forms of the Antibiotic Ciprofloxacin: A Solid-State NMR, X-ray Diffraction, and Computer Simulation Study. J. Am. Chem. Soc. 2012, 134, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Paluch, K.J.; McCabe, T.; Müller-Bunz, H.; Corrigan, O.I.; Healy, A.M.; Tajber, L. Formation and Physicochemical Properties of Crystalline and Amorphous Salts with Different Stoichiometries Formed between Ciprofloxacin and Succinic Acid. Mol. Pharm. 2013, 10, 3640–3654. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, F.; Sui, Y.; She, Z.; Zhai, W.; Deng, Y. Effect of particle size on solubility, dissolution rate, and oral bioavailability: Evaluation using coenzyme Q₁₀ as naked nanocrystals. Int. J. Nanomed. 2012, 7, 5733–5744. [Google Scholar] [CrossRef] [Green Version]
- Mangal, S.; Park, H.; Zeng, L.; Yu, H.H.; Lin, Y.-W.; Velkov, T.; Denman, J.A.; Zemlyanov, D.; Li, J.; Zhou, Q. Composite particle formulations of colistin and meropenem with improved in-vitro bacterial killing and aerosolization for inhalation. Int. J. Pharm. 2018, 548, 443–453. [Google Scholar] [CrossRef]
- Shetty, N.; Ahn, P.; Park, H.; Bhujbal, S.; Zemlyanov, D.; Cavallaro, A.-A.; Mangal, S.; Li, J.; Zhou, Q.T. Improved physical stability and aerosolization of inhalable amorphous ciprofloxacin powder formulations by incorporating synergistic colistin. Mol. Pharm. 2018, 15, 4004–4020. [Google Scholar] [CrossRef]
- Lorian, V. Antibiotics in Laboratory Medicine, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Lázár, V.; Singh, G.P.; Spohn, R.; Nagy, I.; Horváth, B.; Hrtyan, M.; Busa-Fekete, R.; Bogos, B.; Méhi, O.; Csörgő, B.; et al. Bacterial evolution of antibiotic hypersensitivity. Mol. Syst. Biol. 2013, 9, 700. [Google Scholar] [CrossRef]
- Rodriguez de Evgrafov, M.; Gumpert, H.; Munck, C.; Thomsen, T.T.; Sommer, M.O. Collateral resistance and sensitivity modulate evolution of high-level resistance to drug combination treatment in Staphylococcus aureus. Mol. Biol. Evol. 2015, 32, 1175–1185. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Casadesús, J. Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Dawan, J.; Kim, J.C.; Ahn, J. Insights into collateral susceptibility and collateral resistance in Acinetobacter baumannii during antimicrobial adaptation. Lett. Appl. Microbiol. 2021, 73, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Traini, D.; Young, P.M. Delivery of antibiotics to the respiratory tract: An update. Expert Opin. Drug Deliv. 2009, 6, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, S.L. Tobramycin Inhalation Product Approved for Use in Cystic Fibrosis Therapy. JAMA 1998, 279, 645. [Google Scholar] [CrossRef]
- Hickey, A.J.; Durham, P.; Dharmadhikari, A.; Nardell, E. Inhaled drug treatment for tuberculosis: Past progress and future prospects. J. Control. Release 2016, 240, 127–134. [Google Scholar] [CrossRef]
- Fiel, S.B.; Roesch, E.A. The use of tobramycin for Pseudomonas aeruginosa: A review. Expert Rev. Respir. Med. 2022, 16, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, S.; Zou, P.; Chai, G.; Lin, Y.-W.; Velkov, T.; Li, J.; Pan, W.; Zhou, Q.T. Inhalable liposomal powder formulations for co-delivery of synergistic ciprofloxacin and colistin against multi-drug resistant gram-negative lung infections. Int. J. Pharm. 2020, 575, 118915. [Google Scholar] [CrossRef]
