
Derivatives of Amaryllidaceae Alkaloid Ambelline as Selective Inhibitors of Hepatic Stage of Plasmodium berghei Infection In Vitro
 , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
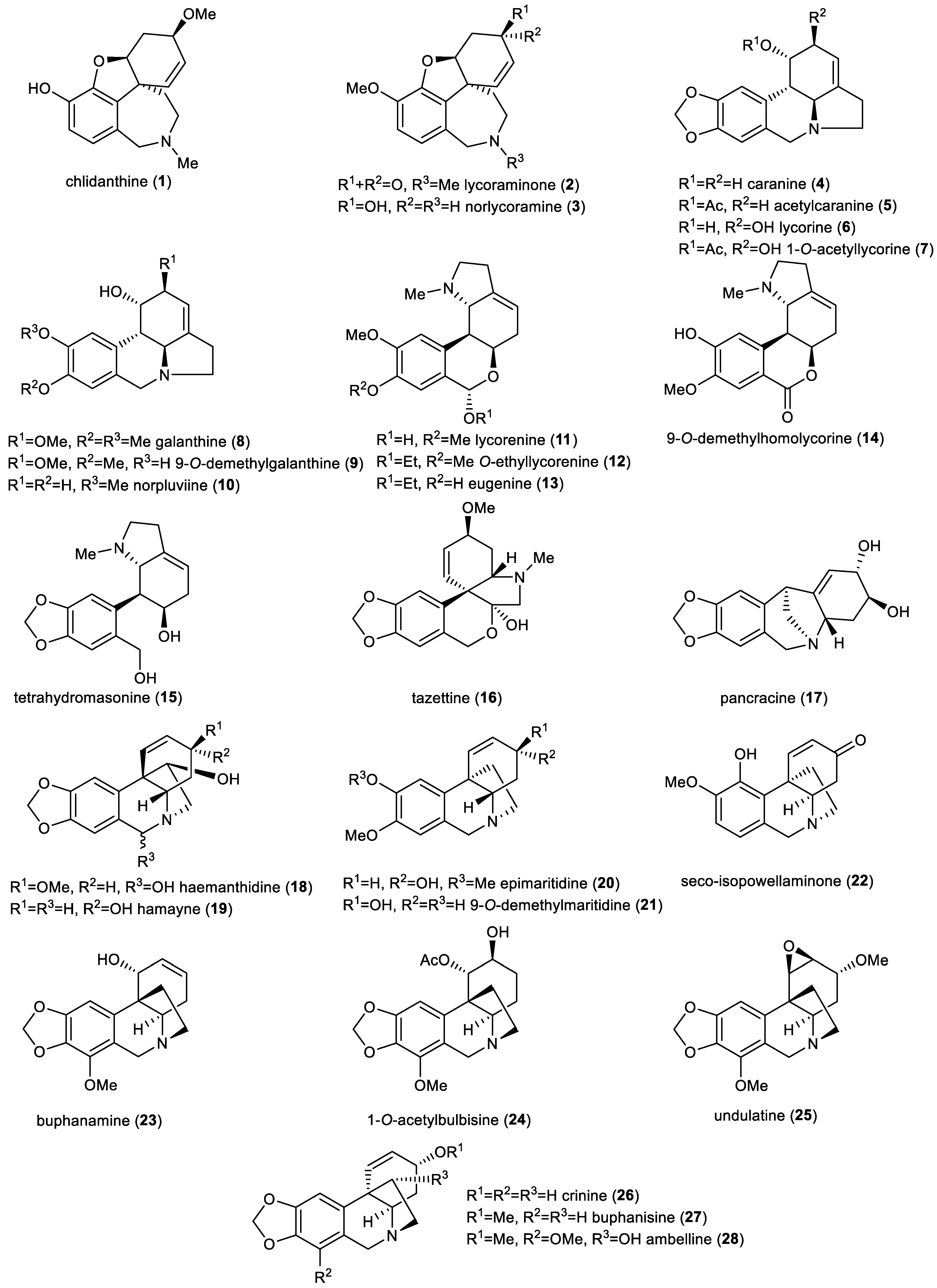
2.2. Amaryllidaceae Alkaloids
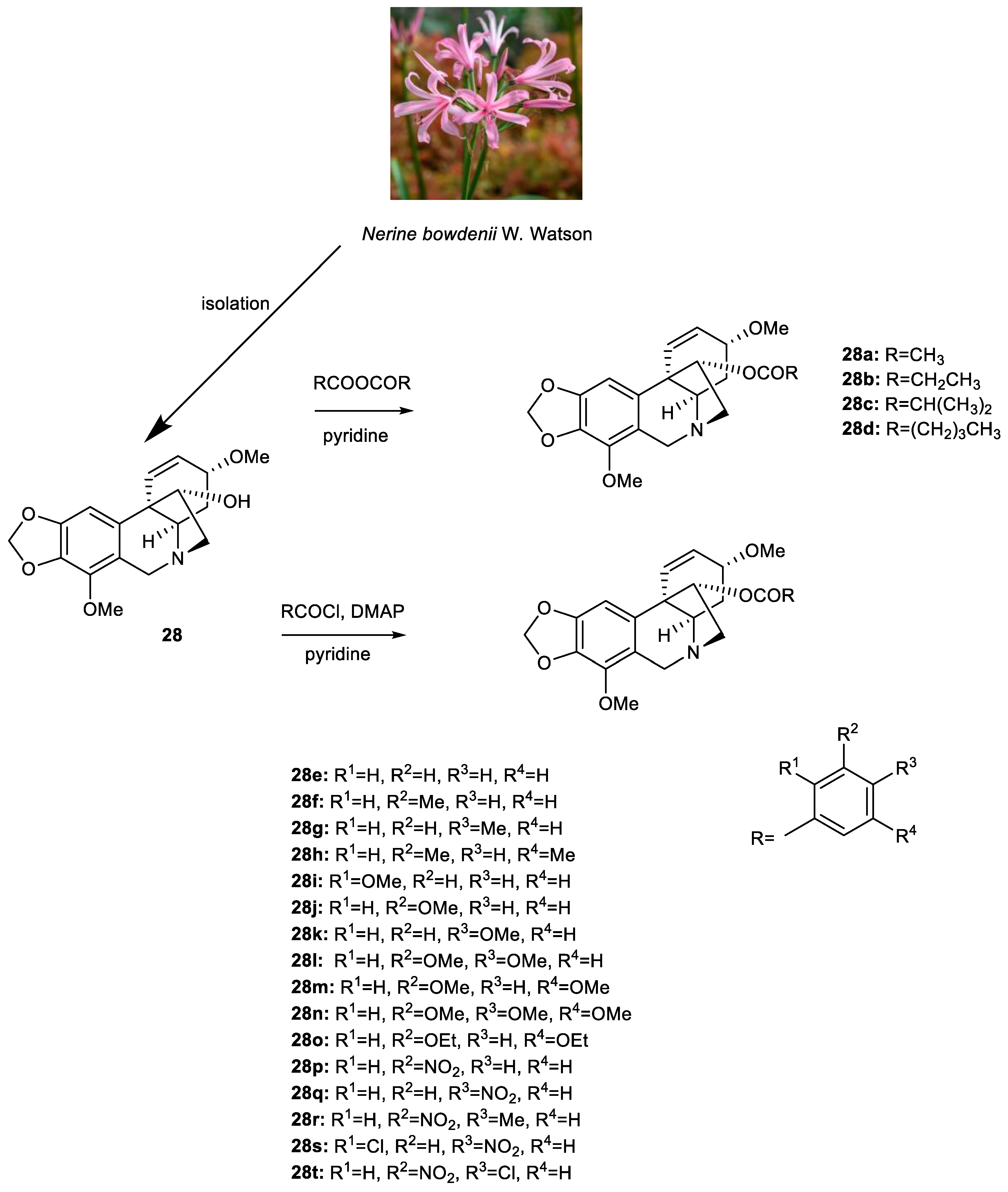
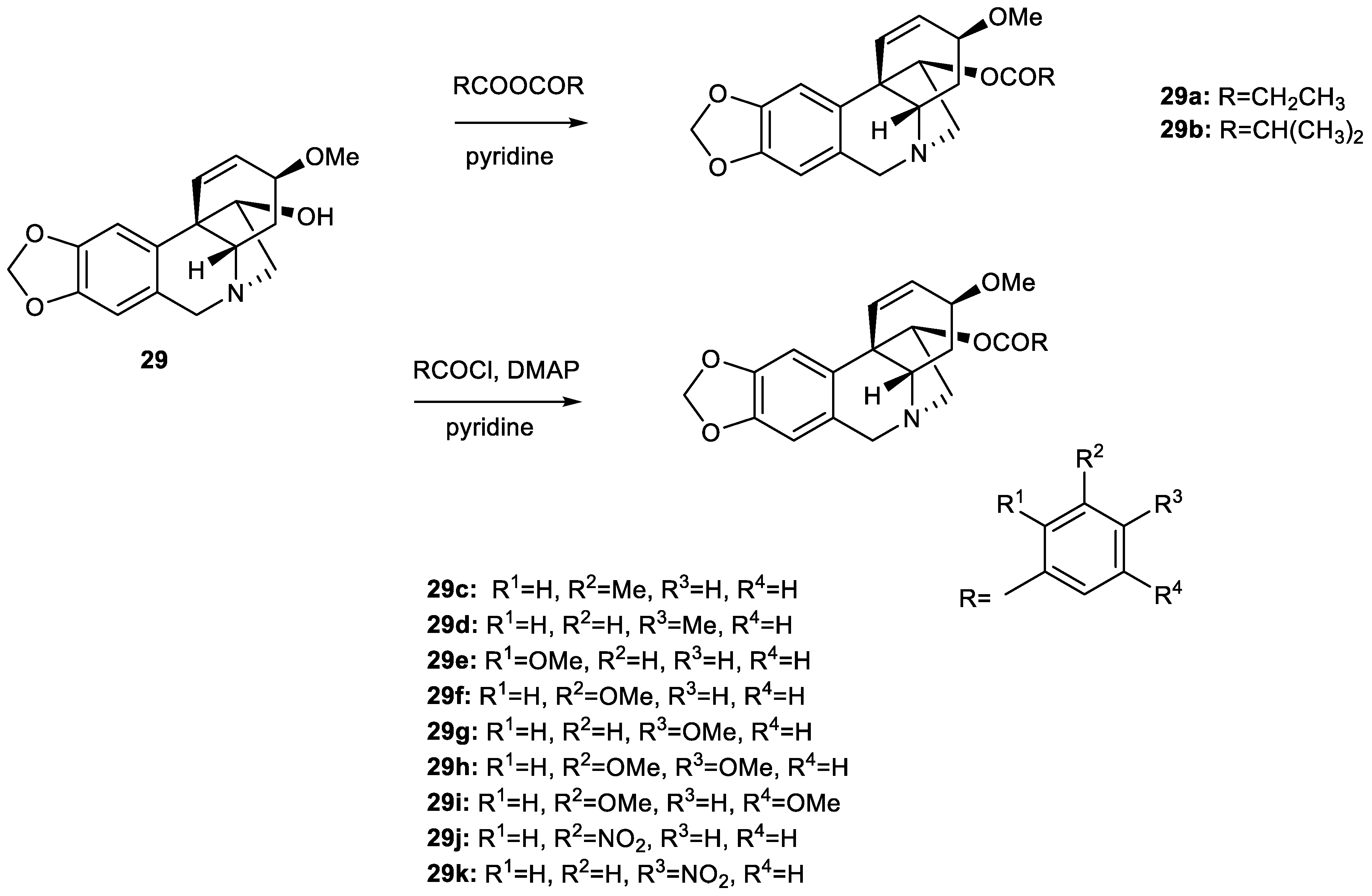
2.3. Preparation of Ambelline (28) and Haemanthamine (29) Derivatives
2.4. NMR Spectra of Newly Synthesized Derivatives
2.4.1. 11-O-(3,5-Dimethylbenzoyl)ambelline (28h)
2.4.2. 11-O-(3,5-Dimethoxybenzoyl)ambelline (28m)
2.4.3. 11-O-(3,4,5-Trimethoxybenzoyl)ambelline (28n)
2.4.4. 11-O-(4-Methyl-3-nitrobenzoyl)ambelline (28r)
2.4.5. 11-O-(2-Chloro-4-nitrobenzoyl)ambelline (28s)
2.4.6. 11-O-(4-Chloro-3-nitrobenzoyl)ambelline (28t)
2.5. In Vitro Activity against P. berghei—Hepatic Stages
2.6. In Vitro Activity against P. falciparum Blood Stages
2.7. Cytotoxicity Assessment In Vitro (MTT Assay)
3. Results and Discussion
3.1. Studied Alkaloids and Their Semisynthetic Derivatives
3.2. Synthesis of Ambelline (28) and Haemanthamine (29) Derivatives
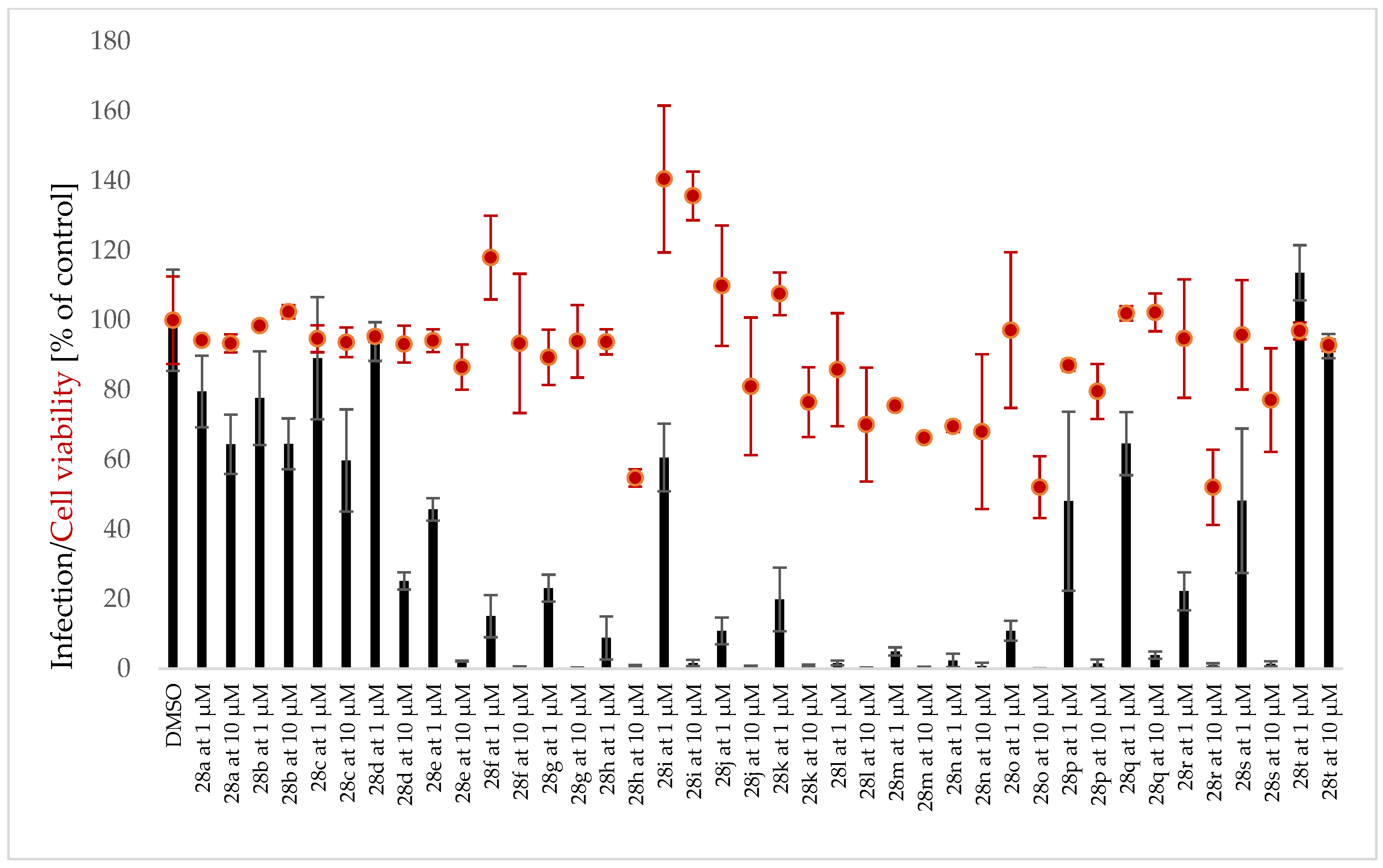
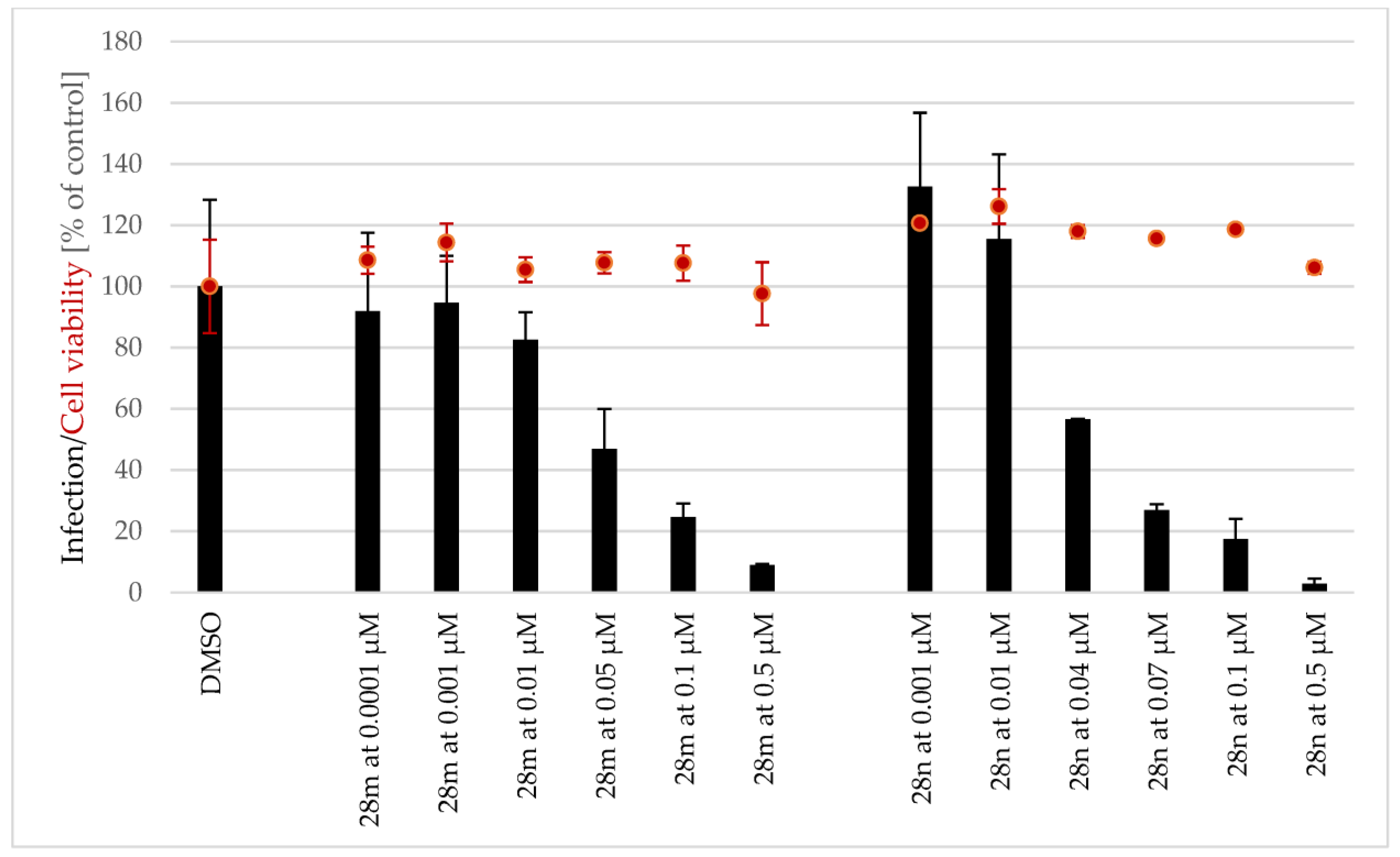
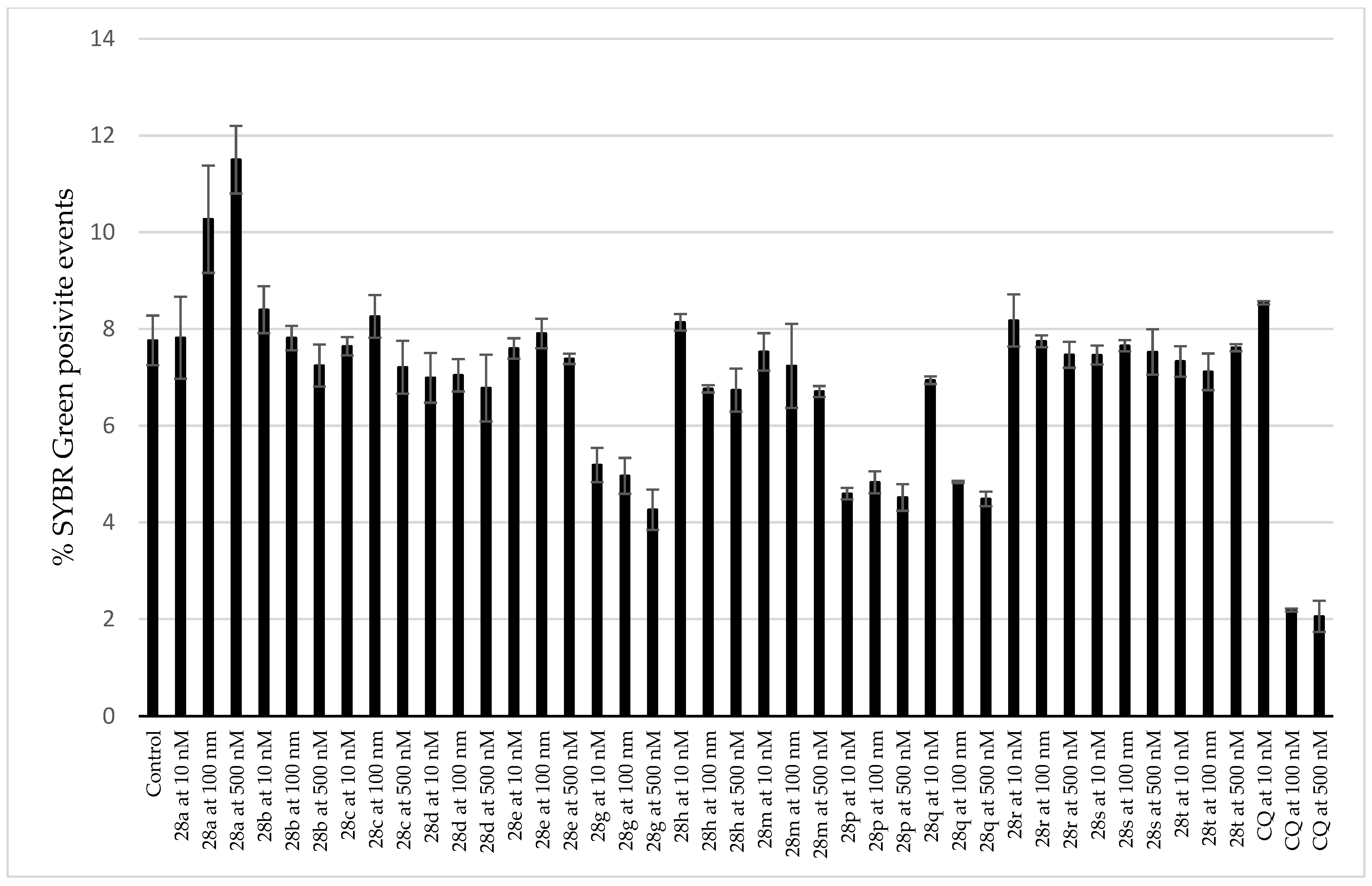
3.3. Antiplasmodial Activity of Selected Alkaloids and Derivatives
3.4. Structure-Activity Relationships
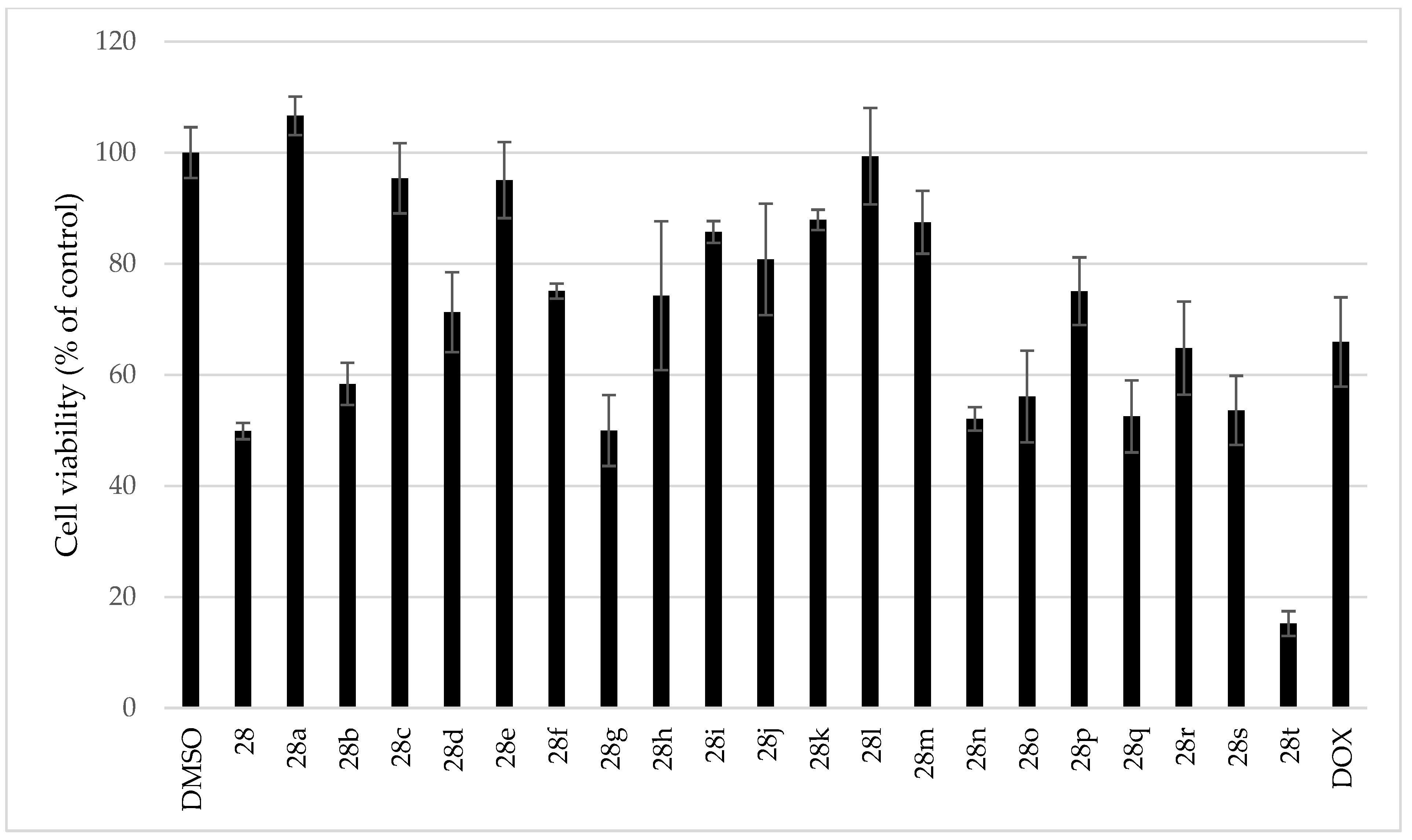
3.5. Cytotoxicity of Tested Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. World Malaria Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Suh, K.N.; Kain, K.C.; Keystone, J.S. Malaria. Can. Med. Assoc. J. 2004, 170, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snow, R.W.; Guerra, C.A.; Noor, A.M.; Myint, H.Y.; Hay, S.I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 2005, 434, 214–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thu, A.M.; Phyo, A.P.; Landier, J.; Parker, D.M.; Nosten, F.H. