
Cell-Friendly Chitosan-Xanthan Gum Membranes Incorporating Hydroxyapatite Designed for Periodontal Tissue Regeneration
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Production of C-X Membranes Containing HA
2.2.1. Synthesis and Processing of Hydroxyapatite
2.2.2. Production of C-X Membranes and C-X-HA
2.3. HA and Membranes Characterization
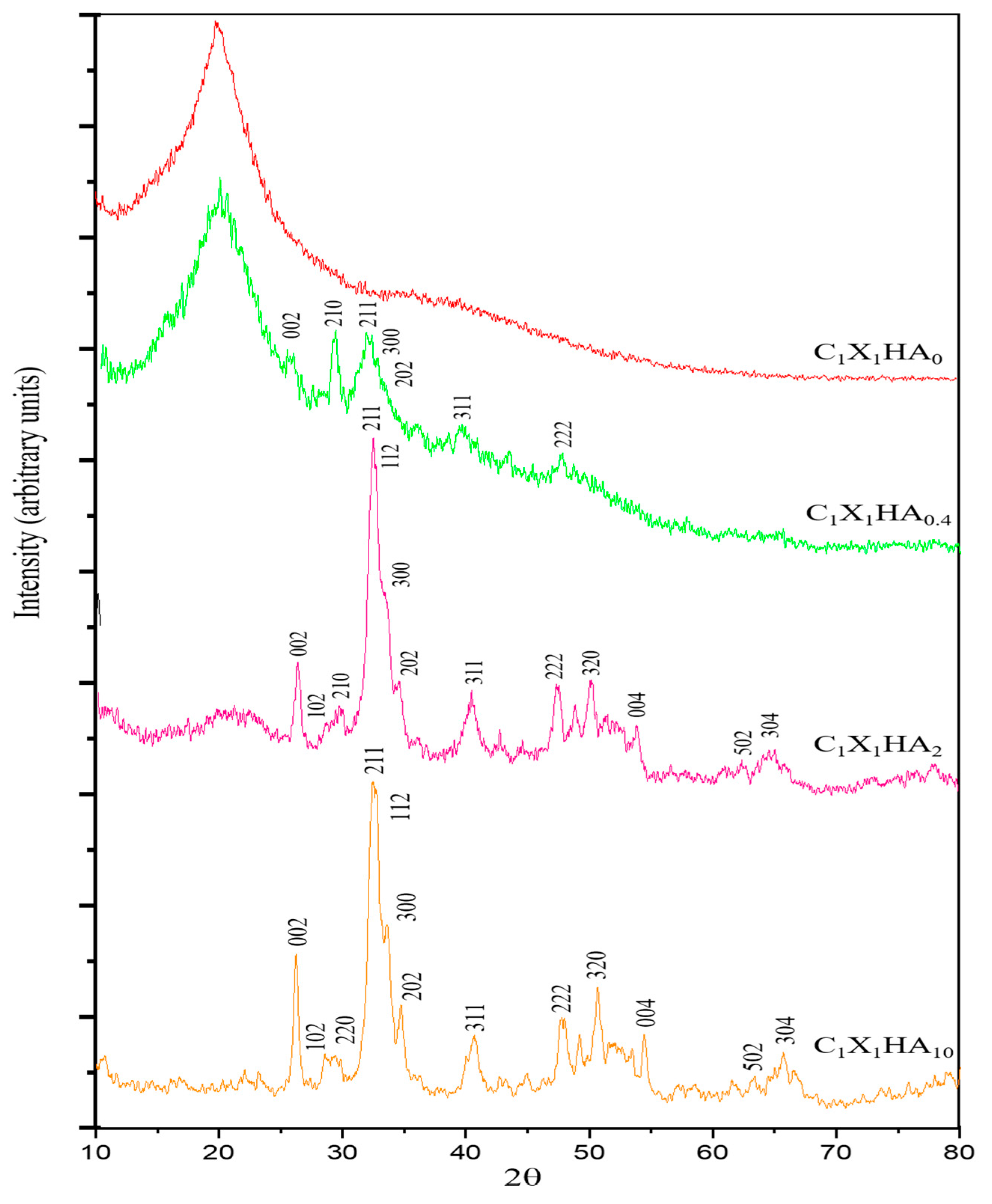
2.3.1. X-ray Diffraction (XRD)
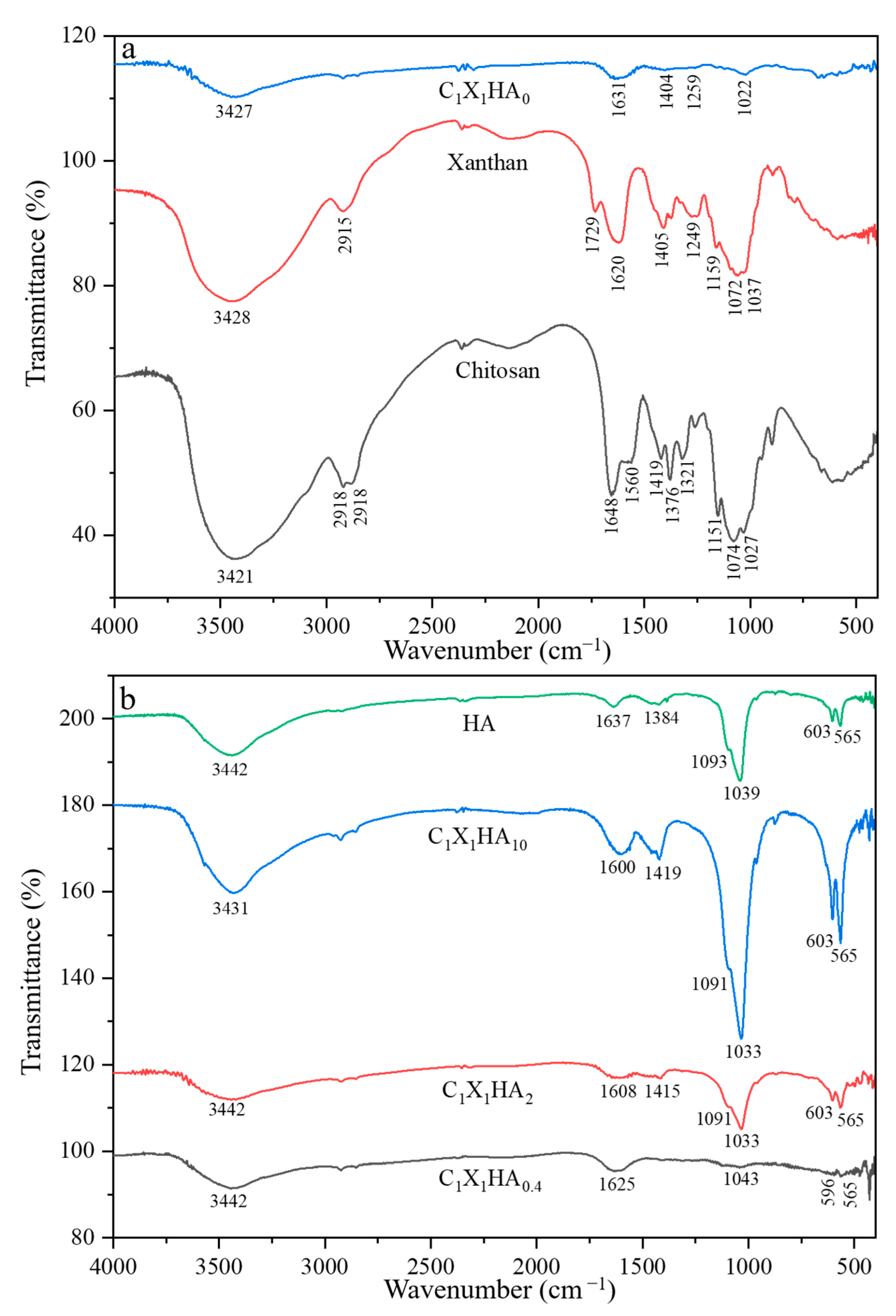
2.3.2. Fourier Transform Infrared Spectroscopy (FTIR)
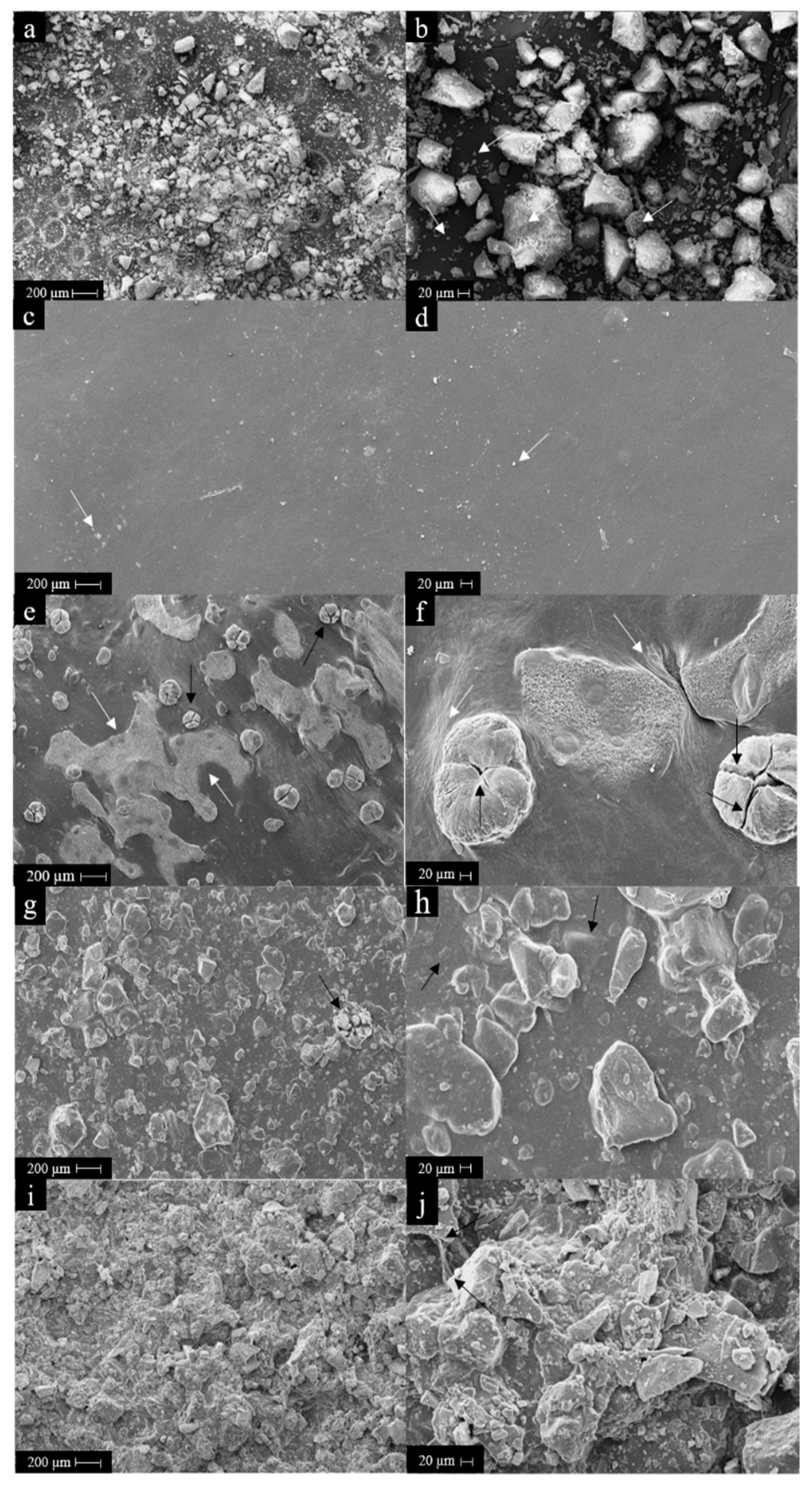
2.3.3. Scanning Electron Microscopy (SEM)
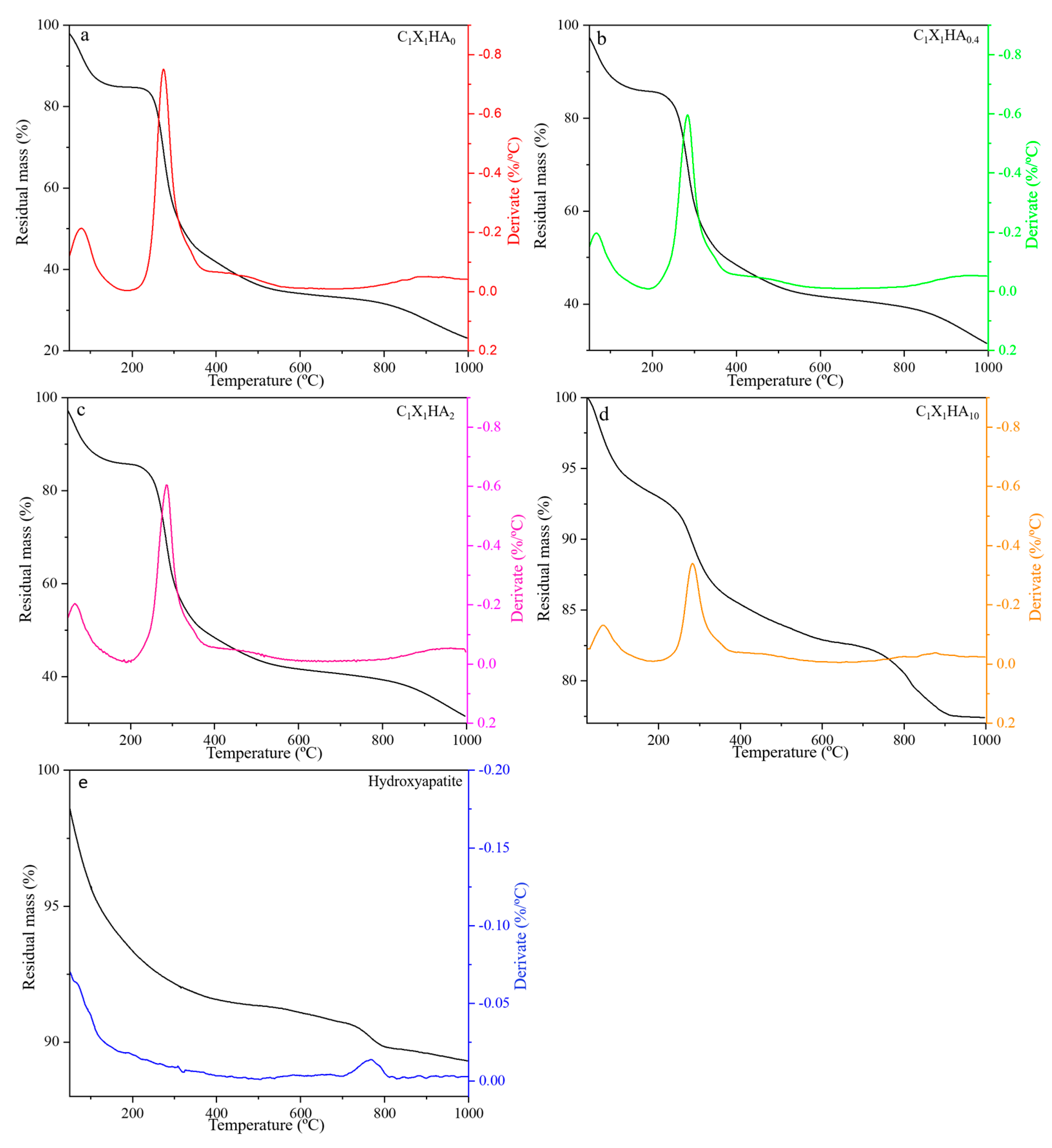
2.3.4. Thermogravimetric Analysis (TGA)
2.3.5. Confocal Laser Microscopy Analysis
2.3.6. Mechanical Properties
2.3.7. Proliferation of DPSC on the Surface of the Membranes
2.4. Statistical Analysis
3. Results and Discussion
3.1. Crystallinity of the Membranes
3.2. Comparative Results of Fourier Transform Infrared Spectroscopy
3.3. Thermal Stability of HA and Membrane Formulations
3.4. Morphology Surface Analysis of HA Powder and Membranes
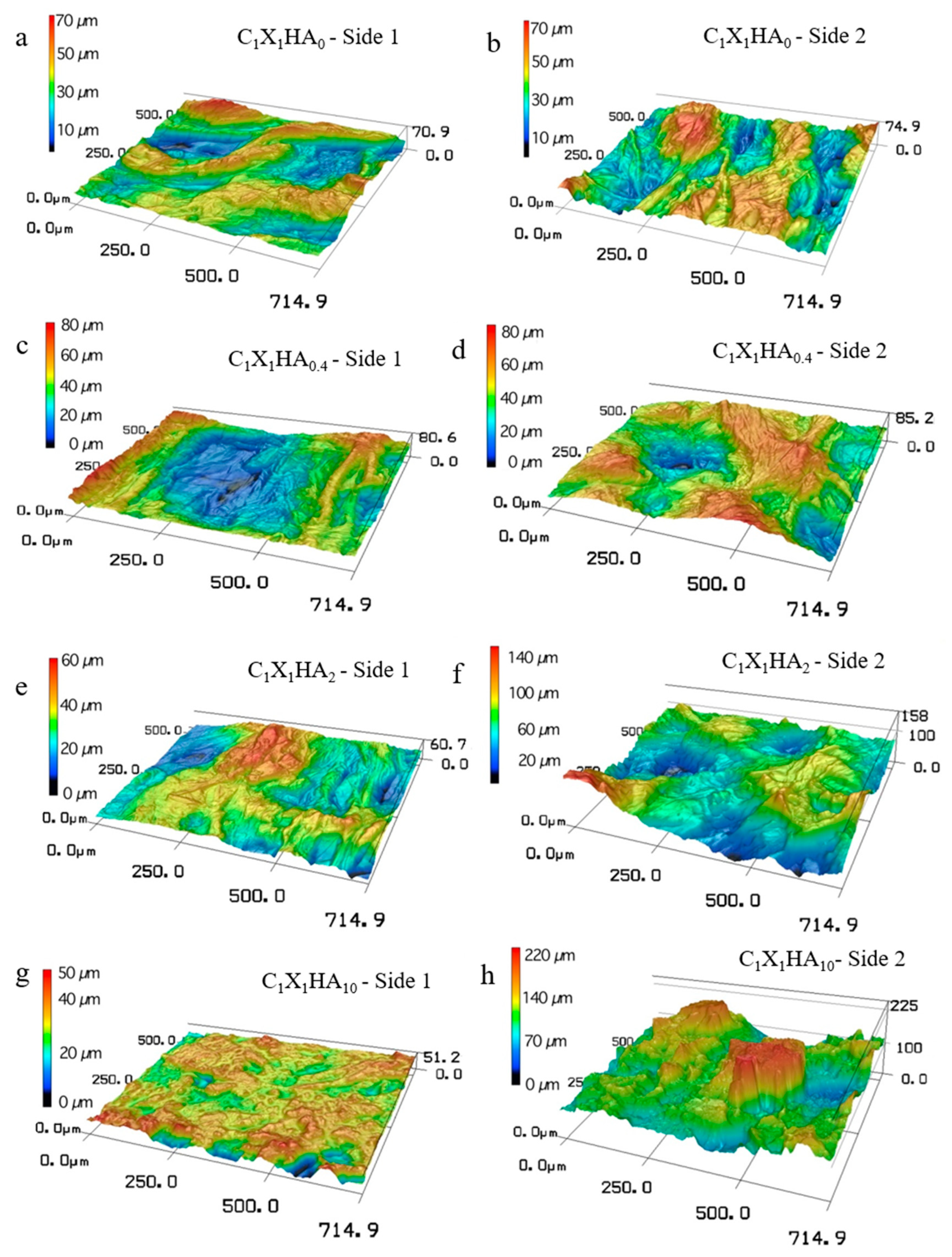
3.5. Membrane Aspect Evaluated by Confocal Microscopy and Roughness
3.6. Mechanical Properties
3.7. In Vitro Cell Proliferation on Both Surfaces of Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amato, M.; Santonocito, S.; Viglianisi, G.; Tatullo, M.; Isola, G. Impact of Oral Mesenchymal Stem Cells Applications as a Promising Therapeutic Target in the Therapy of Periodontal Disease. Int. J. Mol. Sci. 2022, 23, 13419. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E. Shifting the Paradigm from Inhibitors of Inflammation to Resolvers of Inflammation in Periodontitis. J. Periodontol. 2020, 91, S19–S25. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, A.; Gigante, I.; Colucci, S.; Grano, M. Periodontal Disease: Linking the Primary Inflammation to Bone Loss. Clin. Dev. Immunol. 2013, 2013, 503754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H.; Hu, Y.; Jing, Y.; Geng, Z.; Su, J. Bone Repair Biomaterials: A Perspective from Immunomodulation. Adv. Funct. Mater. 2022, 32, 2208639. [Google Scholar] [CrossRef]
