
A Single Injection with Sustained-Release Microspheres and a Prime-Boost Injection of Bovine Serum Albumin Elicit the Same IgG Antibody Response in Mice
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
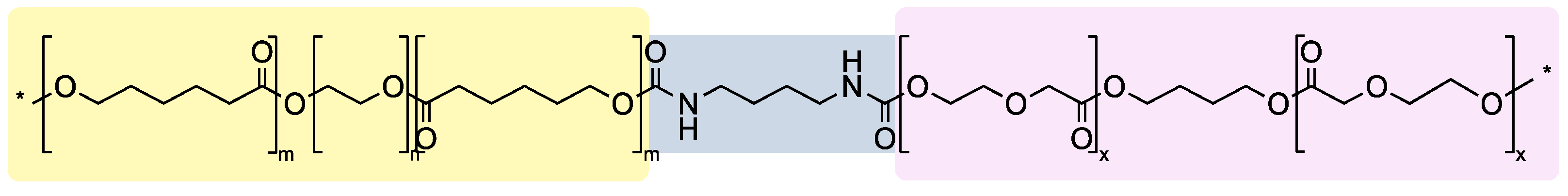
2.2. Polymer Synthesis and Characterization
2.3. Microsphere Production
2.4. Microsphere Size Analysis
2.5. Morphology of Microspheres
2.6. Protein Content of Microspheres
2.7. In Vitro Release of Microspheres
2.8. Residual DCM Content of Microspheres
2.9. Endotoxin Level in Microspheres
2.10. Animal Experiments
2.11. ELISA for BSA-Specific IgG Antibody Titers