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J. 2017, 284, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Perkins, S.L. Malaria’s many mates: Past, present, and future of the systematics of the order Haemosporida. J. Parasitol. 2014, 100, 11–25. [Google Scholar] [CrossRef]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Agop-Nersesian, C.; Niklaus, L.; Wacker, R.; Heussler, V.T. Host cell cytosolic immune response during Plasmodium liver stage development. FEMS Microbiol. Rev. 2018, 42, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Bhardwaj, T.R.; Prasad, D.N.; Singh, R.K. Drug targets for resistant malaria: Historic to future perspectives. Biomed. Pharmacother. 2018, 104, 8–27. [Google Scholar] [CrossRef]
- Frevert, U. Sneaking in through the back entrance: The biology of malaria liver stages. Trends Parasitol. 2004, 20, 417–424. [Google Scholar] [CrossRef]
- Fontinha, D.; Moules, I.; Prudêncio, M. Repurposing drugs to fight hepatic malaria parasites. Molecules 2020, 25, 3409. [Google Scholar] [CrossRef]
- Cowman, A.F.; Healer, J.; Marapana, D.; Marsh, K. Malaria: Biology and disease. Cell 2016, 167, 610–624. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.; Prudêncio, M.; Moreira, R.; Mota, M.M.; Lopes, F. Targeting the liver stage of malaria parasites: A yet unmet goal. J. Med. Chem. 2012, 55, 995–1012. [Google Scholar] [CrossRef]
- Mota, M.M.; Rodriguez, A. New pieces for the malaria liver stage puzzle: Where will they fit? Cell Host Microbe 2008, 3, 63–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.; Goyal, M.; Iqbal, M.S.; Pal, C.; Dey, S.; Bindu, S.; Maity, P.; Bandyopadhyay, U. Novel antimalarial drug targets: Hope for new antimalarial drugs. Expert Rev. Clin. Pharmacol. 2009, 2, 469–489. [Google Scholar] [CrossRef] [PubMed]
