
Influence of the Fatty Acid Metabolism on the Mode of Action of a Cisplatin(IV) Complex with Phenylbutyrate as Axial Ligands
 , , , ,
, , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Cytotoxicity Assays
2.4. ICP-MS Measurements of Cell Uptake
2.5. Animals
2.6. Anticancer Activity In Vivo
2.7. Live-Cell Microscopy
2.8. Cell Cycle Analysis
2.9. Cell Death Analysis
2.10. Nucleus Staining
2.11. Albumin Uptake
2.12. Cellular Albumin Uptake
2.13. Cellular Respiration Experiments
2.14. JC-1 Flow Cytometry Analysis
2.15. Statistical Analysis
3. Results
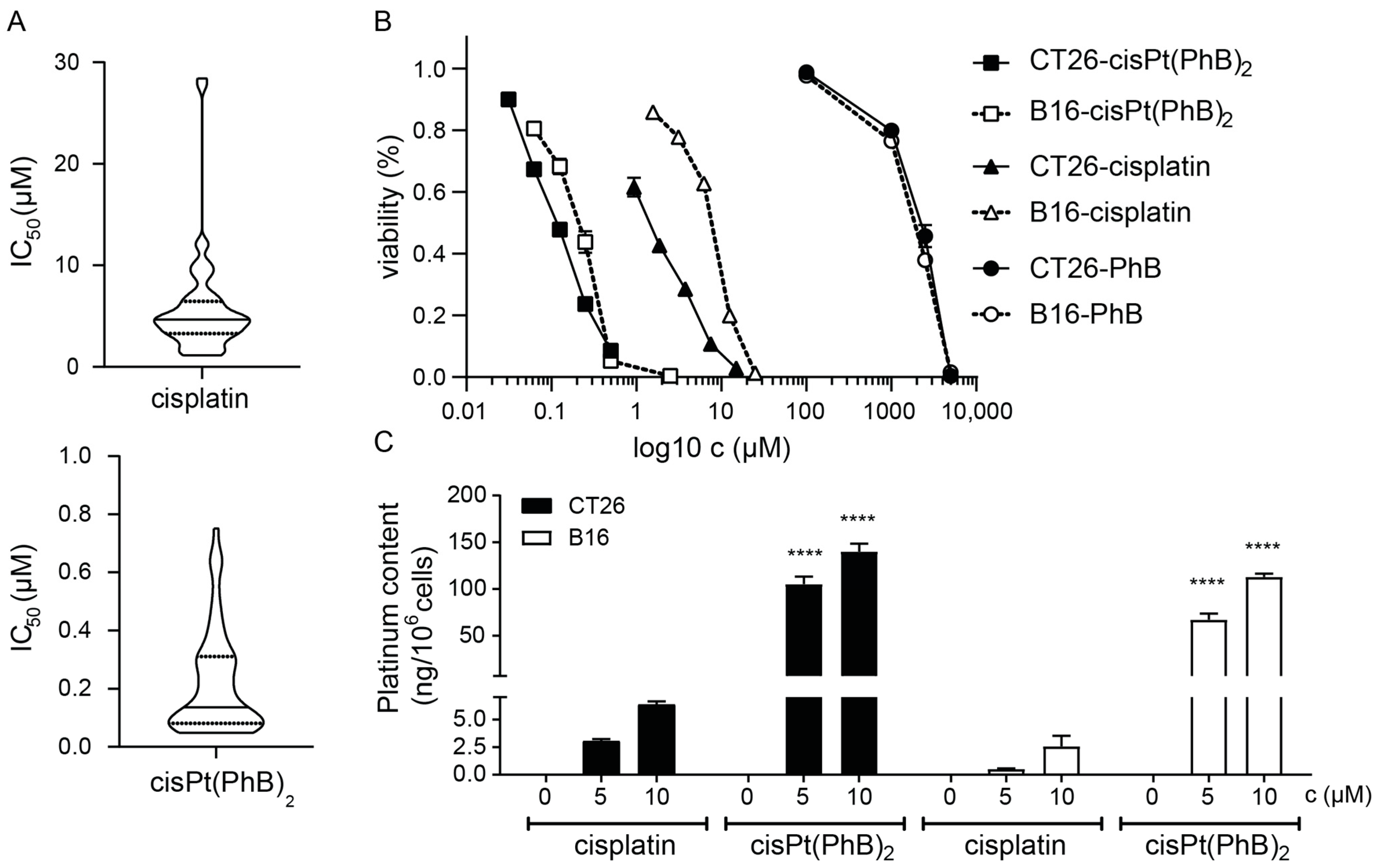
3.1. Anticancer Activity of cisPt(PhB)2 in Platinum-Resistant and -Sensitive Cancer Cells In Vitro
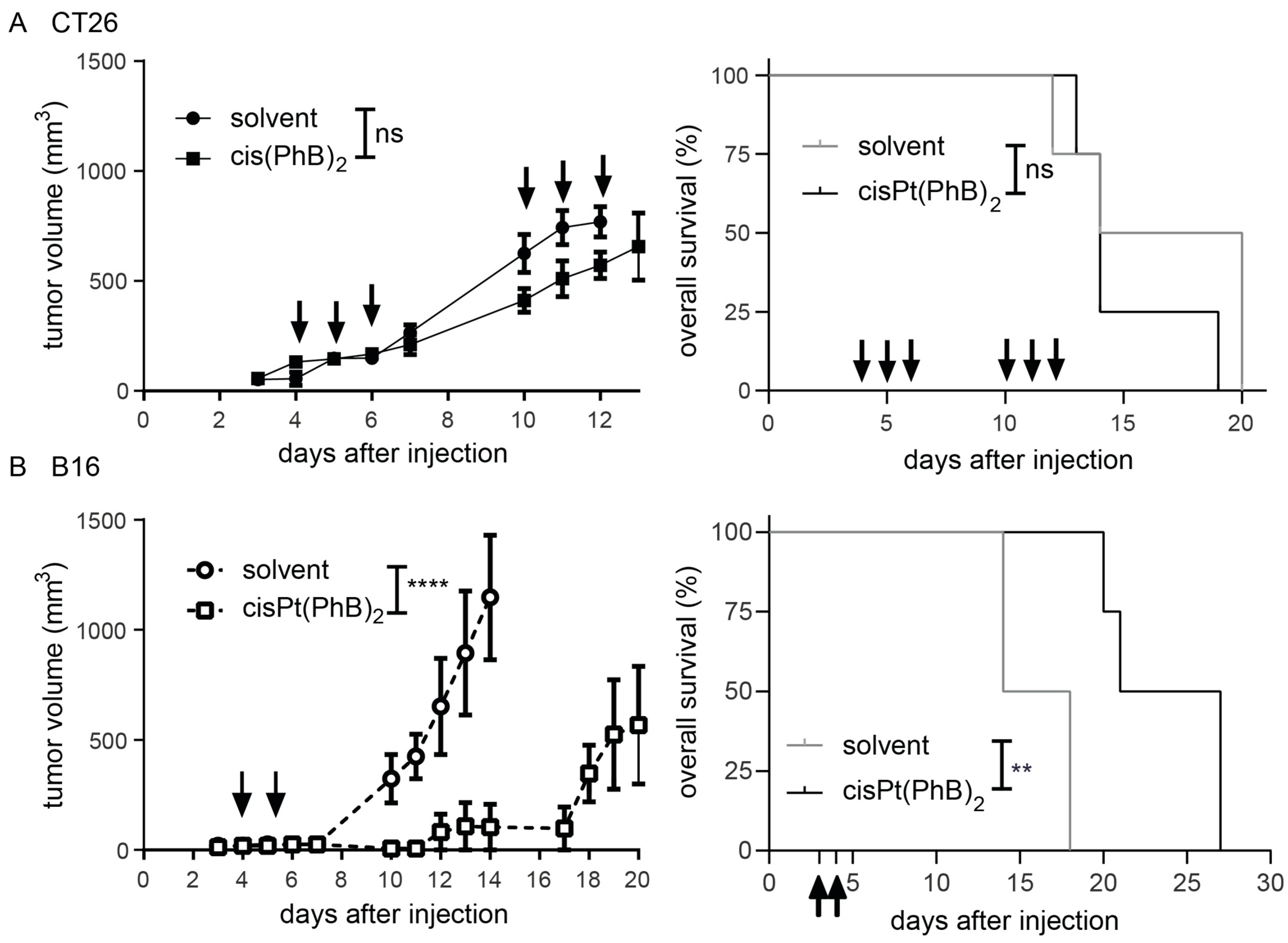
3.2. Anticancer Activity In Vivo
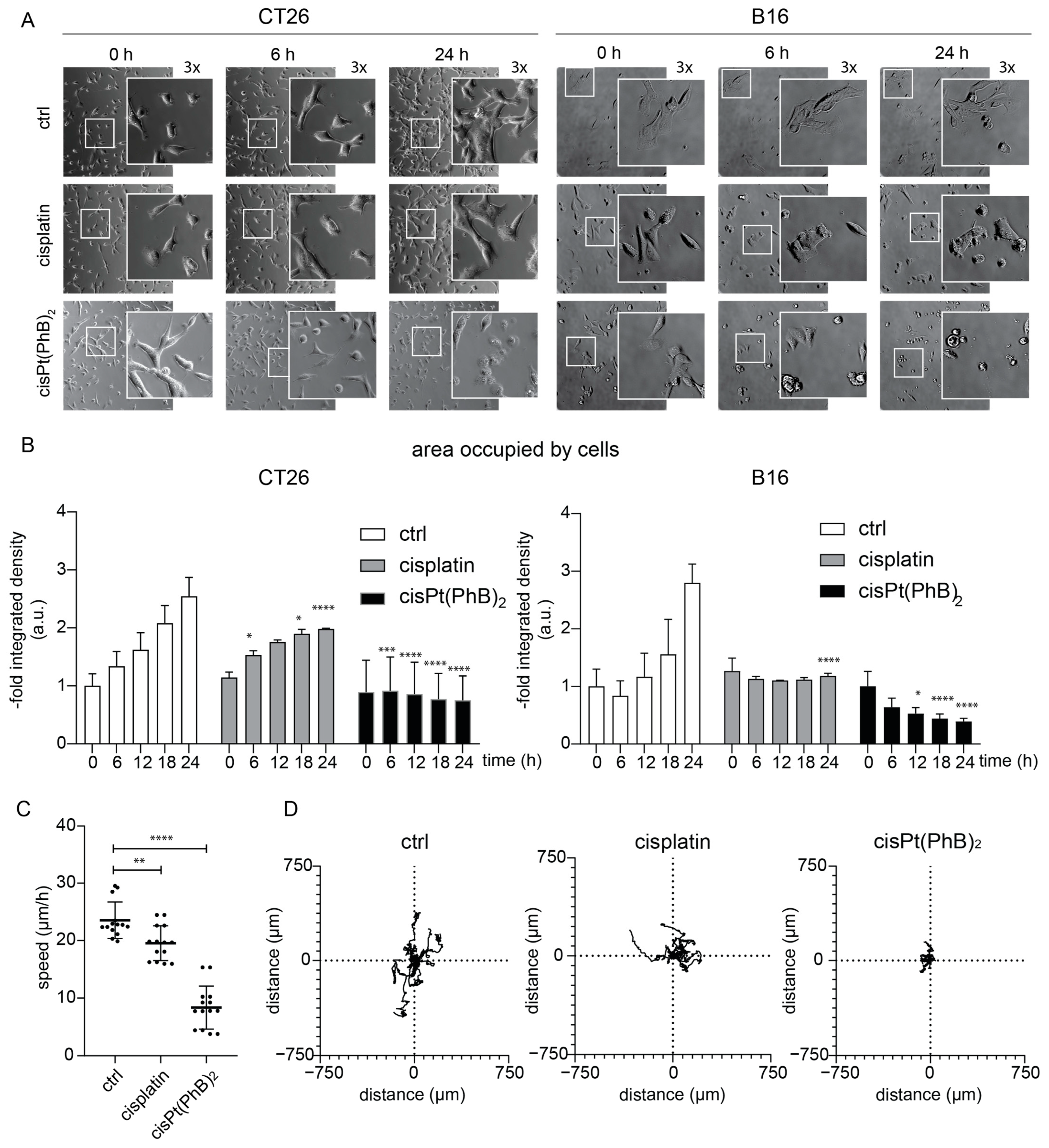
3.3. CisPt(PhB)2 Displays an Earlier Onset of Cytotoxicity than Cisplatin, Which Is More Pronounced in B16
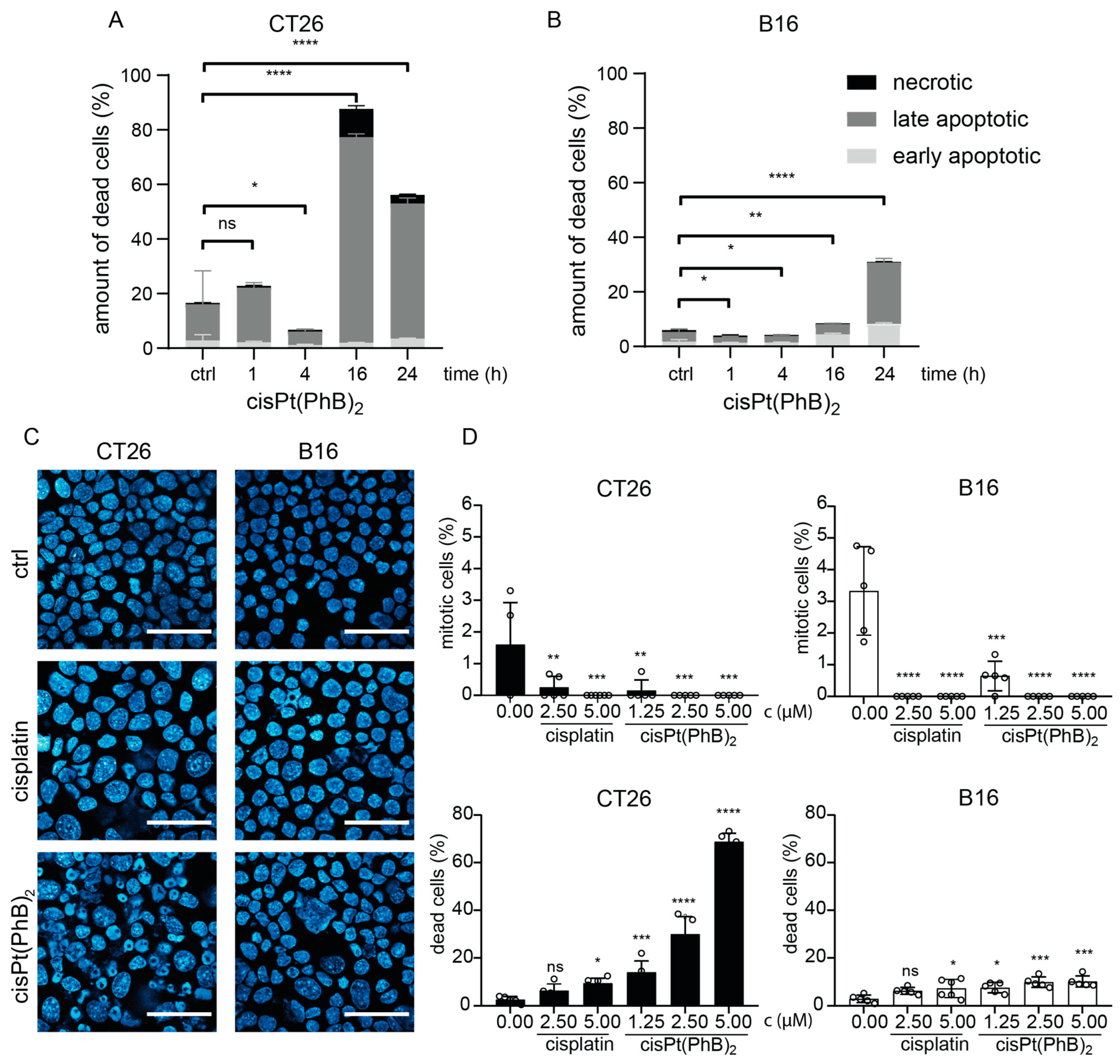
3.4. Differences in Cell Death Induction by cisPt(PhB)2 in CT26 and B16 Cells
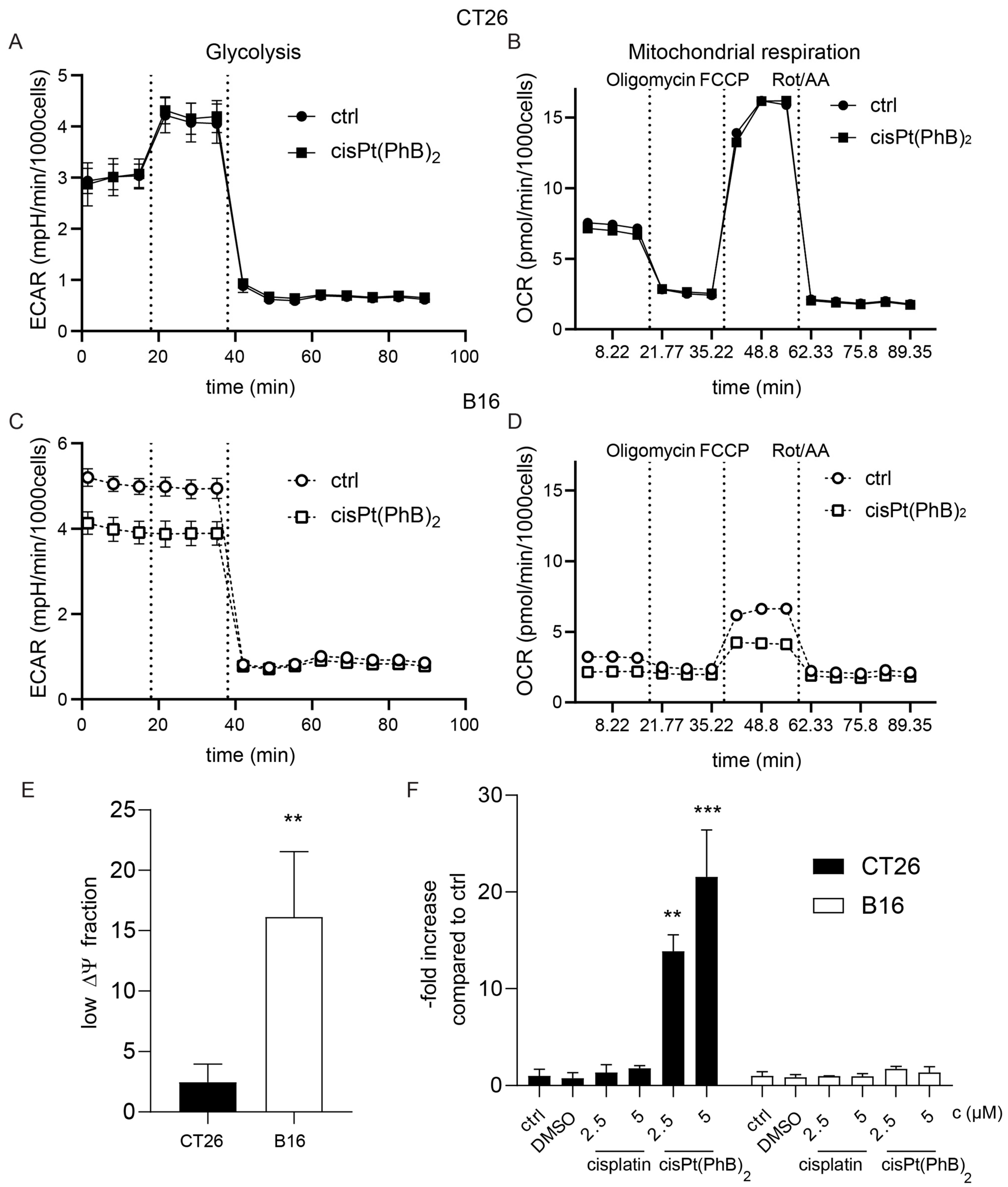
3.5. CT26 and B16 Cells Differ in Their Metabolism and Albumin Homeostasis
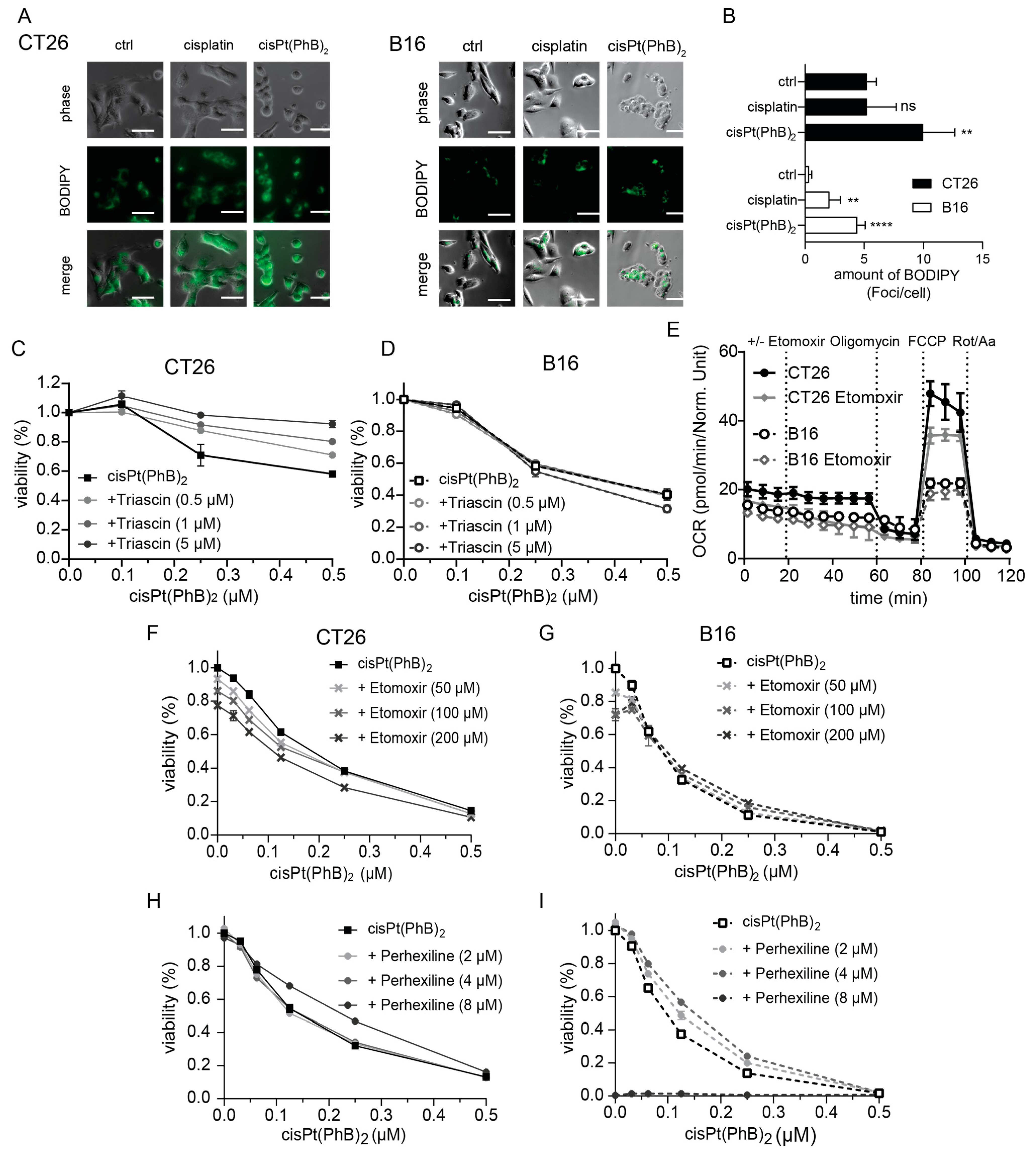
3.6. CisPt(PhB)2 Activity Is Associated with Enhanced Lipid Droplet Formation
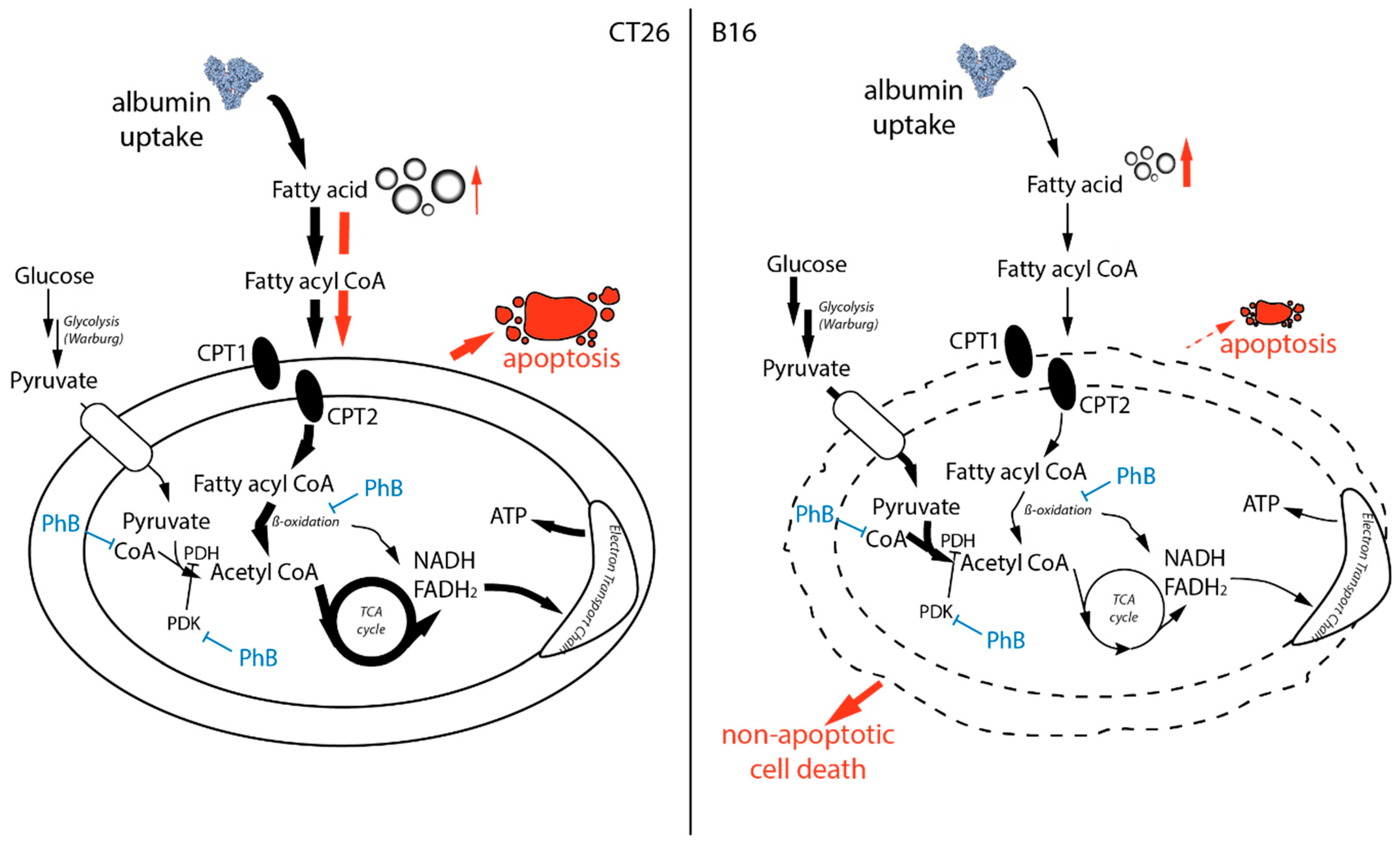
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pötsch, I.; Baier, D.; Keppler, B.K.; Berger, W. CHAPTER 12 Challenges and Chances in the Preclinical to Clinical Translation of Anticancer Metallodrugs. In Metal-Based Anticancer Agents; The Royal Society of Chemistry: London, UK, 2019; pp. 308–347. [Google Scholar]
- Burger, H.; Loos, W.J.; Eechoute, K.; Verweij, J.; Mathijssen, R.H.; Wiemer, E.A. Drug transporters of platinum-based anticancer agents and their clinical significance. Drug Resist. Updates 2011, 14, 22–34. [Google Scholar] [CrossRef]
