
Decreased Levels of Chaperones in Mucopolysaccharidoses and Their Elevation as a Putative Auxiliary Therapeutic Approach
 , , , , and
, , , , and 
Abstract
:1. Introduction

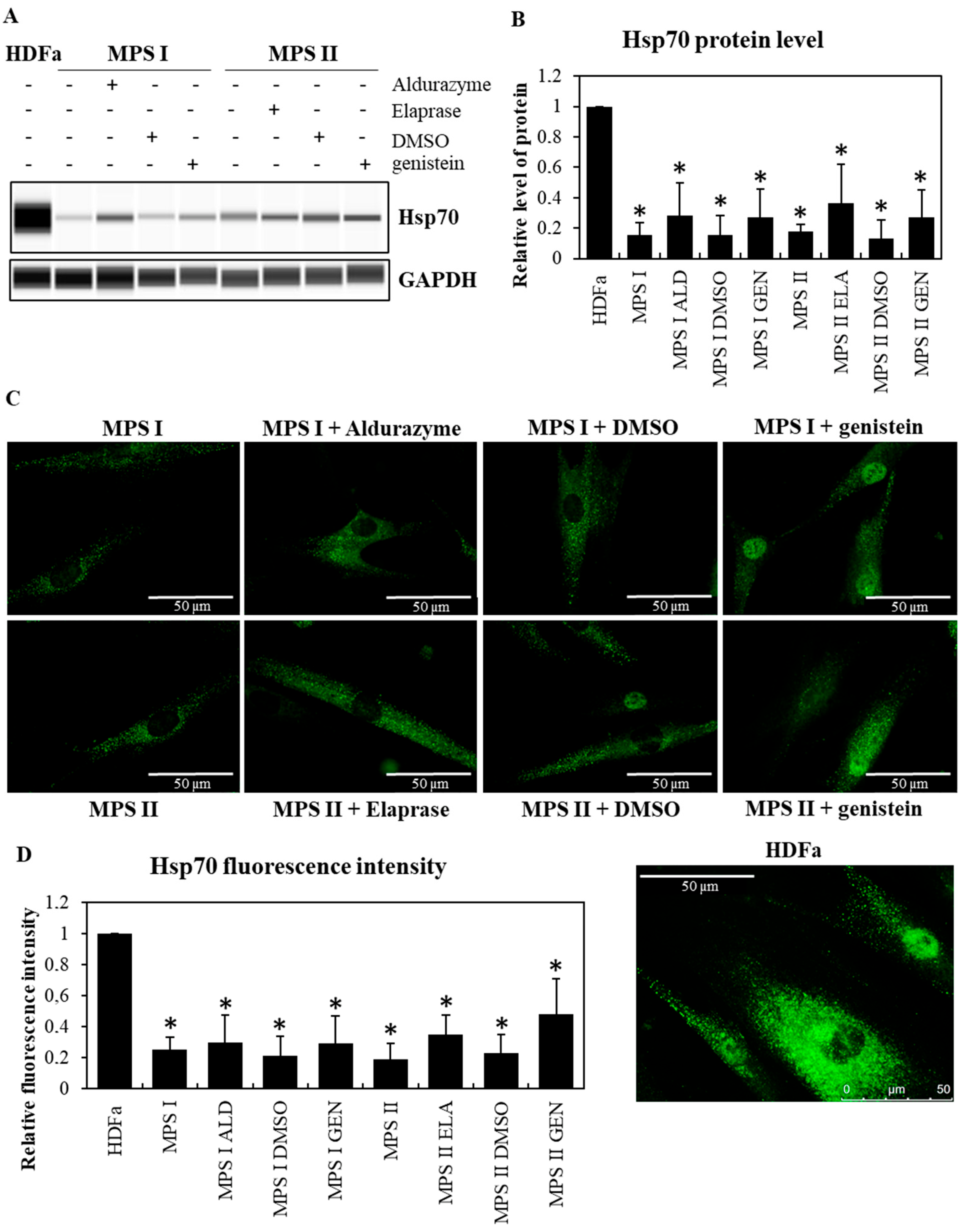{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
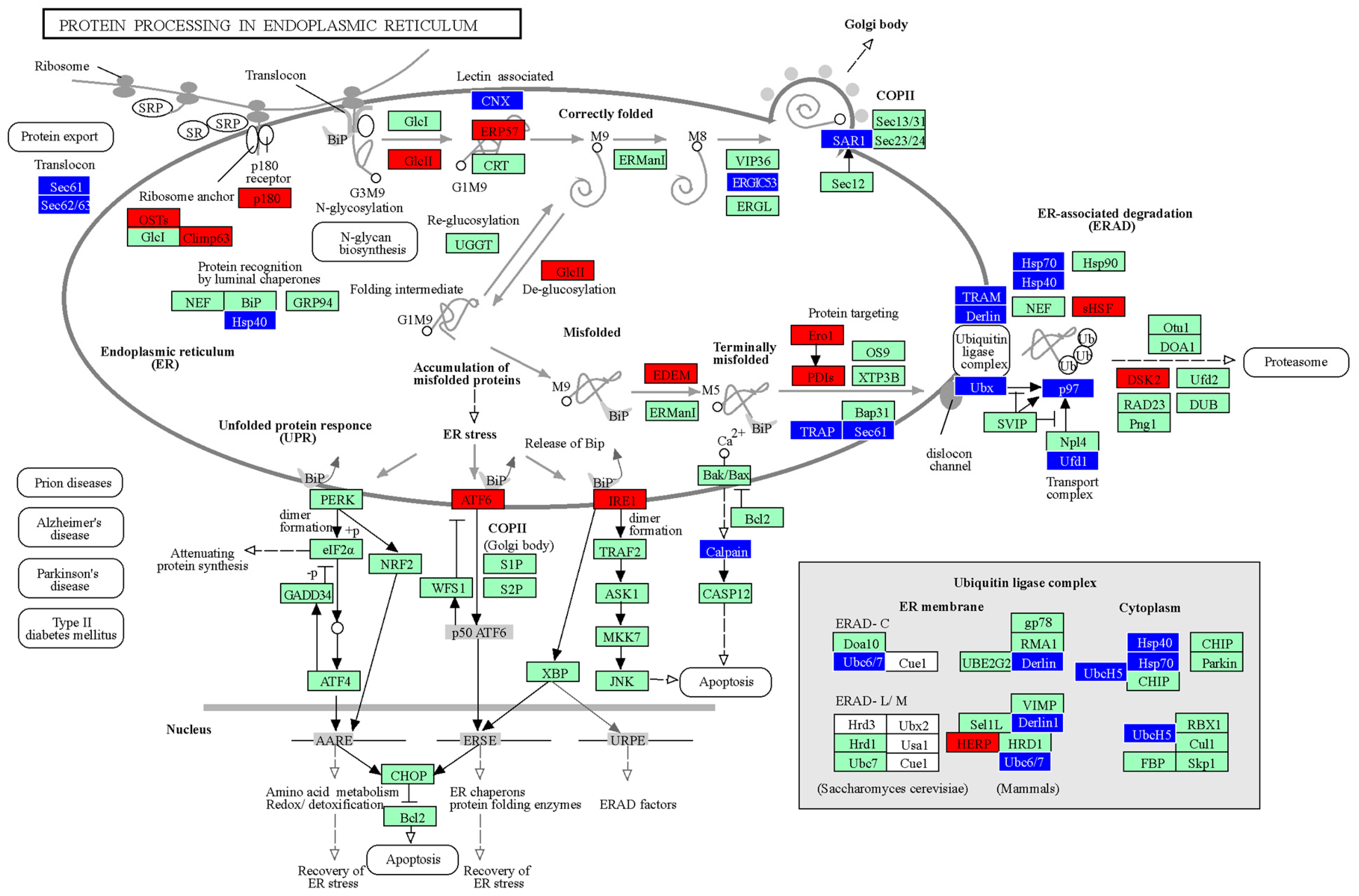{kind=link}
| MPS Type/Subtype | Common Name | Stored GAG | Gene | Defective Enzyme |
|---|---|---|---|---|
| MPS I | Hurler, Scheie, Hurler–Scheie syndrome | HS, DS | IDUA | α-L-iduronidase |
| MPS II | Hunter syndrome | HS, DS | IDS | iduronate sulfatase |
| MPS IIIA | Sanfilippo type A syndrome | HS | SGSH | heparan sulfamidase |
| MPS IIIB | Sanfilippo type B syndrome | HS | NAGLU | N-acetylglucosaminidase |
| MPS IIIC | Sanfilippo type C syndrome | HS | HGSNAT | heparan-α-glucosaminide N-acetyltransferase |
| MPS IIID | Sanfilippo type D syndrome | HS | GNS | N-acetylglucosamine 6-sulfatase |
| MPS IVA | Morquio syndrome | KS, CS | GALNS | galactose-6-sulfate sulfatase |
| MPS IVB | Morquio syndrome | KS | GLB1 | β-galactosidase |
| MPS VI | Maroteaux–Lamy syndrome | DS. | ARSB | N-acetylgalactosamine-4-sulfatase |
| MPS VII | Sly syndrome | HS, DS, CS | GUSB | β-glucuronidase |
| MPS IX | Natowicz syndrome | H | HYAL1 | hyaluronidase |
| MPS X | - | DS | ARSK | arylsulfatase K |
| MPS-PS | MPS Plus syndrome | HS, DS | VPS33A | VPS33A |
2. Materials and Methods
2.1. Fibroblast Lines
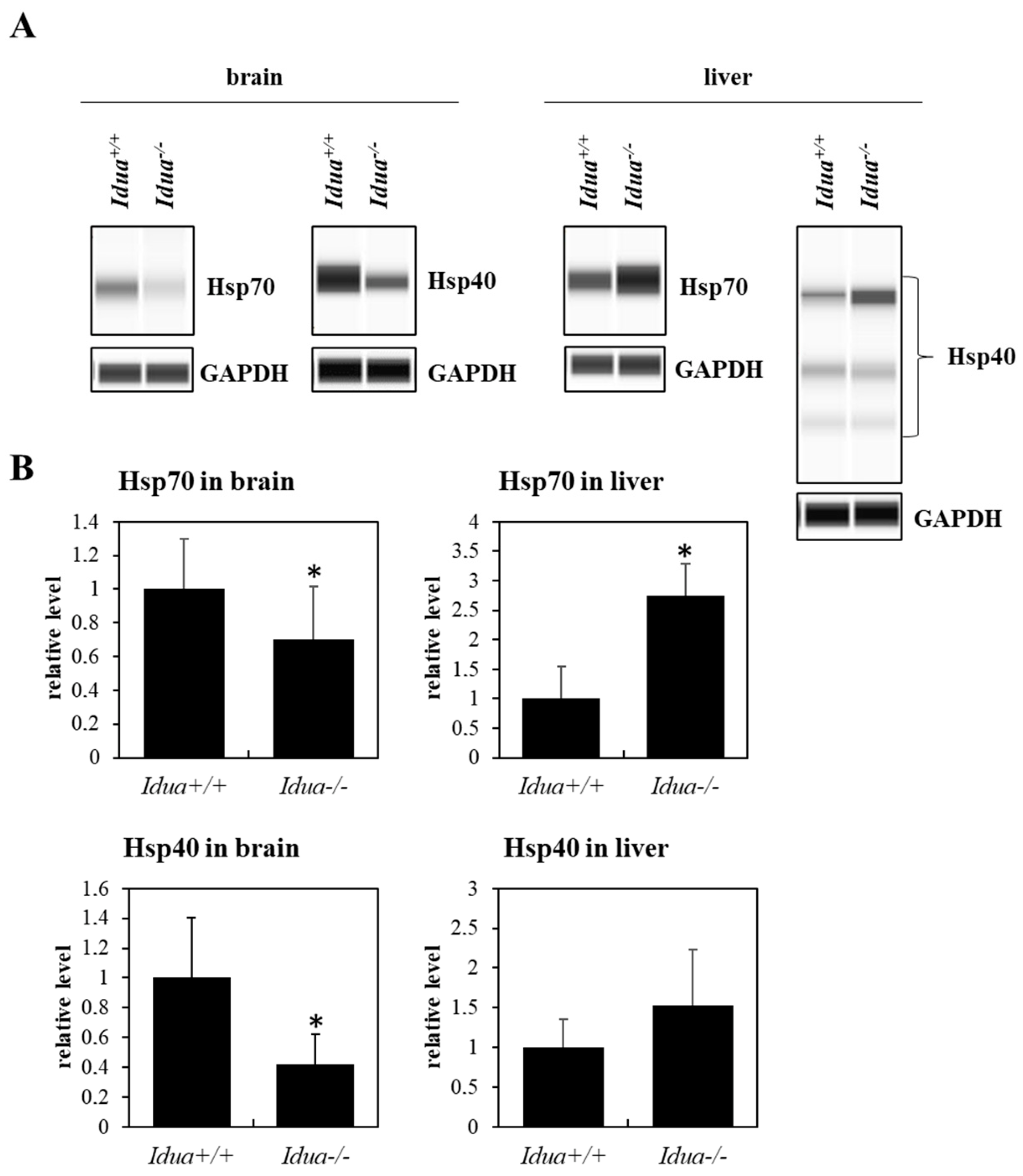
2.2. Mouse Model of MPS Type I
2.3. Enzymes and Genistein
2.4. Western Blotting Analysis
2.5. Fluorescent Microscopy
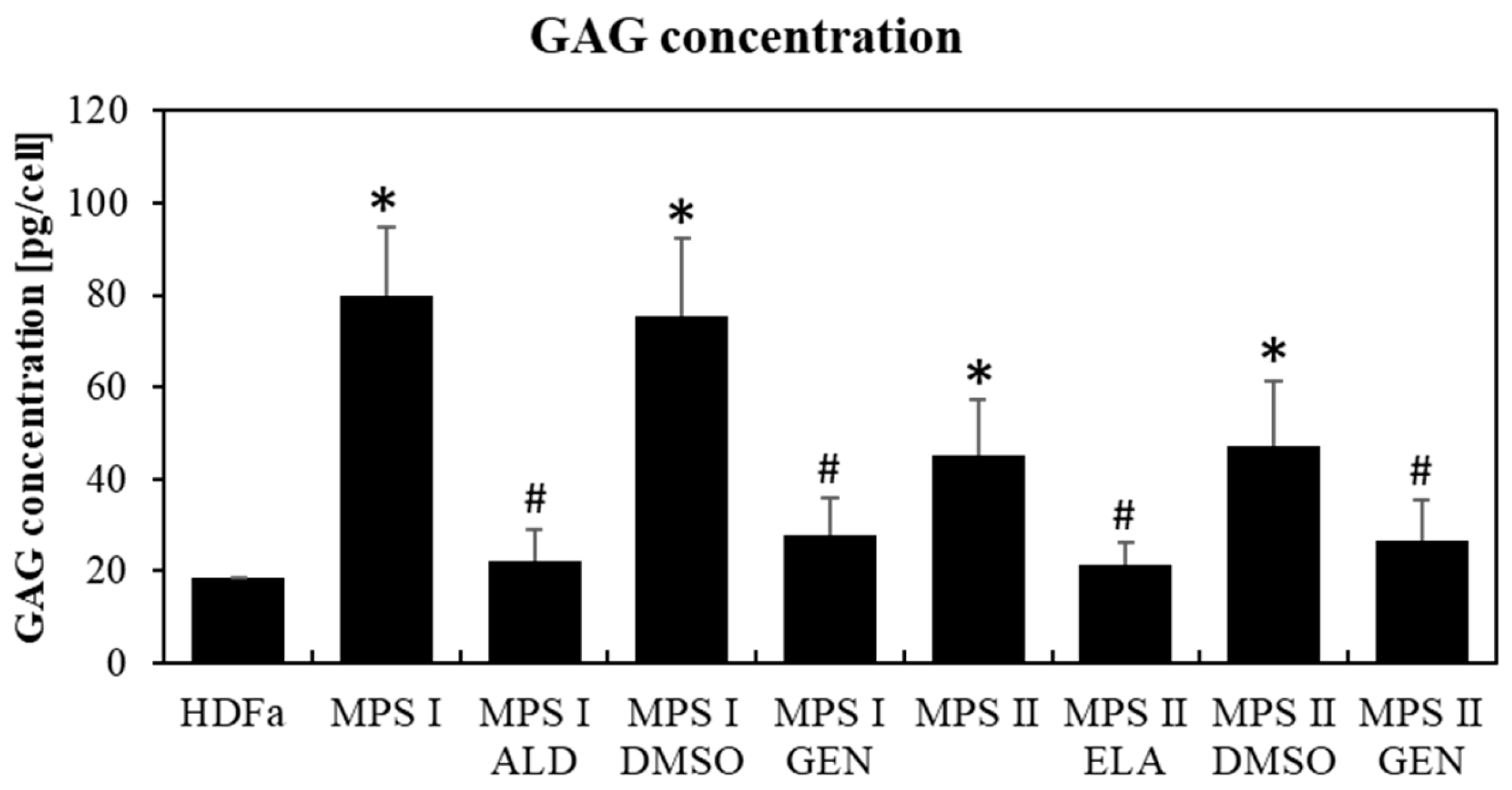
2.6. GAG Level Measurement
2.7. Transcriptomic Analyses
2.8. Statistical Analyses
3. Results
3.1. The Level of Major Chaperone Proteins from the Hsp Family in MPS Cells
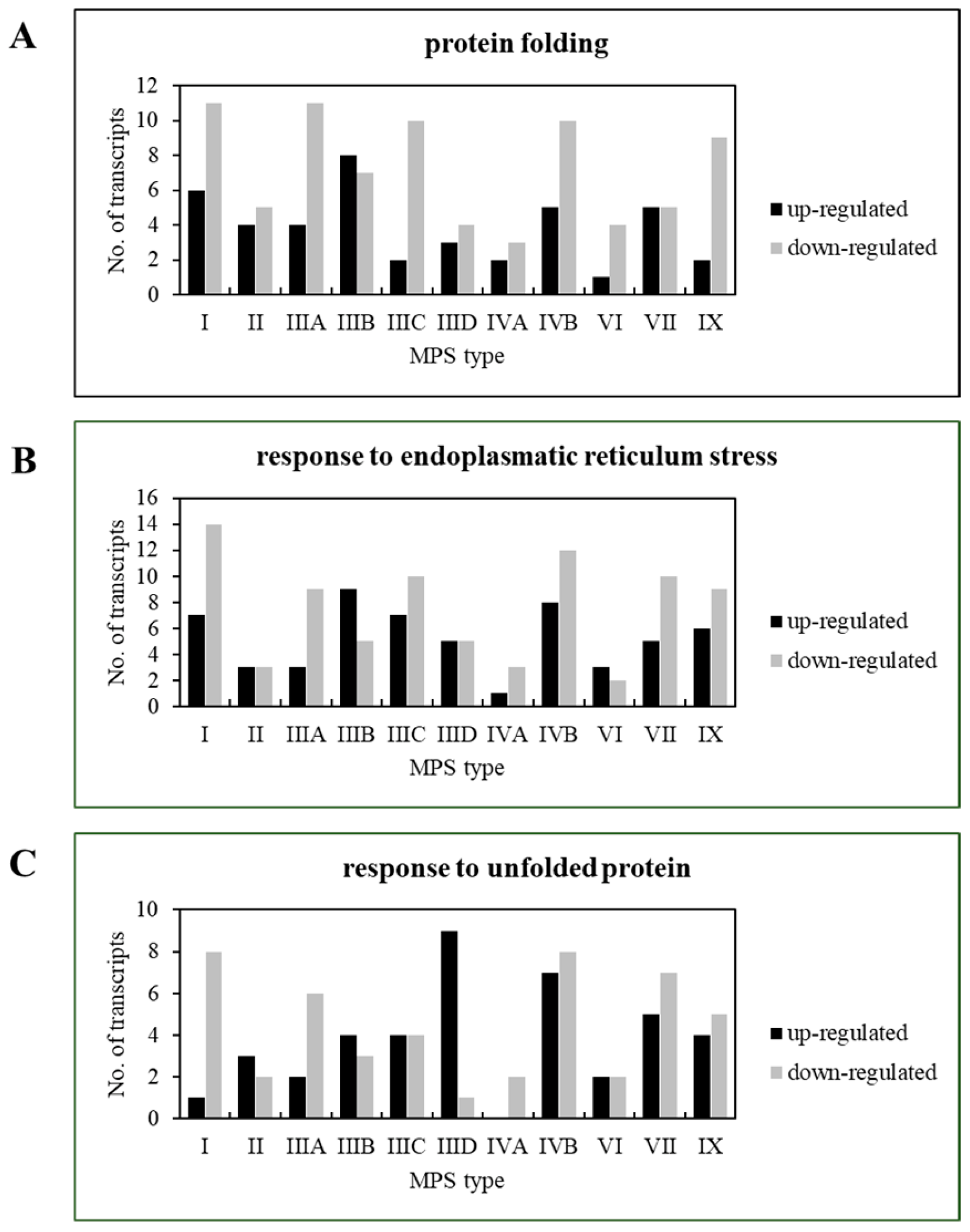
3.2. Transcriptomic Analysis of Changes in the Processes Involving Chaperones in MPS Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Brokowska, J.; Węgrzyn, G. Changes in cellular processes occurring in mucopolysaccharidoses as underestimated pathomechanisms of these diseases. Cell Biol. Int. 2021, 45, 498–506. [Google Scholar] [CrossRef]