- Gottlow, J.; Nyman, S.; Karring, T.; Lindhe, J. New Attachment Formation as the Result of Controlled Tissue Regeneration. J. Clin. Periodontol. 1984, 11, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, R.; Tovar, N.; Anchieta, R.B.; Machado, L.S.; Marin, C.; Teixeira, H.S.; Coelho, P.G. The Combined Effects of Undersized Drilling and Implant Macrogeometry on Bone Healing around Dental Implants: An Experimental Study. Int. J. Oral Maxillofac. Surg. 2014, 43, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Petrescu, N.; Crisan, B.; Aghiorghiesei, O.; Sarosi, C.; Mirica, I.C.; Lucaciu, O.; Iușan, S.A.L.; Dirzu, N.; Apostu, D. Gradual Drug Release Membranes and Films Used for the Treatment of Periodontal Disease. Membranes 2022, 12, 895. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Luan, X.; Liu, X. Recent Advances in Periodontal Regeneration: A Biomaterial Perspective. Bioact. Mater. 2020, 5, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Vani, T.M.S.; Paramashivaiah, R.; Prabhuji, M.L.V.; Peeran, S.W.; Fageeh, H.; Tasleem, R.; Bahamdan, G.K.; Aldosari, L.I.N.; Bhavikatti, S.K.; Scardina, G.A. Regeneration of Intrabony Defects with Nano Hydroxyapatite Graft, Derived from Eggshell along with Periosteum as Barrier Membrane under Magnification—An Interventional Study. Appl. Sci. 2023, 13, 1693. [Google Scholar] [CrossRef]
- dos Santos, V.I.; dos Santos, V.I.; Merlini, C.; Aragones, Á.; Cesca, K.; Fredel, M.C. Influence of Calcium Phosphates Incorporation into Poly(Lactic-Co-Glycolic Acid) Electrospun Membranes for Guided Bone Regeneration. Polym. Degrad. Stab. 2020, 179, 109253. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Tai, H.-Y.; Fu, E.; Don, T.-M. Guided Bone Regeneration Activity of Different Calcium Phosphate/Chitosan Hybrid Membranes. Int. J. Biol. Macromol. 2019, 126, 159–169. [Google Scholar] [CrossRef]
- Camargo, L.G.; de Freitas Rosa Remiro, P.; Rezende, G.S.; Di Carla Santos, S.; Franz-Montan, M.; Moraes, Â.M. Development of Bioadhesive Polysaccharide-Based Films for Topical Release of the Immunomodulatory Agent Imiquimod on Oral Mucosa Lesions. Eur. Polym. J. 2021, 151, 110422. [Google Scholar] [CrossRef]