- Arnesano, F.; Natile, G. Interference between copper transport systems and platinum drugs. Semin. Cancer Biol. 2021, 76, 173–188. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P.; Hager, S.; Pape, V.F.; Pósa, V.; Montsch, B.; et al. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [Green Version]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Gibson, D. Platinum(IV) anticancer agents; are we en route to the holy grail or to a dead end? J. Inorg. Biochem. 2021, 217, 111353. [Google Scholar] [CrossRef]
- Ferraro, M.G.; Piccolo, M.; Misso, G.; Santamaria, R.; Irace, C. Bioactivity and Development of Small Non-Platinum Metal-Based Chemotherapeutics. Pharmaceutics 2022, 14, 954. [Google Scholar] [CrossRef]
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(iv) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Vaishampayan, U.N. Satraplatin: Leading the new generation of oral platinum agents. Expert Opin. Investig. Drugs 2009, 18, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.E.Y.; Lim, Z.C.; Chew, S.L.; Ang, W.H. Strategy for Traceless Codrug Delivery with Platinum(IV) Prodrug Complexes Using Self-Immolative Linkers. Inorg. Chem. 2021, 60, 1823–1831. [Google Scholar] [CrossRef]
- Hambley, T.W. Transporter and protease mediated delivery of platinum complexes for precision oncology. JBIC J. Biol. Inorg. Chem. 2019, 24, 457–466. [Google Scholar] [CrossRef]
- Stiborova, M.; Eckschlager, T.; Poljakova, J.; Hrabeta, J.; Adam, V.; Kizek, R.; Frei, E. The synergistic effects of DNA-targeted chemotherapeutics and histone deacetylase inhibitors as therapeutic strategies for cancer treatment. Curr. Med. Chem. 2012, 19, 4218–4238. [Google Scholar] [CrossRef]