- Daily, J. Malaria 2017: Update on the clinical literature and management. Curr. Infect. Dis. Rep. 2017, 19, 28. [Google Scholar] [CrossRef]
- Uthman, O.A.; Graves, P.M.; Saunders, R.; Gelband, H.; Richardson, M.; Garner, P. Safety of primaquine given to people with G6PD deficiency: Systematic review of prospective studies. Malar. J. 2017, 16, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, L.; Kane, M. Tafenoquine therapy and G6PD genotype. In Medical Genetics Summaries; Pratt, V.M., Scott, S.A., Pirmohamed, M., Eds.; National Center for Biotechnology Information: Bethesda, ML, USA, 2012. [Google Scholar]
- Kappe, S.H.; Duffy, P.E. Malaria liver stage culture: In Vitro veritas? Am. J. Trop. Med. Hyg. 2006, 74, 706–707. [Google Scholar] [CrossRef]
- Antony, H.A.; Parija, S.C. Antimalarial drug resistance: An overview. Trop. Parasitol. 2016, 6, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Mvumbi, D.M.; Bobanga, T.L.; Kayembe, J.-M.N.; Mvumbi, G.L.; Situakibanza, H.N.-T.; Benoit-Vical, F.; Melin, P.; De Mol, P.; Hayette, M.-P. Molecular surveillance of Plasmodium falciparum resistance to artemisinin-based combination therapies in the Democratic Republic of Congo. PLoS ONE 2017, 12, e0179142. [Google Scholar] [CrossRef] [Green Version]
- Septembre-Malaterre, A.; Lalarizo Rakoto, M.; Marodon, C.; Bedoui, Y.; Nakab, J.; Simon, E.; Hoarau, L.; Savriama, S.; Strasberg, D.; Guiraud, P.; et al. Artemisia annua, a traditional plant brought to light. Int. J. Mol. Sci. 2020, 21, 4986. [Google Scholar] [CrossRef]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.-C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef]