- Remiro, P.d.F.R.; Nagahara, M.H.T.; Ghezzi, M.; Filippini, A.; Demurtas, A.; Pescina, S.; Santi, P.; Padula, C.; Moraes, Â.M.; Nicoli, S. An Alternative Device for the Topical Treatment of Oral Cancer: Development and Ex-Vivo Evaluation of Imiquimod-Loaded Polysaccharides Formulations. Pharmaceutics 2022, 14, 2573. [Google Scholar] [CrossRef]
- Veiga, I.G.; Moraes, Â.M. Study of the Swelling and Stability Properties of Chitosan-Xanthan Membranes. J. Appl. Polymer Sci. 2012, 124, E154–E160. [Google Scholar] [CrossRef]
- Bellini, M.Z.; Caliari-Oliveira, C.; Mizukami, A.; Swiech, K.; Covas, D.T.; Donadi, E.A.; Oliva-Neto, P.; Moraes, Â.M. Combining Xanthan and Chitosan Membranes to Multipotent Mesenchymal Stromal Cells as Bioactive Dressings for Dermo-Epidermal Wounds. J. Biomat.s Appl. 2015, 29, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- de Souza, R.F.B.; de Souza, F.C.B.; Thorpe, A.; Mantovani, D.; Popat, K.C.; Moraes, Â.M. Phosphorylation of Chitosan to Improve Osteoinduction of Chitosan/Xanthan-Based Scaffolds for Periosteal Tissue Engineering. Int. J. Biol. Macromol. 2020, 143, 619–632. [Google Scholar] [CrossRef]
- Needleman, I.G.; Smales, F.C.; Martin, G.P. An Investigation of Bioadhesion for Periodontal and Oral Mucosal Drug Delivery. J. Clin. Periodontol. 1997, 24, 394–400. [Google Scholar] [CrossRef]
- Bombaldi de Souza, R.F.; Bombaldi de Souza, F.C.; Westin, C.B.; Barbosa, R.M.; Moraes, Â.M. Xanthan Gum for Regenerative Medicine. In Polysaccharides of Microbial Origin; Springer International Publishing: Cham, Switzerland, 2022; pp. 1133–1160. [Google Scholar]
- Mohammadinejad, R.; Kumar, A.; Ranjbar-Mohammadi, M.; Ashrafizadeh, M.; Han, S.S.; Khang, G.; Roveimiab, Z. Recent Advances in Natural Gum-Based Biomaterials for Tissue Engineering and Regenerative Medicine: A Review. Polymers 2020, 12, 176. [Google Scholar] [CrossRef] [Green Version]
- Busscher, H.J.; Engels, E.; Dijkstra, R.J.B.; van der Mei, H.C. Influence of a Chitosan on Oral Bacterial Adhesion and Growth in Vitro. Eur. J. Oral Sci. 2008, 116, 493–495. [Google Scholar] [CrossRef]
- Anitha, A.; Divya Rani, V.V.; Krishna, R.; Sreeja, V.; Selvamurugan, N.; Nair, S.V.; Tamura, H.; Jayakumar, R. Synthesis, Characterization, Cytotoxicity and Antibacterial Studies of Chitosan, O-Carboxymethyl and N,O-Carboxymethyl Chitosan Nanoparticles. Carbohydr. Polym. 2009, 78, 672–677. [Google Scholar] [CrossRef]