- Al-Keilani, M.S.; Alzoubi, K.H.; Jaradat, S.A. The effect of combined treatment with sodium phenylbutyrate and cisplatin, erlotinib, or gefitinib on resistant NSCLC cells. Clin. Pharmacol. Adv. Appl. 2018, 10, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Al-Keilani, M.S.; Al-Sawalha, N.A. Potential of Phenylbutyrate as Adjuvant Chemotherapy: An Overview of Cellular and Molecular Anticancer Mechanisms. Chem. Res. Toxicol. 2017, 30, 1767–1777. [Google Scholar] [CrossRef]
- Yam, G.H.-F.; Gaplovska-Kysela, K.; Zuber, C.; Roth, J. Sodium 4-Phenylbutyrate Acts as a Chemical Chaperone on Misfolded Myocilin to Rescue Cells from Endoplasmic Reticulum Stress and Apoptosis. Investig. Opthalmology Vis. Sci. 2007, 48, 1683–1690. [Google Scholar] [CrossRef]
- Mai, C.T.; Le, Q.G.; Ishiwata-Kimata, Y.; Takagi, H.; Kohno, K.; Kimata, Y. 4-Phenylbutyrate suppresses the unfolded protein response without restoring protein folding in Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy016. [Google Scholar] [CrossRef] [Green Version]
- Nissar, A.U.; Sharma, L.; Mudasir, M.A.; Nazir, L.A.; Umar, S.A.; Sharma, P.R.; Vishwakarma, R.A.; Tasduq, S.A. Chemical chaperone 4-phenyl butyric acid (4-PBA) reduces hepatocellular lipid accumulation and lipotoxicity through induction of autophagy. J. Lipid Res. 2017, 58, 1855–1868. [Google Scholar] [CrossRef] [Green Version]
- Kormanik, K.; Kang, H.; Cuebas, D.; Vockley, J.; Mohsen, A.-W. Evidence for involvement of medium chain acyl-CoA dehydrogenase in the metabolism of phenylbutyrate. Mol. Genet. Metab. 2012, 107, 684–689. [Google Scholar] [CrossRef] [Green Version]
- King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites 2021, 11, 792. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Phenyl butyrate inhibits pyruvate dehydrogenase kinase 1 and contributes to its anti-cancer effect. Eur. J. Pharm. Sci. 2017, 110, 93–100. [Google Scholar] [CrossRef]