2.12. ELISA for BSA Quantification
2.13. Statistical Analysis
3. Results and Discussion
3.1. Properties of Microspheres of Different Polymer Blend Ratios
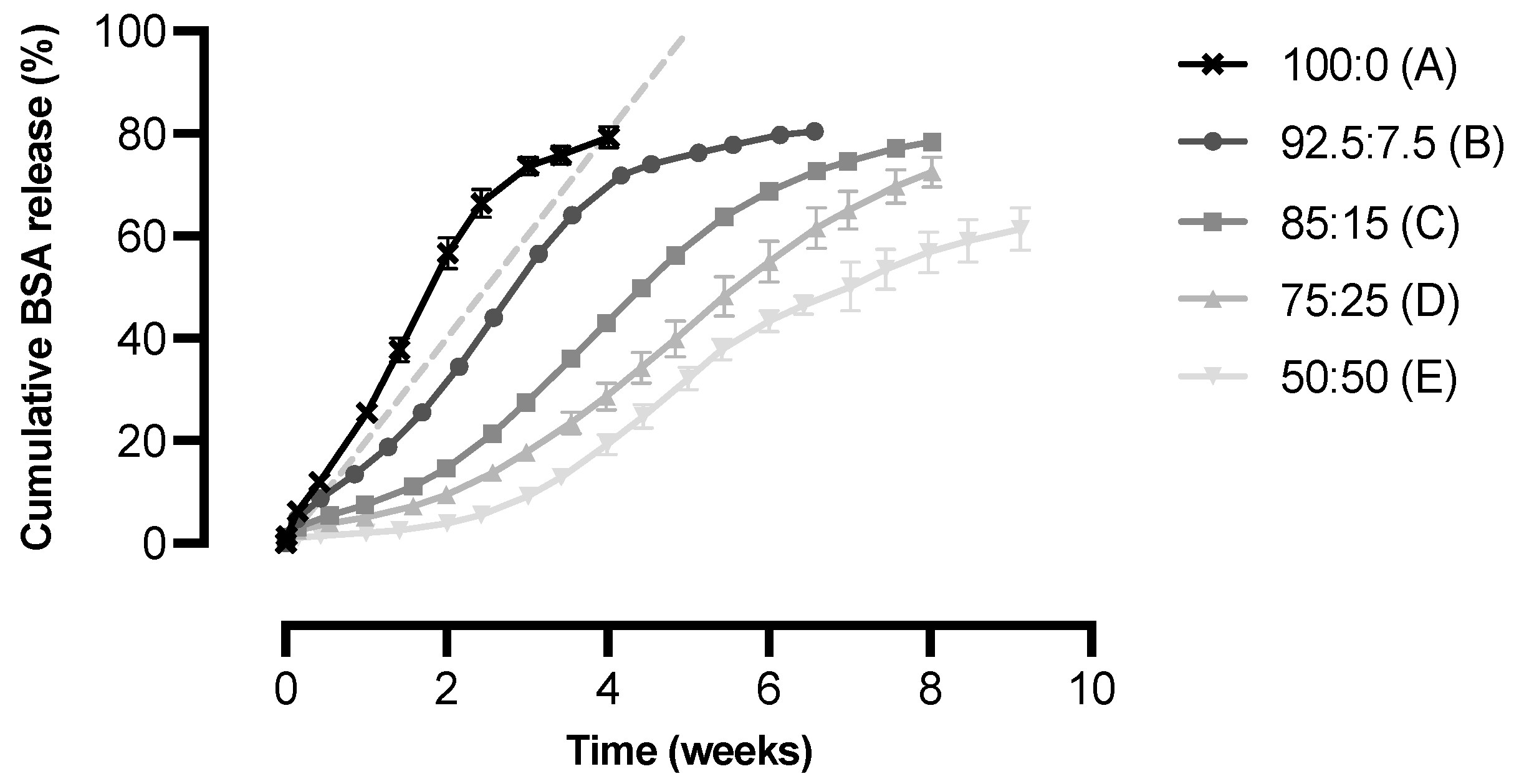
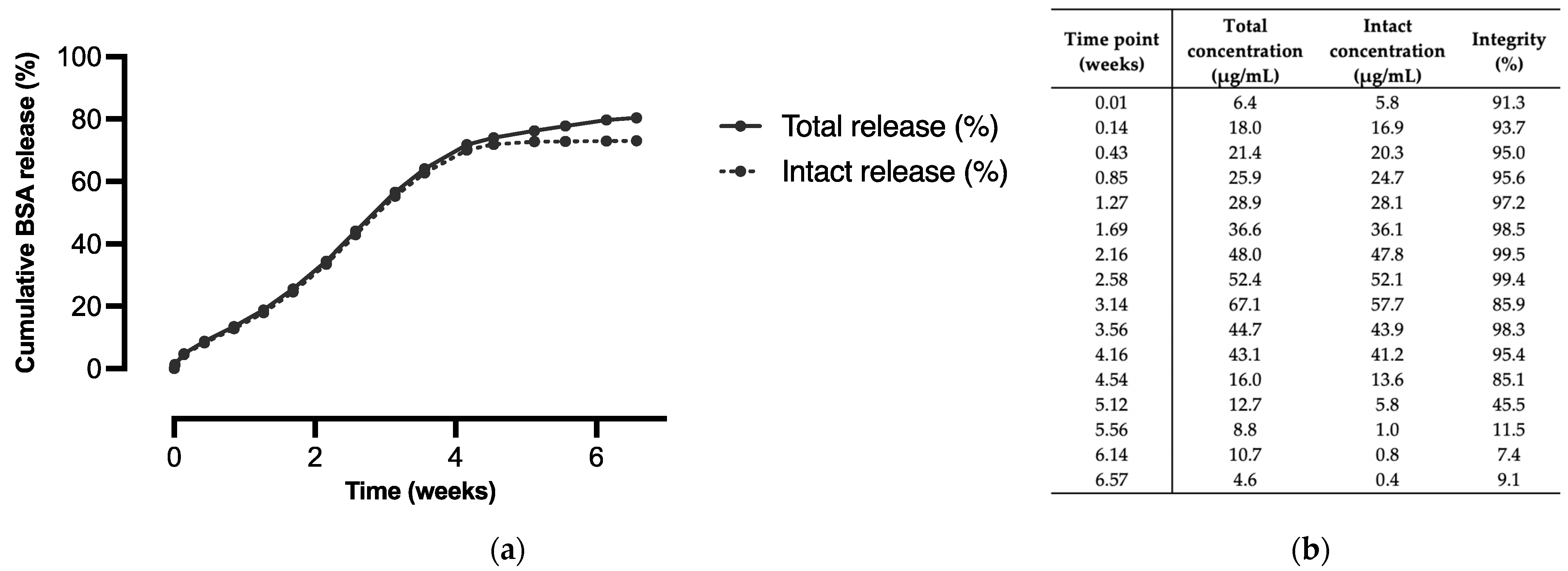