- Zhou, J.; Lin, J.; Leung, W.T.; Wang, L. A basic understanding of mucopolysaccharidosis: Incidence, clinical features, diagnosis, and management. Intractable Rare Dis. Res. 2020, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alden, T.D.; Amartino, H.; Dalla Corte, A.; Lampe, C.; Harmatz, P.R.; Vedolin, L. Surgical management of neurological manifestations of mucopolysaccharidosis disorders. Mol. Genet Metab. 2017, 122S, 41–48. [Google Scholar] [CrossRef]
- Grant, N. Evaluating strategies to manage and endure challenging behaviors in mucopolysaccharidoses. Orphanet J. Rare Dis. 2021, 16, 165. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Hoffmann, S.; Kunzmann, K.; Ries, M. Challenging behavior in mucopolysaccharidoses types I–III and day-to-day coping strategies: A cross sectional explorative study. Orphanet J. Rare Dis. 2020, 15, 275. [Google Scholar] [CrossRef]
- Verheyen, S.; Blatterer, J.; Speicher, M.R.; Bhavani, G.S.; Boons, G.J.; Ilse, M.B.; Andrae, D.; Sproß, J.; Vaz, F.M.; Kircher, S.G.; et al. Novel subtype of mucopolysaccharidosis caused by arylsulfatase K (ARSK) deficiency. J. Med. Genet. 2022, 59, 957–964. [Google Scholar] [CrossRef]
- Vasilev, F.; Sukhomyasova, A.; Otomo, T. Mucopolysaccharidosis-Plus Syndrome. Int. J. Mol. Sci. 2020, 21, 421. [Google Scholar] [CrossRef] [Green Version]
- Faraguna, M.C.; Musto, F.; Crescitelli, V.; Iascone, M.; Spaccini, L.; Tonduti, D.; Fedeli, T.; Kullmann, G.; Canonico, F.; Cattoni, A.; et al. Mucopolysaccharidosis-Plus Syndrome, a Rapidly Progressive Disease: Favorable Impact of a Very Prolonged Steroid Treatment on the Clinical Course in a Child. Genes 2022, 13, 442. [Google Scholar] [CrossRef]
- Hampe, C.S.; Wesley, J.; Lund, T.C.; Orchard, P.J.; Polgreen, L.E.; Eisengart, J.B.; McLoon, L.K.; Cureoglu, S.; Schachern, P.; McIvor, R.S. Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement. Biomolecules 2021, 11, 189. [Google Scholar] [CrossRef]