- Burkitt, K.; Ljungman, M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer 2008, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- E Witzig, T.; Timm, M.; Stenson, M.; A Svingen, P.; Kaufmann, S. Induction of apoptosis in malignant B cells by phenylbutyrate or phenylacetate in combination with chemotherapeutic agents. Clin. Cancer Res. 2000, 6, 681–692. [Google Scholar]
- Han, W.; He, W.; Song, Y.; Zhao, J.; Song, Z.; Shan, Y.; Hua, W.; Sun, Y. Multifunctional platinum(iv) complex bearing HDAC inhibitor and biotin moiety exhibits prominent cytotoxicity and tumor-targeting ability. Dalton Trans. 2022, 51, 7343–7351. [Google Scholar] [CrossRef]
- Taghizadeh, H.; Unseld, M.; Spalt, M.; Mader, R.M.; Müllauer, L.; Fuereder, T.; Raderer, M.; Sibilia, M.; Hoda, M.A.; Aust, S.; et al. Targeted Therapy Recommendations for Therapy Refractory Solid Tumors—Data from the Real-World Precision Medicine Platform MONDTI. J. Pers. Med. 2020, 10, 188. [Google Scholar] [CrossRef]
- Gorelik, R.; Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nat. Protoc. 2014, 9, 1931–1943. [Google Scholar] [CrossRef]
- Romeo, M.A.; Montani, M.S.G.; Benedetti, R.; Garufi, A.; D’Orazi, G.; Cirone, M. PBA Preferentially Impairs Cell Survival of Glioblastomas Carrying mutp53 by Reducing Its Expression Level, Stabilizing wtp53, Downregulating the Mevalonate Kinase and Dysregulating UPR. Biomolecules 2020, 10, 586. [Google Scholar] [CrossRef]
- A Carducci, M.; Nelson, J.B.; Chan-Tack, K.M.; Ayyagari, S.R.; Sweatt, W.H.; Campbell, P.; Nelson, W.G.; Simons, J.W. Phenylbutyrate induces apoptosis in human prostate cancer and is more potent than phenylacetate. Clin. Cancer Res. 1996, 2, 379–387. [Google Scholar]