- Neves, J.G.; Navarro da Rocha, D.; Lopes, C.C.; Barbosa, R.M.; Ferreira, L.F.; Westin, C.B.; Moraes, Â.M.; Calsa, B.; Santamaria, M., Jr.; Correr-Sobrinho, L.; et al. Calcium Phosphates Chitosan-Xanthan Composite Scaffolds Associated with Mesenchymal Stem Cells for Regenerative Dentistry Application. Ceram. Int. 2022, 48, 23088–23095. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, S.; Chen, W.; Hu, Y.; Geng, Z.; Su, J. Bone/Cartilage Targeted Hydrogel: Strategies and Applications. Bioact. Mater. 2023, 23, 156–169. [Google Scholar] [CrossRef]
- da Rocha, D.N.; de Andrade Gobbo, L.; da Silva, M.H.P. Production and Characterization of Niobate Apatite. J. Mater. Res. Technol. 2013, 2, 24–29. [Google Scholar] [CrossRef] [Green Version]
- da Rocha, D.N.; da Silva, M.H.P.; de Campos, J.B.; Santana Blazutti Marçal, R.; Mijares, D.Q.; Coelho, P.G.; Cruz, L.R. Kinetics of Conversion of Brushite Coatings to Hydroxyapatite in Alkaline Solution. J. Mater. Res. Technol. 2018, 7, 479–486. [Google Scholar] [CrossRef]
- da Rocha, D.N.; de Oliveira Cruz, L.R.; de Campos, J.B.; dos Santos, J.L.; Santana Blazutti Marçal, R.; Mijares, D.Q.; Barbosa, R.M.; Coelho, P.G.; da Silva, M.H.P. Bioactivity of Strontium-Monetite Coatings for Biomedical Applications. Ceram. Int. 2019, 45, 7568–7579. [Google Scholar] [CrossRef]
- Zia, I.; Jolly, R.; Mirza, S.; Umar, M.S.; Owais, M.; Shakir, M. Hydroxyapatite Nanoparticles Fortified Xanthan Gum-Chitosan Based Polyelectrolyte Complex Scaffolds for Supporting the Osteo-Friendly Environment. ACS Appl. Bio. Mater. 2020, 3, 7133–7146. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.E.; de O Silva, M.; Rodas, A.C.D.; Bertran, C.A. Mineralized Layered Films of Xanthan and Chitosan Stabilized by Polysaccharide Interactions: A Promising Material for Bone Tissue Repair. Carbohydr. Polym. 2019, 207, 480–491. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Dissolution Mechanism of Calcium Apatites in Acids: A Review of Literature. World J. Methodol. 2012, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Westin, C.B.; Trinca, R.B.; Zuliani, C.; Coimbra, I.B.; Moraes, Â.M. Differentiation of Dental Pulp Stem Cells into Chondrocytes upon Culture on Porous Chitosan-Xanthan Scaffolds in the Presence of Kartogenin. Mater. Sci. Eng. C 2017, 80, 594–602. [Google Scholar] [CrossRef]