- Cyske, Z.; Anikiej-Wiczenbach, P.; Wisniewska, K.; Gaffke, L.; Pierzynowska, K.; Mański, A.; Wegrzyn, G. Sanfilippo Syndrome: Optimizing Care with a Multidisciplinary Approach. J. Multidiscip. Healthc. 2022, 15, 2097–2110. [Google Scholar] [CrossRef]
- Penon-Portmann, M.; Blair, D.R.; Harmatz, P. Current and new therapies for mucopolysaccharidoses. Pediatr. Neonatol. 2022, 64, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Cyr, D.M.; Ramos, C.H. Specification of Hsp70 Function by Hsp40 Co-chaperones. Subcell. Biochem. 2023, 101, 127–139. [Google Scholar] [PubMed]
- Sambri, I.; D’Alessio, R.; Ezhova, Y.; Giuliano, T.; Sorrentino, N.C.; Cacace, V.; De Risi, M.; Cataldi, M.; Annunziato, L.; De Leonibus, E.; et al. Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol. Med. 2017, 9, 112–132. [Google Scholar] [CrossRef] [PubMed]
- Pierzynowska, K.; Podlacha, M.; Gaffke, L.; Majkutewicz, I.; Mantej, J.; Węgrzyn, A.; Osiadły, M.; Myślińska, D.; Węgrzyn, G. Autophagy-dependent mechanism of genistein-mediated elimination of behavioral and biochemical defects in the rat model of sporadic Alzheimer’s disease. Neuropharmacology 2019, 148, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Hoinkis, D.; Rintz, E.; Brokowska, J.; Cyske, Z.; Wegrzyn, G. Underestimated Aspect of Mucopolysaccharidosis Pathogenesis: Global Changes in Cellular Processes Revealed by Transcriptomic Studies. Int. J. Mol. Sci. 2020, 21, 1204. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, A.J.; Chapman, E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J. Med. Chem. 2021, 64, 7060–7082. [Google Scholar] [CrossRef]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef]
- Kim, J.Y.; Han, Y.; Lee, J.E.; Yenari, M.A. The 70-kDa heat shock protein (Hsp70) as a therapeutic target for stroke. Expert Opin. Ther. Targets 2018, 22, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Batulan, Z.; Taylor, D.M.; Aarons, R.J.; Minotti, S.; Doroudchi, M.M.; Nalbantoglu, J.; Durham, H.D. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 24, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kim, Y.J.; Kim, J.Y.; Lee, W.T.; Yenari, M.A.; Giffard, R.G. The 70 kDa heat shock protein suppresses matrix metalloproteinases in astrocytes. Neuroreport 2004, 15, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, F.; Nezafat, N.; Negahdaripour, M.; Dabbagh, F.; Haghghi, A.B.; Kianpour, S.; Banihashemi, M.; Ghasemi, Y. Neuroprotective Effects of Heat Shock Protein70. CNS Neurol. Disord. Drug Targets 2018, 17, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, M.; Yoon, B.W.; Kim, Y.J.; Ma, S.J.; Roh, J.K.; Lee, J.S.; Seo, J.S. Targeted hsp70.1 disruption increases infarction volume after focal cerebral ischemia in mice. Stroke 2001, 32, 2905–2912. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Kim, J.Y.; Yenari, M.A. Anti-inflammatory properties and pharmacological induction of Hsp70 after brain injury. Inflammopharmacology 2012, 20, 177–185. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [Green Version]
- Losada Díaz, J.C.; Cepeda Del Castillo, J.; Rodriguez-López, E.A.; Alméciga-Díaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2019, 21, 232. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y. Chaperone therapy for molecular pathology in lysosomal diseases. Brain Dev. 2021, 43, 45–54. [Google Scholar] [CrossRef]
- Hoshina, H.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Ida, H.; Ohashi, T. Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2018, 123, 118–122. [Google Scholar] [CrossRef]
- Feldhammer, M.; Durand, S.; Pshezhetsky, A.V. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS ONE 2009, 4, e7434. [Google Scholar] [CrossRef]
- Alméciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodríguez-López, A.; Guzman, E.; Garzón, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. J. Med. Chem. 2019, 62, 6175–6189. [Google Scholar] [CrossRef] [PubMed]