- Melchior, S.W.; Brown, L.G.; Figg, W.D.; E Quinn, J.; A Santucci, R.; Brunner, J.; Thuroff, J.W.; Lange, P.H.; Vessella, R.L. Effects of phenylbutyrate on proliferation and apoptosis in human prostate cancer cells in vitro and in vivo. Int. J. Oncol. 1999, 14, 501–509. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.; Jia, Z.; Wu, H.; Gu, K. Oxaliplatin-Based Regimen is Superior to Cisplatin-Based Regimen in Tumour Remission as First-line Chemotherapy for Advanced Gastric Cancer: A Meta-Analysis. J. Cancer 2019, 10, 1923–1929. [Google Scholar] [CrossRef]
- Pichler, V.; Heffeter, P.; Valiahdi, S.M.; Kowol, C.R.; Egger, A.; Berger, W.; Jakupec, M.A.; Galanski, M.S.; Keppler, B.K. Unsymmetric Mono- and Dinuclear Platinum(IV) Complexes Featuring an Ethylene Glycol Moiety: Synthesis, Characterization, and Biological Activity. J. Med. Chem. 2012, 55, 11052–11061. [Google Scholar] [CrossRef] [Green Version]
- Heffeter, P.; Jungwirth, U.; Jakupec, M.; Hartinger, C.; Galanski, M.S.; Elbling, L.; Micksche, M.; Keppler, B.; Berger, W. Resistance against novel anticancer metal compounds: Differences and similarities. Drug Resist. Updat. 2008, 11, 1–16. [Google Scholar] [CrossRef]
- Valente, A.; Podolski-Renić, A.; Poetsch, I.; Filipović, N.; López, Ó.; Turel, I.; Heffeter, P. Metal-and metalloid-based compounds to target and reverse cancer multidrug resistance. Drug Resist. Updates 2021, 58, 100778. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Clemente-Blanco, A. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. Int. J. Mol. Sci. 2020, 21, 446. [Google Scholar] [CrossRef] [Green Version]