- Nosten, F.; White, N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007, 77, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Pazhayam, N.M.; Chhibber-Goel, J.; Sharma, A. New leads for drug repurposing against malaria. Drug Discov. Today 2019, 24, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Mäser, P.; Tadoori, L.P.; Ioset, J.R.; Brun, R. Antiprotozoal activity profiling of approved drugs: A starting point toward drug repositioning. PLoS ONE 2015, 10, e0135556. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [Green Version]
- Nyunt, M.M.; Nguyen, V.K.; Kajubi, R.; Huang, L.; Ssebuliba, J.; Kiconco, S.; Mwima, M.W.; Achan, J.; Aweeka, F.; Parikh, S.; et al. Artemether-lumefantrine pharmacokinetics and clinical response are minimally altered in pregnant ugandan women treated for uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 2015, 60, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Flora Online. Available online: http://worldfloraonline.org/taxon/wfo-7000000018#feedbackModal (accessed on 10 November 2022).
- Berkov, S.; Osorio, E.; Viladomat, F.; Bastida, J. Chemodiversity, chemotaxonomy and chemoecology of Amaryllidaceae alkaloids. In The Alkaloids, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 83, pp. 113–185. [Google Scholar] [CrossRef]
- Nair, J.; van Staden, J. Antiplasmodial studies within the plant family Amaryllidaceae. Nat. Prod. Commun. 2019, 14, 1934578X1987293. [Google Scholar] [CrossRef]
- Soto-Vásquez, M.R.; Horna-Pinedo, M.V.; Tallini, L.R.; Bastida, J. Chemical composition and in vitro antiplasmodial activity of the total alkaloids of the bulbs of two Amaryllidaceae species from northern Peru. Pharmacogn. J. 2021, 13, 1046–1052. [Google Scholar] [CrossRef]
- Cahlíková, L.; Kawano, I.; Řezáčová, M.; Blunden, G.; Hulcová, D.; Havelek, R. The Amaryllidaceae alkaloids haemanthamine, haemanthidine and their semisynthetic derivatives as potential drugs. Phytochem. Rev. 2021, 20, 303–323. [Google Scholar] [CrossRef]