3.2. In Vitro Release of BSA from Microspheres of Different Polymer Blend Ratios
3.3. Residual DCM Content and Endotoxin Analysis of Microspheres Intended for the In Vivo Study
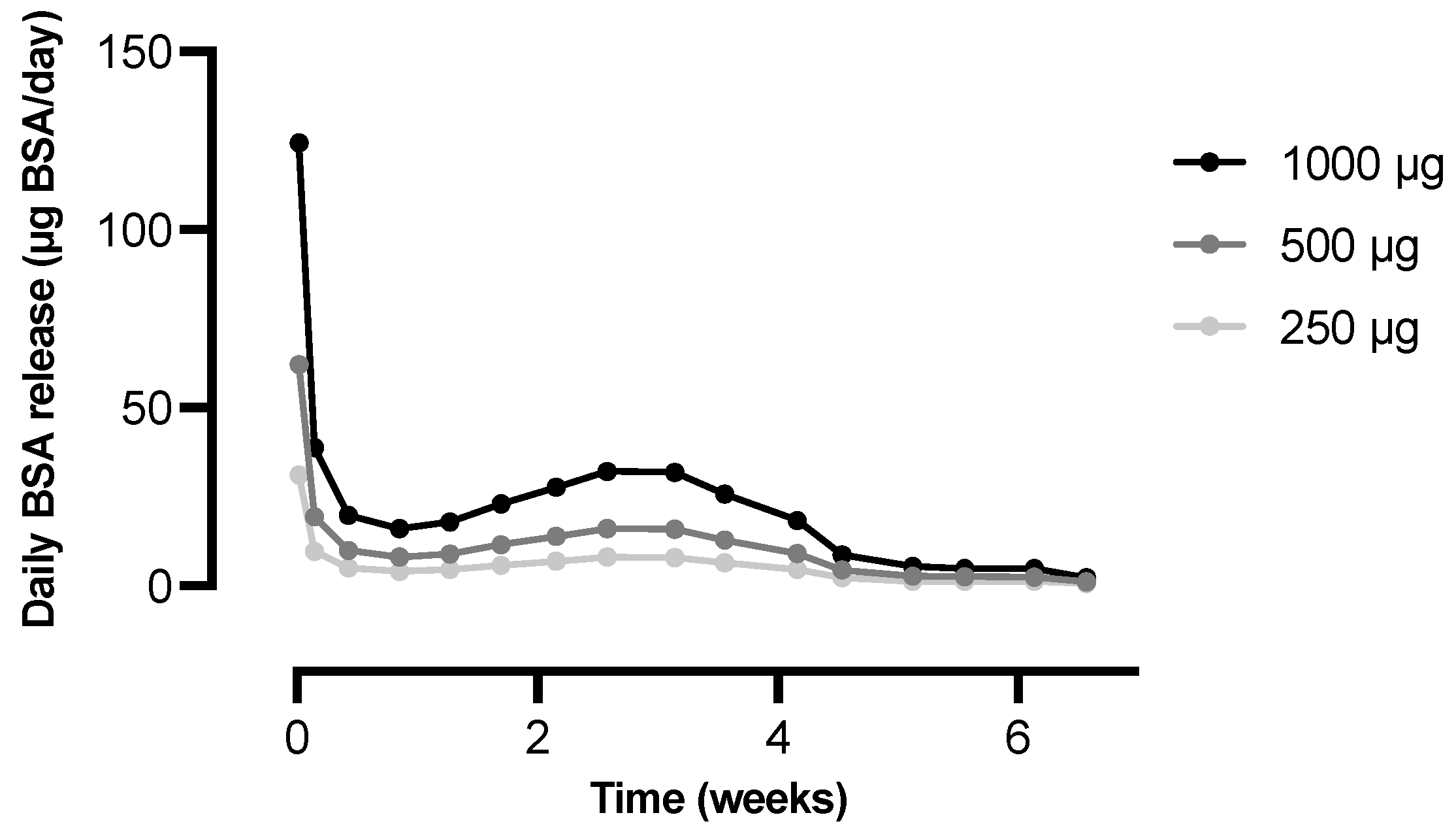
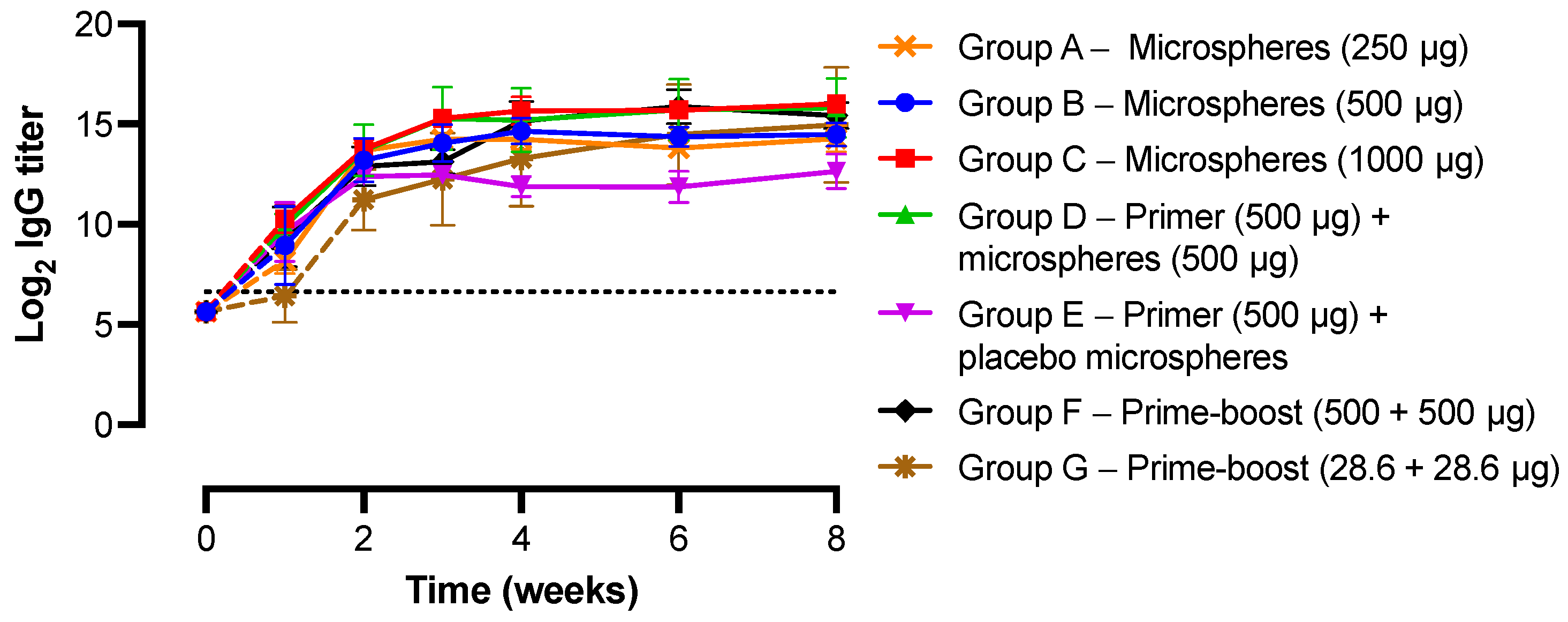
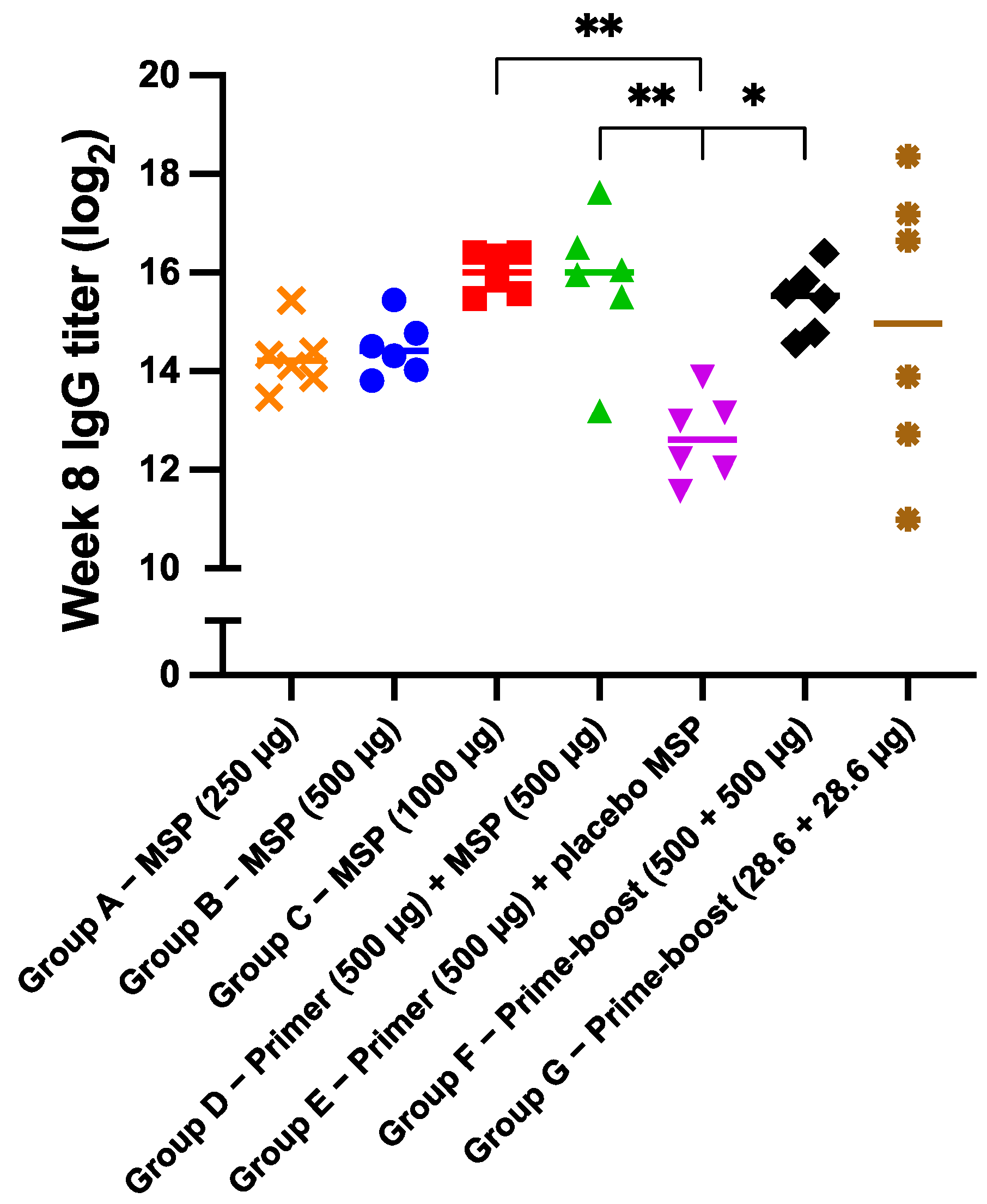
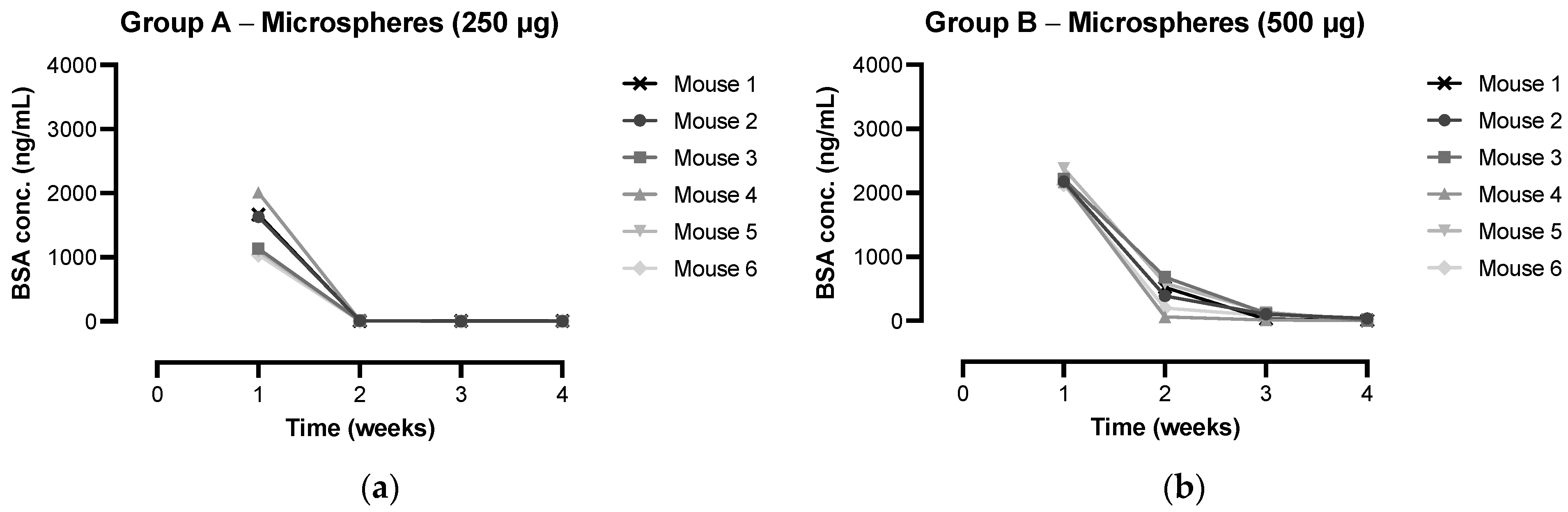