- Yu, S.; Pu, X.; Ahmed, M.U.; Yu, H.H.; Mutukuri, T.T.; Li, J.; Zhou, Q.T. Spray-freeze-dried inhalable composite microparticles containing nanoparticles of combinational drugs for potential treatment of lung infections caused by Pseudomonas aeruginosa. Int. J. Pharm. 2021, 610, 121160. [Google Scholar] [CrossRef]
- Lin, Y.; Quan, D.; Chang, R.Y.K.; Chow, M.Y.; Wang, Y.; Li, M.; Morales, S.; Britton, W.J.; Kutter, E.; Li, J.; et al. Synergistic activity of phage PEV20-ciprofloxacin combination powder formulation—A proof-of-principle study in a P. aeruginosa lung infection model. Eur. J. Pharm. Biopharm. 2021, 158, 166–171. [Google Scholar] [CrossRef]
- Chai, G.; Hassan, A.; Meng, T.; Lou, L.; Ma, J.; Simmers, R.; Zhou, L.; Rubin, B.K.; Zhou, Q.; Longest, P.W.; et al. Dry powder aerosol containing muco-inert particles for excipient enhanced growth pulmonary drug delivery. Nanomed. Nanotechnol. Biol. Med. 2020, 29, 102262. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotics | MICs (µg/mL) | MIC Break Points (µg/mL) | |
|---|---|---|---|
| S≤ | R> | ||
| Aztreonam | 64 | N | N |
| Ciprofloxacin | 32 | 0.25 | 0.5 |
| Polymyxin B * | 1 | – | – |
| Tobramycin | 256 | 4 | 4 |
| Dry Powders | Abbreviation | Solid Contents of Feeding Solution | |
|---|---|---|---|
| PMB Solution (mg/mL) | CIP Suspension (mg/mL) | ||
| PMB spray–dried powder | PMB–SD | 9.6 | – |
| CIP spray–dried powder | CIP–SD | – | 9.8 |
| PMB–CIP co–spray–dried powder | PMB–CIP–SD | 0.31 | 9.8 |
| Code | Name | Treatment | Concentration (µg/mL) | |
|---|---|---|---|---|
| CIP | PMB | |||
| a | Control | no antibiotic | – | – |
| b | PMB–SD | add PMB–SD at time zero | – | 1 |
| c | CIP–SD | add CIP–SD at time zero | 32 | – |
| d | PMB–CIP–SD | add PMB–SD and CIP–SD at time zero | 32 | 1 |
| e | PMB–SD–CIP–SD | add PMB–SD at the beginning then add CIP–SD added after 24 h | 32 | 1 |
| Sample Name | Diameter (µm) | Span (µm) | ||
|---|---|---|---|---|
| Dv10 | Dv50 | Dv90 | ||
| PMB–SD | 1.2 ± 0.1 | 2.7 ± 0.1 | 5.7 ± 0.3 | 1.6 ± 0.1 |
| CIP–SD | 0.7 ± 0.1 | 2.6 ± 0.2 | 6.4 ± 0.3 | 2.2 ± 0.1 |
| PMB–CIP SD | 0.7 ± 0.1 | 2.7 ± 0.1 | 6.3 ± 0.1 | 2.0 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Stegger, M.; Moodley, A.; Yang, M. Drug Combination of Ciprofloxacin and Polymyxin B for the Treatment of Multidrug–Resistant Acinetobacter baumannii Infections: A Drug Pair Limiting the Development of Resistance. Pharmaceutics 2023, 15, 720. https://doi.org/10.3390/pharmaceutics15030720
Wang J, Stegger M, Moodley A, Yang M. Drug Combination of Ciprofloxacin and Polymyxin B for the Treatment of Multidrug–Resistant Acinetobacter baumannii Infections: A Drug Pair Limiting the Development of Resistance. Pharmaceutics. 2023; 15(3):720. https://doi.org/10.3390/pharmaceutics15030720
Chicago/Turabian StyleWang, Junwei, Marc Stegger, Arshnee Moodley, and Mingshi Yang. 2023. "Drug Combination of Ciprofloxacin and Polymyxin B for the Treatment of Multidrug–Resistant Acinetobacter baumannii Infections: A Drug Pair Limiting the Development of Resistance" Pharmaceutics 15, no. 3: 720. https://doi.org/10.3390/pharmaceutics15030720