- Palir, N.; Ruiter, J.P.N.; Wanders, R.J.A.; Houtkooper, R.H. Identification of enzymes involved in oxidation of phenylbutyrate. J. Lipid Res. 2017, 58, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Crossland, H.; Smith, K.; Idris, I.; Phillips, B.E.; Atherton, P.J.; Wilkinson, D.J. Phenylbutyrate, a branched-chain amino acid keto dehydrogenase activator, promotes branched-chain amino acid metabolism and induces muscle catabolism in C2C12 cells. Exp. Physiol. 2021, 106, 585–592. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef] [Green Version]
- Schueffl, H.; Theiner, S.; Hermann, G.; Mayr, J.; Fronik, P.; Groza, D.; van Schonhooven, S.; Galvez, L.; Sommerfeld, N.S.; Schintlmeister, A.; et al. Albumin-targeting of an oxaliplatin-releasing platinum(iv) prodrug results in pronounced anticancer activity due to endocytotic drug uptake in vivo. Chem. Sci. 2021, 12, 12587–12599. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Qiu, B.; Simon, M.C. BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio-Protoc. 2016, 6, e1912. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Zhou, Y.-T.; Levi, M.; Unger, R.H. Fatty acid-induced β cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, W.; Devarakonda, T.; Zhou, H.; Wang, X.-Y.; Salloum, F.N.; Spiegel, S.; Fang, X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci. Rep. 2020, 10, 1450. [Google Scholar] [CrossRef] [Green Version]
- Almotairy, A.R.Z.; Montagner, D.; Morrison, L.; Devereux, M.; Howe, O.; Erxleben, A. Pt(IV) pro-drugs with an axial HDAC inhibitor demonstrate multimodal mechanisms involving DNA damage and apoptosis independent of cisplatin resistance in A2780/A2780cis cells. J. Inorg. Biochem. 2020, 210, 111125. [Google Scholar] [CrossRef]