- Osaki, Y.; Saito, A.; Kanemoto, S.; Kaneko, M.; Matsuhisa, K.; Asada, R.; Masaki, T.; Orii, K.; Fukao, T.; Tomatsu, S.; et al. Shutdown of ER-associated degradation pathway rescues functions of mutant iduronate 2-sulfatase linked to mucopolysaccharidosis type II. Cell Death Dis. 2018, 9, 808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuhisa, K.; Imaizumi, K. Loss of Function of Mutant IDS Due to Endoplasmic Reticulum-Associated Degradation: New Therapeutic Opportunities for Mucopolysaccharidosis Type II. Int. J. Mol. Sci. 2021, 22, 12227. [Google Scholar] [CrossRef]
- Meijer, O.L.M.; Welling, L.; Valstar, M.J.; Hoefsloot, L.H.; Brüggenwirth, H.T.; van der Ploeg, A.T.; Ruijter, G.J.G.; Wagemans, T.; Wijburg, F.A.; van Vlies, N. Residual N-acetyl-α-glucosaminidase activity in fibroblasts correlates with disease severity in patients with mucopolysaccharidosis type IIIB. J. Inherit. Metab. Dis. 2016, 39, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, D.; Kovács, M.; Ahmed, S.; Barna, J. Functional diversification of heat shock factors. Biol. Futur. 2022, 73, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Brooks, D.A.; Hopwood, J.J.; Li, P.; Thompson, J.N.; Bedwell, D.M. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum. Mol. Genet. 2001, 10, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Mu, T.W.; Fowler, D.M.; Kelly, J.W. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biol. 2008, 6, e26. [Google Scholar] [CrossRef]
- Rintz, E.; Gaffke, L.; Podlacha, M.; Brokowska, J.; Cyske, Z.; Węgrzyn, G.; Pierzynowska, K. Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 3194. [Google Scholar] [CrossRef]
- Khalid, O.; Vera, M.U.; Gordts, P.L.; Ellinwood, N.M.; Schwartz, P.H.; Dickson, P.I.; Esko, J.D.; Wang, R.Y. Immune-Mediated Inflammation May Contribute to the Pathogenesis of Cardiovascular Disease in Mucopolysaccharidosis Type I. PLoS ONE 2016, 11, e0150850. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, J.V.; Bravo, S.B.; Chantada-Vázquez, M.P.; Barbosa-Gouveia, S.; Colón, C.; López-Suarez, O.; Tomatsu, S.; Otero-Espinar, F.J.; Couce, M.L. Plasma Proteomic Analysis in Morquio A Disease. Int. J. Mol. Sci. 2021, 22, 6165. [Google Scholar] [CrossRef]