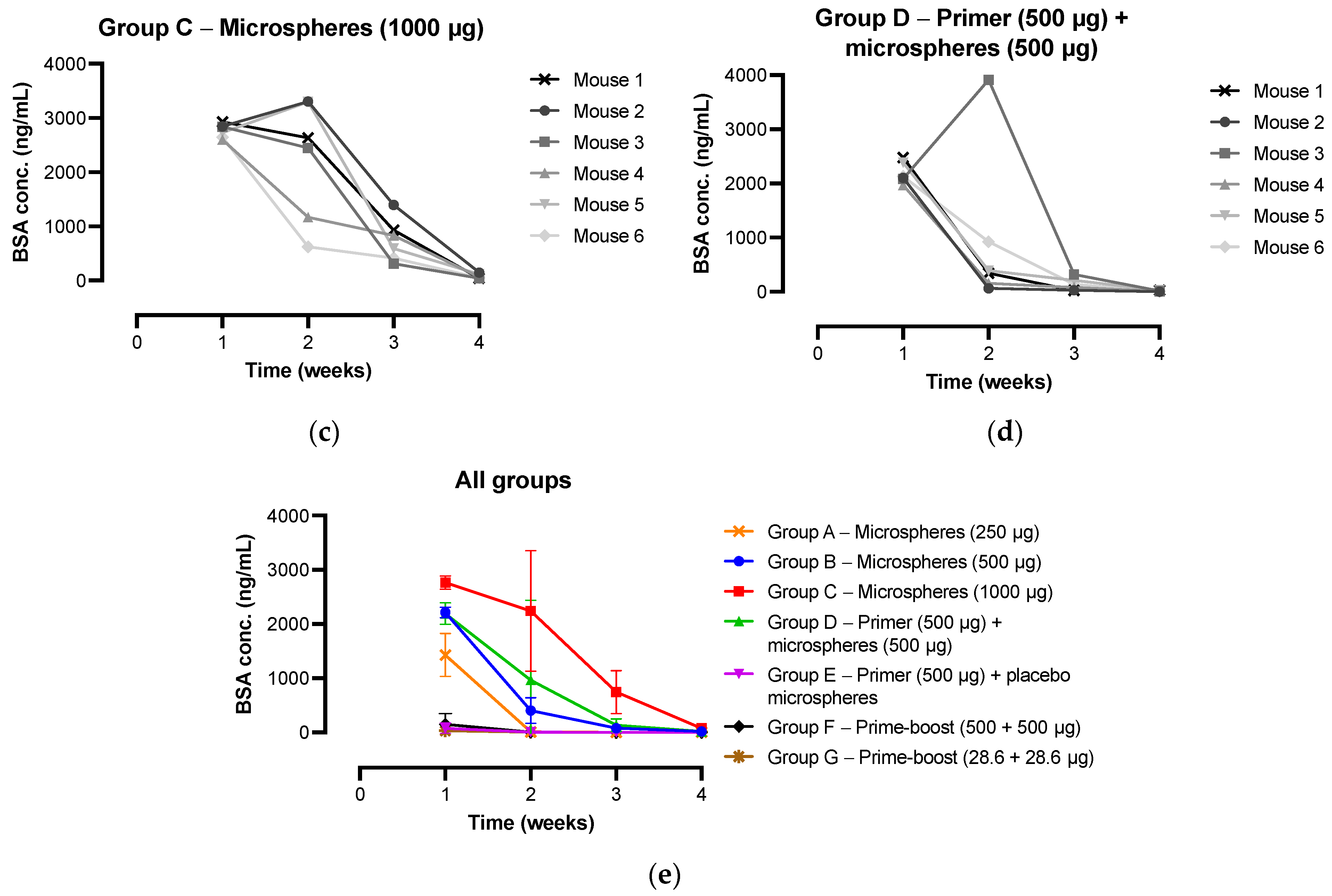
3.4. IgG Antibody Response and Kinetics of Microspheres In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Immunization Coverage. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/immunization-coverage (accessed on 14 December 2022).