- Gonring-Salarini, K.; Conti, R.; Andrade, J.P.; Borges, B.J.P.; Aguiar, A.C.; de Souza, J.O.; Zanini, C.L.; Oliva, G.; Tenorio, J.C.; Ellena, J.; et al. In vitro antiplasmodial activities of alkaloids isolated from roots of Worsleya procera (Lem.) Traub (Amaryllidaceae). J. Braz. Chem. Soc. 2019, 30, 1624–1633. [Google Scholar] [CrossRef]
- Cho, N.; Du, Y.; Valenciano, A.L.; Fernández-Murga, M.L.; Goetz, M.; Clement, J.; Cassera, M.B.; Kingston, D.G.I. Antiplasmodial alkaloids from bulbs of Amaryllis belladonna Steud. Bioorg. Med. Chem. Lett. 2018, 28, 40–42. [Google Scholar] [CrossRef]
- Akinyele, S.T.; Elusiyan, C.A.; Omisore, N.O.; Adewunmi, C.O. Antimalarial activities and alkaloids from Crinum jagus (Thomps) DANDY. J. Ethnopharmacol. 2022, 296, 115359. [Google Scholar] [CrossRef]
- Tajuddeen, N.; Van Heerden, F.R. Antiplasmodial natural products: An update. Malar. J. 2019, 18, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaněčková, N.; Hošťálková, A.; Šafratová, M.; Kuneš, J.; Hulcová, D.; Hrabinová, M.; Doskočil, I.; Štěpánková, Š.; Opletal, L.; Nováková, L.; et al. Isolation of Amaryllidaceae alkaloids from Nerine bowdenii W. Watson and their biological activities. RSC Adv. 2016, 6, 80114–80120. [Google Scholar] [CrossRef]
- Šafratová, M.; Hošťálková, A.; Hulcová, D.; Breiterová, K.; Hrabcová, V.; Machado, M.; Fontinha, D.; Prudêncio, M.; Kuneš, J.; Chlebek, J.; et al. Alkaloids from Narcissus poeticus cv. Pink Parasol of various structural types and their biological activity. Arch. Pharm. Res. 2018, 41, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Hulcová, D.; Maříková, J.; Korábečný, J.; Hošťálková, A.; Jun, D.; Kuneš, J.; Chlebek, J.; Opletal, L.; De Simone, A.; Nováková, L.; et al. Amaryllidaceae alkaloids from Narcissus pseudonarcissus L. cv. Dutch Master as potential drugs in treatment of Alzheimer’s disease. Phytochemistry 2019, 165, 112055. [Google Scholar] [CrossRef]