- Villani, G.R.; Chierchia, A.; Di Napoli, D.; Di Natale, P. Unfolded protein response is not activated in the mucopolysaccharidoses but protein disulfide isomerase 5 is deregulated. J. Inherit. Metab. Dis. 2012, 35, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.; Fraldi, A. Protein aggregation and autophagy dysfunction: New lessons from mucopolysaccharidoses. Autophagy 2021, 17, 3875–3876. [Google Scholar] [CrossRef]
- Nguyen, V.C.; Deck, C.A.; Pamenter, M.E. Naked mole-rats reduce the expression of ATP-dependent but not ATP-independent heat shock proteins in acute hypoxia. J. Exp. Biol. 2019, 222, jeb211243. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Saito, K.; Ohmi, Y.; Fujie, N.; Ohtsuka, K. Paeoniflorin, a novel heat shock protein-inducing compound. Cell Stress Chaperones 2004, 9, 378–389. [Google Scholar] [CrossRef] [Green Version]







| MPS Type/Subtype | Sex | Age * | Mutated Gene and Mutations | Cat. No. # |
|---|---|---|---|---|
| MPS I | Female | 1 | IDUA, p.Trp402X/p.Trp402X | GM00798 |
| MPS II | Male | 3 | IDS, p.His70ProfsX29/- | GM13203 |
| MPS IIIA | Female | 3 | SGSH, p.Glu447Lys/p.Arg245His | GM00879 |
| MPS IIIB | Male | 7 | NAGLU, p.Arg626Ter/p.Arg626Ter | GM00156 |
| MPS IIIC | Male | 8 | HGSNAT, p.Gly262Arg/pArg509Asp | GM05157 |
| MPS IIID | Male | 7 | GNS, p.Arg355Ter/p.Arg355Ter | GM05093 |
| MPS IVA | Female | 7 | GALNS, p.Arg386Cys/p.Phe285Ter | GM00593 |
| MPS IVB | Female | 4 | GLB1, p.Trp273Leu/p.Trp509Cys | GM03251 |
| MPS VI | Female | 3 | ARSB, ND | GM03722 |
| MPS VII | Male | 3 | GUSB, p.Trp627Cys/p.Arg356X | GM00121 |
| MPS IX | Female | 14 | HYAL1, ND | GM17494 |
| Transcript | log2FC of Selected Transcripts’ Levels in Particular MPS Type vs. HDFa Line | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | IIIA | IIIB | IIIC | IIID | IVA | IVB | VI | VII | IX | |
| Protein folding (GO:0006457) | |||||||||||
| HSPB6 | 1.90 | 2.03 | 0.14 | 2.29 | 2.27 | 0.38 | 1.42 | 2.37 | 1.47 | 1.55 | 1.57 |