- Hodge, H.C.; LeFevre, M.L.; Bale, W.F. Chemical and X-ray Diffraction Studies of Calcium Phosphates. Ind. Eng. Chem. Anal. Ed. 1938, 10, 156–161. [Google Scholar] [CrossRef]
- Neves, J.G.; Navarro da Rocha, D.; Lopes, C.C.; Prado da Silva, M.H.; Sinhoreti, M.A.C.; Correr-Sobrinho, L.; Fraga, M.A.A.; Correr, A.B. Effect of PH Level and Calcination on the Production of Calcium Phosphates by Acidic Route of Wet Precipitation. Ceramica 2021, 67, 236–243. [Google Scholar] [CrossRef]
- Dumitriu, S.; Magny, P.; Montané, D.; Vidal, P.F.; Chornet, E. Polyionic Hydrogels Obtained by Complexation between Xanthan and Chitosan: Their Properties as Supports for Enzyme Immobilization. J. Bioact. Compat. Polym. 1994, 9, 184–209. [Google Scholar] [CrossRef]
- de Souza, R.F.B.; de Souza, F.C.B.; Moraes, Â.M. Polysaccharide-Based Membranes Loaded with Erythromycin for Application as Wound Dressings. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Mauricio-Sánchez, R.A.; Salazar, R.; Gabriel Luna-Bárcenas, J.; Mendoza-Galván, A. FTIR Spectroscopy Studies on the Spontaneous Neutralization of Chitosan Acetate Films by Moisture Conditioning. Vib. Spectrosc. 2018, 94, 1–6. [Google Scholar] [CrossRef]
- Pawlicka, A.; Tavares, F.C.; Dörr, D.S.; Cholant, C.M.; Ely, F.; Santos, M.J.L.; Avellaneda, C.O. Dielectric Behavior and FTIR Studies of Xanthan Gum-Based Solid Polymer Electrolytes. Electrochim. Acta 2019, 305, 232–239. [Google Scholar] [CrossRef]
- Popa, N.; Novac, O.; Profire, L.; Lupusoru, C.E.; Popa, M.I. Hydrogels Based on Chitosan–Xanthan for Controlled Release of Theophylline. J. Mater. Sci. Mater. Med. 2010, 21, 1241–1248. [Google Scholar] [CrossRef]
- Ćirić, A.; Medarević, Đ.; Čalija, B.; Dobričić, V.; Mitrić, M.; Djekic, L. Study of Chitosan/Xanthan Gum Polyelectrolyte Complexes Formation, Solid State and Influence on Ibuprofen Release Kinetics. Int. J. Biol. Macromol. 2020, 148, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Azaman, F.A.; Daubiné, F.; Lebatard, A.; Brennan Fournet, M.E.; Devine, D.M. Chitosan/Hydroxyapatite Scaffolds with P28 as a Promising Osteoinductive Scaffold for Bone Healing Applications. Micro 2023, 3, 118–142. [Google Scholar] [CrossRef]