- Breiterová, K.; Koutová, D.; Maříková, J.; Havelek, R.; Kuneš, J.; Majorošová, M.; Opletal, L.; Hošťálková, A.; Jenčo, J.; Řezáčová, M.; et al. Amaryllidaceae alkaloids of different structural types from Narcissus L. cv. Professor Einstein and their cytotoxic activity. Plants 2020, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Maříková, J.; Ritomská, A.; Korábečný, J.; Peřinová, R.; Al Mamun, A.; Kučera, T.; Kohelová, E.; Hulcová, D.; Kobrlová, T.; Kuneš, J.; et al. Aromatic esters of the crinane amaryllidaceae alkaloid ambelline as selective inhibitors of butyrylcholinesterase. J. Nat. Prod. 2020, 83, 1359–1367. [Google Scholar] [CrossRef]
- Viladomat, F.; Codina, C.; Bastida, J.; Mathee, S.; Campbell, W.E. Further alkaloids from Brunsvigia josephinae. Phytochemistry 1995, 40, 961–965. [Google Scholar] [CrossRef]
- Kohelová, E.; Peřinová, R.; Maafi, N.; Korábečný, J.; Hulcová, D.; Maříková, J.; Kučera, T.; Martínez González, L.; Hrabinová, M.; Vorčáková, K.; et al. Derivatives of the β-crinane amaryllidaceae alkaloid haemanthamine as multi-target directed ligands for alzheimer’s disease. Molecules 2019, 24, 1307. [Google Scholar] [CrossRef] [Green Version]
- Peřinová, R.; Maafi, N.; Korábečný, J.; Kohelová, E.; De Simone, A.; Al Mamun, A.; Hulcová, D.; Marková, J.; Kučera, T.; Jun, D.; et al. Functionalized aromatic esters of the Amaryllidaceae alkaloid haemanthamine and their in vitro and in silico biological activity connected to Alzheimer’s disease. Bioorg. Chem. 2020, 100, 103928. [Google Scholar] [CrossRef]