| PDIA3 | 0.46 | 0.39 | 0.31 | 0.50 | 0.30 | 0.38 | 0.37 | 0.36 | 0.20 | 0.32 | −0.13 |
| CLU | 2.69 | 2.98 | 1.59 | 1.75 | 2.95 | 1.14 | 1.73 | 3.46 | 2.58 | 2.08 | 3.44 |
| PRKCSH | 0.42 | 0.45 | 0.52 | 0.50 | 0.38 | 0.33 | 0.50 | 0.51 | 0.22 | 0.39 | 0.09 |
| DNAJC3 | −0.58 | −0.58 | −0.56 | −0.60 | −0.86 | −0.37 | −0.58 | −0.71 | −0.55 | −0.15 | −0.63 |
| Response to endoplasmic reticulum stress (GO:0034976) | |||||||||||
| PDIA3 | 0.46 | 0.39 | 0.31 | 0.50 | 0.30 | 0.38 | 0.37 | 0.36 | 0.20 | 0.32 | −0.13 |
| TRIM25 | 0.60 | 0.39 | 0.29 | 0.88 | 0.51 | 0.69 | 0.27 | 1.00 | 0.29 | 0.79 | 0.94 |
| CLU | 2.69 | 2.97 | 1.59 | 1.75 | 2.95 | 1.14 | 1.73 | 3.46 | 2.58 | 2.08 | 3.44 |
| TMX1 | −0.75 | −0.47 | −0.86 | −0.94 | −0.62 | −0.84 | −0.54 | −0.51 | −0.51 | −1.06 | −1.07 |
| DNAJC3 | −0.58 | −0.58 | −0.56 | −0.60 | −0.86 | −0.37 | −0.58 | −0.71 | −0.55 | −0.15 | −0.63 |
| UFD1 | −0.50 | −0.38 | −0.61 | −0.44 | −0.59 | −0.62 | −0.66 | −0.51 | −0.40 | −0.55 | −0.78 |
| Response to unfolded protein (GO:0006986) | |||||||||||
| DNAJC3 | −0.58 | −0.58 | −0.56 | −0.60 | −0.86 | −0.37 | −0.58 | −0.71 | −0.55 | −0.15 | −0.63 |
| Transcript | log2FC > 2 or <−2 of Selected Transcripts’ Levels in Particular MPS Type vs. HDFa Line | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | IIIA | IIIB | IIIC | IIID | IVA | IVB | VI | VII | IX | |
| Protein folding (GO:0006457) | |||||||||||
| CLU | 2.69 | 2.97 | 1.59 | 1.75 | 2.95 | 1.14 | 1.73 | 3.46 | 2.58 | 2.08 | 3.44 |
| HSPB6 | 1.90 | 2.03 | 0.14 | 2.29 | 2.27 | 0.38 | 1.42 | 2.37 | 1.47 | 1.55 | 1.57 |
| FKBP1C | −0.14 | −0.03 | −2.13 | −0.27 | −0.93 | 0.13 | 0.01 | −0.01 | −0.05 | −0.55 | −0.19 |
| Response to endoplasmic reticulum stress (GO:0034976) | |||||||||||
| LMNA | 3.03 | 2.39 | 2.56 | 2.28 | 2.58 | 2.38 | 2.55 | 2.27 | 2.40 | 1.58 | 2.29 |
| CLU | 2.69 | 2.97 | 1.59 | 1.75 | 2.95 | 1.14 | 1.73 | 3.46 | 2.58 | 2.08 | 3.44 |
| KDELR3 | 0.29 | 0.28 | 0.55 | 1.53 | 1.62 | 2.03 | 0.82 | 1.75 | 1.28 | 0.99 | 0.81 |