- Martins, A.F.; de Oliveira, D.M.; Pereira, A.G.B.; Rubira, A.F.; Muniz, E.C. Chitosan/TPP Microparticles Obtained by Microemulsion Method Applied in Controlled Release of Heparin. Int. J. Biol. Macromol. 2012, 51, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Dorozhkin, S. Calcium Orthophosphate-Based Bioceramics. Materials 2013, 6, 3840–3942. [Google Scholar] [CrossRef] [Green Version]
- Othman, R.; Mustafa, Z.; Kien, P.T.; Ishak, N.F.; Shaaban, A.; Noor, A.M. Parameters Affecting the Synthesis of b-tricalcium Phosphate Powder Using a Wet Precipitation Method. J. Mech. Eng. Sci. 2017, 11, 3197–3205. [Google Scholar] [CrossRef]
- Pathak, T.S.; Yun, J.-H.; Lee, J.; Paeng, K.-J. Effect of Calcium Ion (Cross-Linker) Concentration on Porosity, Surface Morphology and Thermal Behavior of Calcium Alginates Prepared from Algae (Undaria Pinnatifida). Carbohydr. Polym. 2010, 81, 633–639. [Google Scholar] [CrossRef]
- Khan, I.M.; Ahmad, A.; Ullah, M.F. Synthesis, Crystal Structure, Antimicrobial Activity and DNA-Binding of Hydrogen-Bonded Proton-Transfer Complex of 2,6-Diaminopyridine with Picric Acid. J. Photochem. Photobiol. B. Biol. 2011, 103, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lim, L.C. Effect of Particle Size Distribution on Sintering of Agglomerate-Free Submicron Alumina Powder Compacts. J. Eur. Ceram. Soc. 2002, 22, 2197–2208. [Google Scholar] [CrossRef]
- Szcześ, A.; Yan, Y.; Chibowski, E.; Hołysz, L.; Banach, M. Properties of Natural and Synthetic Hydroxyapatite and Their Surface Free Energy Determined by the Thin-Layer Wicking Method. Appl. Surf. Sci. 2018, 434, 1232–1238. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Sathe, S.R.; Yim, E.K.F. From Nano to Micro: Topographical Scale and Its Impact on Cell Adhesion, Morphology and Contact Guidance. J. Phys. Condens Matter. 2016, 28, 183001. [Google Scholar] [CrossRef]
- de Souza, R.F.B.; de Souza, F.C.B.; Rodrigues, C.; Drouin, B.; Popat, K.C.; Mantovani, D.; Moraes, Â.M. Mechanically-Enhanced Polysaccharide-Based Scaffolds for Tissue Engineering of Soft Tissues. Mater. Sci. Eng. C 2019, 94, 364–375. [Google Scholar] [CrossRef]