- Ali, H.A.; Hartner, A.M.; Echeverria-Londono, S.; Roth, J.; Li, X.; Abbas, K.; Portnoy, A.; Vynnycky, E.; Woodruff, K.; Ferguson, N.M.; et al. Vaccine equity in low and middle income countries: A systematic review and meta-analysis. Int. J. Equity Health 2022, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- WHO. Immunization Agenda 2030. 2021. Available online: https://www.who.int/docs/default-source/immunization/strategy/ia2030/ia2030-document-en.pdf (accessed on 25 October 2022).
- McHugh, K.J.; Guarecuco, R.; Langer, R.; Jaklenec, A. Single-injection vaccines: Progress, challenges, and opportunities. J. Control. Release 2015, 219, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.L.; Lim, A.; Barrón, L.; Duenas, E.T.; Powell, M.F. Development of a single-shot subunit vaccine for HIV-I: Part 4. Optimizing microencapsulation and pulsatile release of MN rgp120 from biodegradable microspheres. J. Control. Release 1997, 47, 135–150. [Google Scholar] [CrossRef]
- van der Kooij, R.S.; Steendam, R.; Zuidema, J.; Frijlink, H.; Hinrichs, W.L.J. Microfluidic production of polymeric core-shell microspheres for the delayed pulsatile release of bovine serum albumin as a model antigen. Pharmaceutics 2021, 13, 1854. [Google Scholar] [CrossRef] [PubMed]
- Amssoms, K.; Born, P.A.; Beugeling, M.; De Clerck, B.; Van Gulck, E.; Hinrichs, W.L.J.; Frijlink, H.W.; Grasmeijer, N.; Kraus, G.; Sutmuller, R.; et al. Ovalbumin-containing core-shell implants suitable to obtain a delayed IgG1 antibody response in support of a biphasic pulsatile release profile in mice. PLoS One 2018, 13, e0202961. [Google Scholar] [CrossRef] [PubMed]
- Guarecuco, R.; Lu, J.; McHugh, K.J.; Norman, J.J.; Thapa, L.S.; Lydon, E.; Langer, R.; Jaklenec, A. Immunogenicity of pulsatile-release PLGA microspheres for single-injection vaccination. Vaccine 2018, 36, 3161–3168. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.L. Single-administration vaccines: Controlled-release technology to mimic repeated immunizations. Trends Biotechnol. 1999, 17, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Qi, X.R.; Zhou, X.J.; Maitani, Y.; Wang, S.C.; Jiang, Y.; Nagai, T. Pharmaceutical and immunological evaluation of a single-dose hepatitis B vaccine using PLGA microspheres. J. Control. Release 2006, 112, 35–42. [Google Scholar] [CrossRef]
- Singh, M.; Singh, A.; Talwar, G.P. Controlled delivery of diphtheria toxoid using biodegradable poly(d,l-lactide) microcapsules. Pharm. Res. 1991, 8, 958–961. [Google Scholar] [CrossRef]
- Du, G.; Sun, X. Current advances in sustained release microneedles. Pharm. Front. 2020, 2, e11–e22. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Meng, J.; Ma, X.; Lin, J.; Lu, X. Advanced materials for the delivery of vaccines for infectious diseases. Biosaf. Health 2022, 4, 95–104. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Duque, L.; Körber, M.; Bodmeier, R. Improving release completeness from PLGA-based implants for the acid-labile model protein ovalbumin. Int. J. Pharm. 2018, 538, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Giteau, A.; Venier-Julienne, M.C.; Aubert-Pouëssel, A.; Benoit, J.P. How to achieve sustained and complete protein release from PLGA-based microparticles? Int. J. Pharm. 2008, 350, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Houchin, M.L.; Topp, E.M. Chemical degradation of peptides and proteins in PLGA: A review of reactions and mechanisms. J. Pharm. Sci. 2008, 97, 2395–2404. [Google Scholar] [CrossRef] [PubMed]
- Stanković, M.; de Waard, H.; Steendam, R.; Hiemstra, C.; Zuidema, J.; Frijlink, H.W.; Hinrichs, W.L.J. Low temperature extruded implants based on novel hydrophilic multiblock copolymer for long-term protein delivery. Eur. J. Pharm. Sci. 2013, 49, 578–587. [Google Scholar] [CrossRef]
- Stanković, M.; Tomar, J.; Hiemstra, C.; Steendam, R.; Frijlink, H.W.; Hinrichs, W.L.J. Tailored protein release from biodegradable poly(ε-caprolactone-PEG)-b-poly(ε-caprolactone) multiblock-copolymer implants. Eur. J. Pharm. Biopharm. 2014, 87, 329–337. [Google Scholar] [CrossRef]
- Teekamp, N.; Van Dijk, F.; Broesder, A.; Evers, M.; Zuidema, J.; Steendam, R.; Post, E.; Hillebrands, J.L.; Frijlink, H.W.; Poelstra, K.; et al. Polymeric microspheres for the sustained release of a protein-based drug carrier targeting the PDGFβ-receptor in the fibrotic kidney. Int. J. Pharm. 2017, 534, 229–236. [Google Scholar] [CrossRef]