| BBC3 | 1.55 | 1.32 | 1.39 | 1.32 | 1.34 | 2.19 | 1.36 | 1.16 | 1.91 | 1.53 | 2.46 |
| CXCL8 | −4.35 | −2.46 | −5.08 | −0.33 | −1.28 | −0.12 | −2.24 | −1.09 | −2.28 | −4.04 | −0.76 |
| CAV1 | −4.74 | −2.67 | −3.79 | −4.75 | −3.60 | −1.53 | −2.95 | −4.74 | −2.57 | −1.26 | −3.28 |
| Response to unfolded proteins (GO:0006986) | |||||||||||
| LMNA | 3.03 | 2.39 | 2.56 | 2.28 | 2.58 | 2.38 | 2.55 | 2.27 | 2.40 | 1.58 | 2.29 |
| HSPB8 | 0.02 | 0.28 | −1.47 | 2.03 | 0.17 | 0.78 | 0.29 | 0.42 | 0.33 | 0.77 | 0.84 |
| COMP | 2.33 | 3.24 | 0.50 | 1.27 | 3.17 | 4.75 | 2.58 | 1.67 | 3.60 | 0.00 | 2.18 |
| HSPB7 | −0.31 | 1.87 | 0.16 | 1.03 | 0.44 | 2.70 | 1.46 | 0.22 | 1.42 | 1.85 | 1.47 |
| KDELR3 | 0.29 | 0.28 | 0.55 | 1.53 | 1.62 | 2.03 | 0.82 | 1.75 | 1.28 | 0.99 | 0.81 |
| HSPB7 | −0.44 | 1.79 | −0.14 | 0.96 | 0.37 | 2.38 | 1.01 | 0.07 | 1.43 | 1.72 | 1.81 |
| BBC3 | 1.55 | 1.32 | 1.39 | 1.32 | 1.34 | 2.19 | 1.36 | 1.16 | 1.91 | 1.53 | 2.45 |
| CXCL8 | −4.35 | −2.46 | −5.08 | −0.33 | −1.28 | −0.12 | −2.24 | −1.09 | −2.28 | −4.04 | −0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żabińska, M.; Gaffke, L.; Bielańska, P.; Podlacha, M.; Rintz, E.; Cyske, Z.; Węgrzyn, G.; Pierzynowska, K. Decreased Levels of Chaperones in Mucopolysaccharidoses and Their Elevation as a Putative Auxiliary Therapeutic Approach. Pharmaceutics 2023, 15, 704. https://doi.org/10.3390/pharmaceutics15020704
Żabińska M, Gaffke L, Bielańska P, Podlacha M, Rintz E, Cyske Z, Węgrzyn G, Pierzynowska K. Decreased Levels of Chaperones in Mucopolysaccharidoses and Their Elevation as a Putative Auxiliary Therapeutic Approach. Pharmaceutics. 2023; 15(2):704. https://doi.org/10.3390/pharmaceutics15020704
Chicago/Turabian StyleŻabińska, Magdalena, Lidia Gaffke, Patrycja Bielańska, Magdalena Podlacha, Estera Rintz, Zuzanna Cyske, Grzegorz Węgrzyn, and Karolina Pierzynowska. 2023. "Decreased Levels of Chaperones in Mucopolysaccharidoses and Their Elevation as a Putative Auxiliary Therapeutic Approach" Pharmaceutics 15, no. 2: 704. https://doi.org/10.3390/pharmaceutics15020704