- Lavenus, S.; Pilet, P.; Guicheux, J.; Weiss, P.; Louarn, G.; Layrolle, P. Behaviour of Mesenchymal Stem Cells, Fibroblasts and Osteoblasts on Smooth Surfaces. Acta Biomater. 2011, 7, 1525–1534. [Google Scholar] [CrossRef]
- Rougerie, P.; dos Santos, R.S.; Farina, M.; Anselme, K. Molecular Mechanisms of Topography Sensing by Osteoblasts: An Update. Appl. Sci. 2021, 11, 1791. [Google Scholar] [CrossRef]
- Elgali, I.; Omar, O.; Dahlin, C.; Thomsen, P. Guided Bone Regeneration: Materials and Biological Mechanisms Revisited. Eur. J. Oral Sci. 2017, 125, 315–337. [Google Scholar] [CrossRef] [Green Version]
- de Souza, R.F.B.; de Souza, R.F.B.; de Souza, F.C.B.; Moraes, Â.M. Analysis of the Performance of Polysaccharide Membranes in Aqueous Media as a Tool to Assist Wound-Dressing Selection. J. Appl. Polym. Sci. 2017, 134, 45386. [Google Scholar] [CrossRef]
- Prakash, J.; Prema, D.; Venkataprasanna, K.S.; Balagangadharan, K.; Selvamurugan, N.; Devanand Venkatasubbu, G. Nanocomposite Chitosan Film Containing Graphene Oxide/Hydroxyapatite/Gold for Bone Tissue Engineering. Int. J. Biol. Macromol. 2020, 154, 62–71. [Google Scholar] [CrossRef]
- Salim, S.A.; Loutfy, S.A.; El-Fakharany, E.M.; Taha, T.H.; Hussien, Y.; Kamoun, E.A. Influence of Chitosan and Hydroxyapatite Incorporation on Properties of Electrospun PVA/HA Nanofibrous Mats for Bone Tissue Regeneration: Nanofibers Optimization and in-Vitro Assessment. J. Drug Deliv. Sci. Technol. 2021, 62, 102417. [Google Scholar] [CrossRef]
- Discher, D.E. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, S.; Zibandeh, N.; Genc, D.; Ozcan, E.M.; Goker, K.; Akkoc, T. The Comparison of the Immunologic Properties of Stem Cells Isolated from Human Exfoliated Deciduous Teeth, Dental Pulp, and Dental Follicles. Stem Cells Int. 2016, 2016, 4682875. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, K.; Kidoaki, S. Cellular Durotaxis Revisited: Initial-Position-Dependent Determination of the Threshold Stiffness Gradient to Induce Durotaxis. Langmuir 2019, 35, 7478–7486. [Google Scholar] [CrossRef]
- Mehrabani, D.; Mahdiyar, P.; Torabi, K.; Robati, R.; Zare, S.; Dianatpour, M.; Tamadon, A. Growth Kinetics and Characterization of Human Dental Pulp Stem Cells: Comparison between Third Molar and First Premolar Teeth. J. Clin. Exp. Dent. 2017, 9, e172–e177. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Root Mean Square Roughness (µm) | |