- Scheiner, K.C.; Maas-Bakker, R.F.; Nguyen, T.T.; Duarte, A.M.; Hendriks, G.; Sequeira, L.; Duffy, G.P.; Steendam, R.; Hennink, W.E.; Kok, R.J. Sustained release of vascular endothelial growth factor from poly(ε-caprolactone-PEG-ε-caprolactone)-b-poly(l-lactide) multiblock copolymer microspheres. ACS Omega 2019, 4, 11481–11492. [Google Scholar] [CrossRef] [Green Version]
- Igartua, M.; Hernández, R.M.; Esquisabel, A.; Gascón, A.R.; Calvo, M.B.; Pedraz, J.L. Enhanced immune response after subcutaneous and oral immunization with biodegradable PLGA microspheres. J. Control. Release 1998, 56, 63–73. [Google Scholar] [CrossRef]
- Conway, B.R.; Eyles, J.; Alpar, H.O. A comparative study on the immune responses to antigens in PLA and PHB microspheres. J. Control. Release 1997, 49, 1–9. [Google Scholar] [CrossRef]
- Lemperle, G. Biocompatibility of injectable microspheres. Biomed. J. Sci. Tech. Res. 2018, 2, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Kim, S.; Park, K. Issues in long-term protein delivery using biodegradable microparticles. J. Control. Release 2010, 146, 241–260. [Google Scholar] [CrossRef]
- Vladisavljević, G.T. Structured microparticles with tailored properties produced by membrane emulsification. Adv. Colloid Interface Sci. 2015, 225 (Suppl. C), 53–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Wu, J.; Fan, Q.; He, F.; Tian, G.; Yang, T.; Ma, G.; Su, Z. Preparation of uniform-sized exenatide-loaded PLGA microspheres as long-effective release system with high encapsulation efficiency and bio-stability. Colloids Surfaces B Biointerfaces 2013, 112, 492–498. [Google Scholar] [CrossRef]
- Bodmeier, R.; McGinity, J.W. Solvent selection in the preparation of poly(dl-lactide) microspheres prepared by the solvent evaporation method. Int. J. Pharm. 1988, 43, 179–186. [Google Scholar] [CrossRef]
- Rafati, H.; Coombes, A.G.A.; Adler, J.; Holland, J.; Davis, S.S. Protein-loaded poly(dl-lactide-co-glycolide) microparticles for oral administration: Formulation, structural and release characteristics. J. Control. Release 1997, 43, 89–102. [Google Scholar] [CrossRef]
- Mao, S.; Xu, J.; Cai, C.; Germershaus, O.; Schaper, A.; Kissel, T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. Int. J. Pharm. 2007, 334, 137–148. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Preferential interactions of proteins with salts in concentrated solutions. Biochemistry 1982, 21, 6545–6552. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- van Dijkhuizen-Radersma, R.; Métairie, S.; Roosma, J.R.; de Groot, K.; Bezemer, J.M. Controlled release of proteins from degradable poly(ether-ester) multiblock copolymers. J. Control. Release 2005, 101, 175–186. [Google Scholar] [CrossRef]
- Van Dijk, F.; Teekamp, N.; Beljaars, L.; Post, E.; Zuidema, J.; Steendam, R.; Kim, Y.O.; Frijlink, H.W.; Schuppan, D.; Poelstra, K.; et al. Pharmacokinetics of a sustained release formulation of PDGFβ-receptor directed carrier proteins to target the fibrotic liver. J. Control. Release 2018, 269, 258–265. [Google Scholar] [CrossRef]
- Haitjema, H.; Steendam, R.; Hiemstra, C.; Zuidema, J.; Doornbos, A.; Nguyen, T. Biodegradable, Phase Separated, Thermoplastic Multi-Block Copolymer. PCT Patent No. WO 2021/066650 A1, 8 April 2021. [Google Scholar]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef] [PubMed]
- Heene, S.; Thoms, S.; Kalies, S.; Wegner, N.; Peppermüller, P.; Born, N.; Walther, F.; Scheper, T.; Blume, C.A. Vascular network formation on macroporous polydioxanone scaffolds. Tissue Eng. Part A 2021, 27, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, M.; Nelson, K.D.; Eberhart, R.C. Mechanical properties and in vitro degradation of bioresorbable fibers and expandable fiber-based stents. J. Biomed. Mater. Res. Part B Appl. Biomater. 2005, 74B, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Kotcharat, P.; Chuysinuan, P.; Thanyacharoen, T.; Techasakul, S.; Ummartyotin, S. Development of bacterial cellulose and polycaprolactone (PCL) based composite for medical material. Sustain. Chem. Pharm. 2021, 20, 100404. [Google Scholar] [CrossRef]
- European Medicines Agency ICH Guideline Q3C (R8) on Impurities: Guideline for Residual Solvents. 2021. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ich-guideline-q3c-r8-impurities-guideline-residual-solvents-step-5_en.pdf (accessed on 25 October 2022).