- Kovalev, R.A.; Shtam, T.A.; Karelov, D.V.; Burdakov, V.S.; Volnitskiy, A.V.; Makarov, E.M.; Filatov, M.V. Histone deacetylase inhibitors cause the TP53-dependent induction of p21/Waf1 in tumor cells carrying mutations in TP53. Tsitologiia 2015, 57, 204–211. [Google Scholar]
- Sonnemann, J.; Marx, C.; Becker, S.; Wittig, S.; Palani, C.D.; Krämer, O.H.; Beck, J.F. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br. J. Cancer 2014, 110, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A. Drivers of the Warburg Phenotype. Cancer J. 2015, 21, 56–61. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2020, 599, 1745–1757. [Google Scholar] [CrossRef]
- Lee, H.; Woo, S.M.; Jang, H.; Kang, M.; Kim, S.-Y. Cancer depends on fatty acids for ATP production: A possible link between cancer and obesity. Semin. Cancer Biol. 2022, 86, 347–357. [Google Scholar] [CrossRef]
- Dechandt, C.R.P.; Zuccolotto-Dos-Reis, F.H.; Teodoro, B.G.; Fernandes, A.M.A.P.; Eberlin, M.N.; Kettelhut, I.C.; Curti, C.; Alberici, L.C. Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. J. Bioenerg. Biomembr. 2017, 49, 399–411. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.A.; Unger, S.A.; Horowitz, J.D. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem. Pharmacol. 1996, 52, 273–280. [Google Scholar] [CrossRef]
- Huang, C.; Freter, C. Lipid Metabolism, Apoptosis and Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018, 28, 490–503.e7. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Entities | IC50 (µM) Mean ± SD | Resistance Ratio | ||||
|---|---|---|---|---|---|---|
| Cell Line | Cisplatin | cis(PhB)2 | Ratio | Cisplatin | cisPt(PhB)2 | |
| colorectal | HCT116 | 5.8 ± 0.9 | 0.08 ± 0.01 | 71.5 | 2.2 | 1.8 |
| HCT116p53KO | 13.0 ± 2.6 | 0.14 ± 0.00 | 91.9 | |||
| RKO | 1.5 ± 0.0 | 0.06 ± 0.00 | 25.4 | 4.6 | 1.1 | |
| RKOp53KO | 7.0 ± 0.0 | 0.06 ± 0.00 | 108.6 | |||