|---|---|---|
| Side 1 | Side 2 | |
| C1X1HA0 | 9.97 ± 1.82 a,A | 10.23 ± 3.21 a,A |
| C1X1HA0.4 | 9.98 ± 1.25 a,A | 11.15 ± 2.22 a,A |
| C1X1HA2 | 8.03 ± 4.98 a,A,B | 21.30 ± 2.56 b,B |
| C1X1HA10 | 4.51 ± 1.05 a,B | 27.78 ± 12.57 b,B |
| Formulation | Tension at Break (kPa) | Young’s Modulus (kPa) |
|---|---|---|
| C1X1HA0 | 288.17 ± 76.55 a | 5.27 ± 0.02 a |
| C1X1HA0.4 | 50.51 ± 11.17 b | 7.29 ± 0.23 b |
| C1X1HA2 | 211.59 ± 16.12 c | 5.35 ± 0.01 a |
| C1X1HA10 | 83.28 ± 9.64 b | 3.43 ± 0.02 c |
| Time (h) | Cell Proliferation on Side 1 (%) | |||
| C1X1HA0 | C1X1HA.4 | C1X1HA2 | C1X1HA10 | |
| 24 | 41.5 ± 3.7 a,A | 39.4 ± 4.6 a,b,A | 39.9 ± 2.0 a,b,A | 32.3 ± 1.7 b,A |
| 48 | 56.2 ± 2.0 a,B | 40.5 ± 4.0 b,A | 39.0 ± 2.6 b,A | 41.6 ± 5.5 b,A |
| 72 | 40.9 ± 1.0 a,A | 43.5 ± 2.1 a,A | 49.6 ± 8.6 a,A | 76.9 ± 12.9 b,B |
| Cell proliferation on Side 2 (%) | ||||
| C1X1HA0 | C1X1HA0.4 | C1X1HA2 | C1X1HA10 | |
| 24 | 41.5 ± 1.4 a,A,B | 35.9 ± 2.5 a,A | 37.2 ± 3.1 a,A | 53.1 ± 3.3 b,A |
| 48 | 36.0 ± 4.3 a,A | 35.2 ± 4.0 a,A | 37.1 ± 3.4 a,A | 55.1 ± 3.5 b,A |
| 72 | 50.8 ± 7.1 a,B | 66.0 ± 8.9 a,B | 59.5 ± 5.2 a,B | 91.6 ± 7.3 b,B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa, R.M.; da Rocha, D.N.; Bombaldi de Souza, R.F.; Santos, J.L.; Ferreira, J.R.M.; Moraes, Â.M. Cell-Friendly Chitosan-Xanthan Gum Membranes Incorporating Hydroxyapatite Designed for Periodontal Tissue Regeneration. Pharmaceutics 2023, 15, 705. https://doi.org/10.3390/pharmaceutics15020705
Barbosa RM, da Rocha DN, Bombaldi de Souza RF, Santos JL, Ferreira JRM, Moraes ÂM. Cell-Friendly Chitosan-Xanthan Gum Membranes Incorporating Hydroxyapatite Designed for Periodontal Tissue Regeneration. Pharmaceutics. 2023; 15(2):705. https://doi.org/10.3390/pharmaceutics15020705
Chicago/Turabian StyleBarbosa, Rafael Maza, Daniel Navarro da Rocha, Renata Francielle Bombaldi de Souza, Jheison Lopes Santos, José Ricardo M. Ferreira, and Ângela Maria Moraes. 2023. "Cell-Friendly Chitosan-Xanthan Gum Membranes Incorporating Hydroxyapatite Designed for Periodontal Tissue Regeneration" Pharmaceutics 15, no. 2: 705. https://doi.org/10.3390/pharmaceutics15020705