- Prudêncio, M.; Mota, M.M.; Mendes, A.M. A toolbox to study liver stage malaria. Trends Parasitol. 2011, 27, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Ploemen, I.H.J.; Prudêncio, M.; Douradinha, B.G.; Ramesar, J.; Fonager, J.; van Gemert, G.-J.; Luty, A.J.F.; Hermsen, C.C.; Saurwein, R.W.; Baptista, F.G.; et al. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, L.; Fontinha, D.; Francisco, D.; Mendes, A.M.; Prudêncio, M.; Singh, K. Molecular design and synthesis of ivermectin hybrids targeting hepatic and erythrocytic stages of Plasmodium parasites. J. Med. Chem. 2020, 63, 1750–1762. [Google Scholar] [CrossRef] [PubMed]
- Havelek, R.; Muthna, D.; Tomšík, P.; Královec, K.; Seifrtová, M.; Cahlíková, L.; Hošťálková, A.; Šafratová, M.; Perwein, M.; Čermáková, E.; et al. Anticancer potential of Amaryllidaceae alkaloids evaluated by screening with a panel of human cells, real-time cellular analysis and Ehrlich tumor-bearing mice. Chem. Biol. Interact. 2017, 275, 121–132. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 P. berghei [µM] | HepG2 [%] 1 (c = 10 µM) |
|---|---|---|
| Ambelline (28) | >10 | 50 ± 1 |
| 11-O-Acetylambelline (28a) | >10 | 107 ± 3 |
| 11-O-Propionylambelline (28b) | >10 | 58 ± 4 |
| 11-O-Isobutanoylambelline (28c) | >10 | 95 ± 6 |
| 11-O-Pentanoylambelline (28d) | 4.08 ± 1.78 | 71 ± 7 |
| 11-O-Benzoylambelline (28e) | 0.726 ± 0.126 | 95 ± 7 |
| 11-O-(3-Methylbenzoyl)ambelline (28f) | 0.317 ± 0.118 | 75 ± 1 |
| 11-O-(4-Methylbenzoyl)ambelline (28g) | 0.402 ± 0.081 | 50 ± 6 |
| 11-O-(3,5-Dimethylbenzoyl)ambelline (28h) | 0.100 ± 0.018 | 74 ± 13 |
| 11-O-(2-Methoxybenzoyl)ambelline (28i) | 2.40 ± 0.77 | 86 ± 2 |
| 11-O-(3-Methoxybenzoyl)ambelline (28j) | 0.261 ± 0.090 | 81 ± 10 |
| 11-O-(4-Methoxybenzoyl)ambelline (28k) | 0.451 ± 0.007 | 88 ± 2 |
| 11-O-(3,4-Dimethoxybenzoyl)ambelline (28l) | 0.147 ± 0.018 | 99 ± 9 |
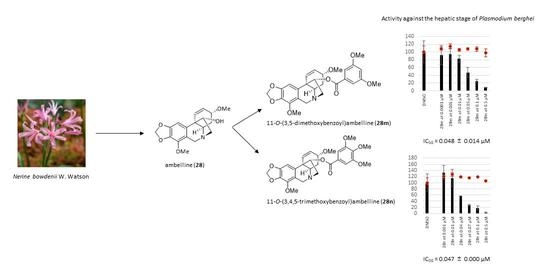
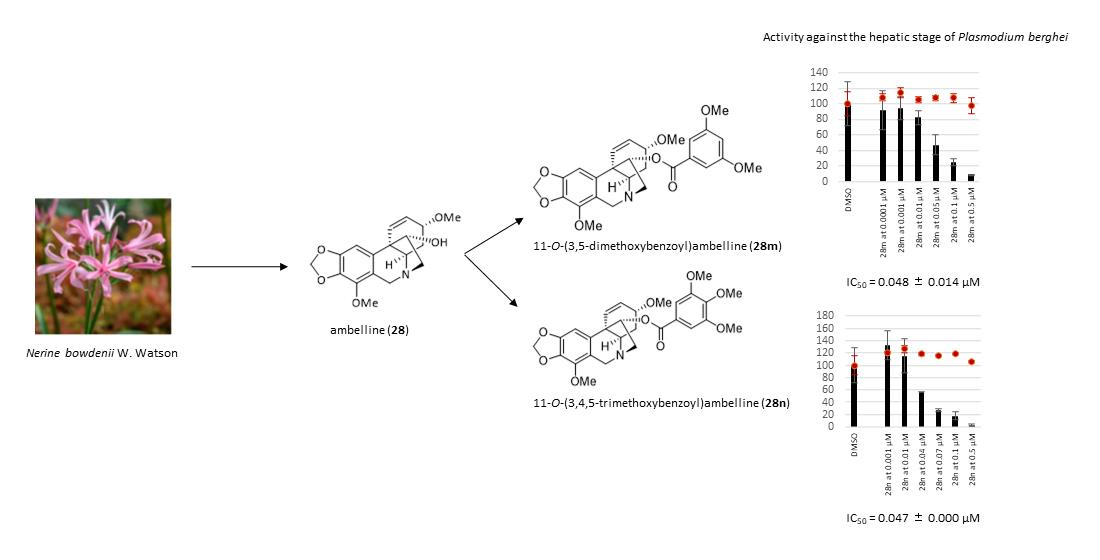