| SW480 | 7.1 ± 0.4 | 0.10 ± 0.00 | 74.0 | |||
| CT26 * | 1.4 ± 0.2 | 0.13 ± 0.03 | 10.8 | |||
| MC38 * | 28.0 ± 0.6 | 0.60 ± 0.08 | 46.6 | |||
| ovarian | A2780 | 1.6 ± 0.6 | 0.07 ± 0.01 | 22.1 | 5.7 | 1.2 |
| A2780/cisR | 9.0 ± 0.8 | 0.09 ± 0.01 | 101.8 | |||
| pancreas | SKOV3 | 3.7 ± 1.0 | 0.34 ± 0.07 | 10.9 | ||
| Capan-1 | 3.7 ± 1.2 | 0.06 ± 0.01 | 60.7 | |||
| PANC-1 | 10.7 ± 1.6 | 0.25 ± 0.04 | 42.9 | |||
| melanoma | VM1 | 4.0 ± 0.7 | 0.22 ± 0.09 | 18.1 | ||
| B16 * | 8.6 ± 1.4 | 0.24 ± 0.06 | 35.7 | |||
| mesothelium | p31 | 9.7 ± 0.7 | 0.57 ± 0.10 | 17.0 | 3.2 | 1.4 |
| p31/cisR | 30.9 ± 8.6 | 0.80 ± 0.14 | 38.6 | |||
| breast | MDA-MB-231 | 4.9 ± 0.7 | 0.17 ± 0.03 | 28.7 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendrina, T.; Poetsch, I.; Schueffl, H.; Baier, D.; Pirker, C.; Ries, A.; Keppler, B.K.; Kowol, C.R.; Gibson, D.; Grusch, M.; et al. Influence of the Fatty Acid Metabolism on the Mode of Action of a Cisplatin(IV) Complex with Phenylbutyrate as Axial Ligands. Pharmaceutics 2023, 15, 677. https://doi.org/10.3390/pharmaceutics15020677
Mendrina T, Poetsch I, Schueffl H, Baier D, Pirker C, Ries A, Keppler BK, Kowol CR, Gibson D, Grusch M, et al. Influence of the Fatty Acid Metabolism on the Mode of Action of a Cisplatin(IV) Complex with Phenylbutyrate as Axial Ligands. Pharmaceutics. 2023; 15(2):677. https://doi.org/10.3390/pharmaceutics15020677
Chicago/Turabian StyleMendrina, Theresa, Isabella Poetsch, Hemma Schueffl, Dina Baier, Christine Pirker, Alexander Ries, Bernhard K. Keppler, Christian R. Kowol, Dan Gibson, Michael Grusch, and et al. 2023. "Influence of the Fatty Acid Metabolism on the Mode of Action of a Cisplatin(IV) Complex with Phenylbutyrate as Axial Ligands" Pharmaceutics 15, no. 2: 677. https://doi.org/10.3390/pharmaceutics15020677