| 11-O-(3,5-Dimethoxybenzoyl)ambelline (28m) | 0.048 ± 0.014 | 87 ± 6 |
| 11-O-(3,4,5-Trimethoxybenzoyl)ambelline (28n) | 0.047 ± 0.000 | 52 ± 2 |
| 11-O-(3,5-Diethoxybenzoyl)ambelline (28o) | 0.316 ± 0.013 | 56 ± 8 |
| 11-O-(3-Nitrobenzoyl)ambelline (28p) | 1.53 ± 0.22 | 75 ± 6 |
| 11-O-(4-Nitrobenzoyl)ambelline (28q) | 1.20 ± 0.18 | 53 ± 6 |
| 11-O-(4-Methyl-3-nitrobenzoyl)ambelline (28r) | 0.410 ± 0.054 | 65 ± 8 |
| 11-O-(2-Chloro-4-nitrobenzoyl)ambelline (28s) | not tested | 54 ± 6 |
| 11-O-(4-Chloro-3-nitrobenzoyl)ambelline (28t) | not tested | 15 ± 2 |
| Primaquine 2 | 5.74 ± 0.86 | - |
| Doxorubicine 2 | - | 66 ± 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breiterová, K.H.; Ritomská, A.; Fontinha, D.; Křoustková, J.; Suchánková, D.; Hošťálková, A.; Šafratová, M.; Kohelová, E.; Peřinová, R.; Vrabec, R.; et al. Derivatives of Amaryllidaceae Alkaloid Ambelline as Selective Inhibitors of Hepatic Stage of Plasmodium berghei Infection In Vitro. Pharmaceutics 2023, 15, 1007. https://doi.org/10.3390/pharmaceutics15031007
Breiterová KH, Ritomská A, Fontinha D, Křoustková J, Suchánková D, Hošťálková A, Šafratová M, Kohelová E, Peřinová R, Vrabec R, et al. Derivatives of Amaryllidaceae Alkaloid Ambelline as Selective Inhibitors of Hepatic Stage of Plasmodium berghei Infection In Vitro. Pharmaceutics. 2023; 15(3):1007. https://doi.org/10.3390/pharmaceutics15031007
Chicago/Turabian StyleBreiterová, Kateřina Hradiská, Aneta Ritomská, Diana Fontinha, Jana Křoustková, Daniela Suchánková, Anna Hošťálková, Marcela Šafratová, Eliška Kohelová, Rozálie Peřinová, Rudolf Vrabec, and et al. 2023. "Derivatives of Amaryllidaceae Alkaloid Ambelline as Selective Inhibitors of Hepatic Stage of Plasmodium berghei Infection In Vitro" Pharmaceutics 15, no. 3: 1007. https://doi.org/10.3390/pharmaceutics15031007