- Serota, D.G.; Thakur, A.; Ulland, B.; Kirschman, J.; Brown, N.; Coots, R.H. A two-year drinking-water study of dichloromethane in rodents. II. Mice. Food Chem. Toxicol. 1986, 24, 959–963. [Google Scholar] [CrossRef]
- Dresser, D.W.; Gowland, G. Immunological paralysis induced in adult rabbits by small amounts of a protein antigen. Nature 1964, 203, 733–736. [Google Scholar] [CrossRef]
- Dixon, F.J.; Maurer, P.H. Immunologic unresponsiveness induced by protein antigens. J. Exp. Med. 1955, 101, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, N.A. Induction of immunological paralysis in two zones of dosage. Proc. R. Soc. Lond. B 1964, 161, 275–292. [Google Scholar]
- Lofthouse, S. Immunological aspects of controlled antigen delivery. Adv. Drug Deliv. Rev. 2002, 54, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, E.C.; Yeh, M.K.; Coombes, A.G.A.; Davis, S.S. The stability and immunogenicity of a protein antigen encapsulated in biodegradable microparticles based on blends of lactide polymers and polyethylene glycol. Vaccine 1999, 17, 512–529. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Rahman, D.; McGee, J.P.; Jeffery, H.; Davies, M.C.; Williams, P.; Davis, S.S.; Challacombe, S.J. Biodegradable microparticles as controlled release antigen delivery systems. Immunology 1991, 73, 239–242. [Google Scholar] [PubMed]
- Heesters, B.A.; van der Poel, C.E.; Das, A.; Carroll, M.C. Antigen presentation to B cells. Trends Immunol. 2016, 37, 844–854. [Google Scholar] [CrossRef]
- Leo, O.; Cunningham, A.; Stern, P.L. Vaccine immunology. Perspect. Vaccinol. 2011, 1, 25–59. [Google Scholar] [CrossRef] [Green Version]
- Sandor, M.; Harris, J.; Mathiowitz, E. A novel polyethylene depot device for the study of PLGA and P(FASA) microspheres in vitro and in vivo. Biomaterials 2002, 23, 4413–4423. [Google Scholar] [CrossRef]
- Tracy, M.A.; Ward, K.L.; Firouzabadian, L.; Wang, Y.; Dong, N.; Qian, R.; Zhang, Y. Factors affecting the degradation rate of poly(lactide-co-glycolide) microspheres in vivo and in vitro. Biomaterials 1999, 20, 1057–1062. [Google Scholar] [CrossRef]
- Nguyen, A.; Reyes, A.E.; Zhang, M.; McDonald, P.; Wong, W.L.T.; Damico, L.A.; Dennis, M.S. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng. Des. Sel. 2006, 19, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.; Eyre, R.; Bull, R. Adduction of hemoglobin and albumin in vivo by metabolites of trichloroethylene, trichloroacetate, and dichloroacetate in rats and mice. Fundam. Appl. Toxicol. 1992, 19, 336–342. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer A1 | Polymer A2 | Polymer B | |
|---|---|---|---|
| 20[PCL-PEG3000-PCL]-80[PDO] | 50[PCL-PEG1000-PCL]-20[PDO] | ||
| Molar caprolactate/PEG ratio (1H NMR) | 6.8 (6.4 in-weight) | 6.4 (6.4 in-weight) | 8.3 (8.6 in-weight) |
| Molar dioxanonate/PEG ratio (1H NMR) | 141.6 (147.6 in-weight) | 155.2 (147.5 in-weight) | 18.5 (18.9 in-weight) |
| Weight ratio PCL-PEG-PCL/PDO block (1H NMR) | 21.2/78.8 | 19.6/80.4 | 49.9/50.1 |
| Mn (×104 g/mol) | 2.8 | 1.5 | 3.6 |
| Mw (×104 g/mol) | 4.3 | 4.5 | 6.7 |
| Intrinsic viscosity (dL/g) | 0.70 | 0.69 | 0.73 |
| 1,4-dioxane content (ppm) | <18 | <18 | <18 |
| Tg (°C) | −15 | −14 | −57 and −23 |
| Tm (°C) | 34 and 88 | 34 and 89 | 88 |
| Formulation Parameters | Formulation | ||||||
|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | ||
| Weight ratio polymer A:polymer B 1 | 100:0 | 92.5:7.5 | 85:15 | 75:25 | 50:50 | 92.5:7.5 | |
| Target BSA loading (wt-%) | 5 | 5 | 5 | 5 | 5 | n.a. 2 | |
| Batch size (g) | 1.5 | 3.5 | 3.5 | 3.5 | 1.5 | 3.5 | |
| Ultra-Turrax® | Speed (rpm) | 21,000 | 25,000 | 25,000 | 25,000 | 21,000 | 25,000 |
| Time (s) | 40 | 60 | 60 | 60 | 40 | 60 | |
| Extraction | Vessel size (L) | 2 | 5 | 5 | 5 | 2 | 5 |
| Stirrer type | Anchor-type stirring shaft | Stirring bar (10.8 × 2.6 cm) | Stirring bar (10.8 × 2.6 cm) | Stirring bar (10.8 × 2.6 cm) | Anchor-type stirring shaft | Stirring bar (10.8 × 2.6 cm) | |
| Stirrer speed (rpm) | 200 | 75 | 75 | 75 | 200 | 75 | |
| Airflow (L/min) | 5 | 10 | 10 | 10 | 5 | 10 | |
| Time (h) | 3 | 4 | 4 | 4 | 3 | 4 | |
| Ratio Polymer A: Polymer B | Total PEG (wt-%) | PEG3000 (wt-%) | PEG1000 (wt-%) | PCL (wt-%) | PDO (wt-%) | BDO (wt-%) | BDI (wt-%) |
|---|---|---|---|---|---|---|---|
| 100:0 | 15 | 15 | 0 | 4 | 73 | 3 | 5 |
| 92.5:7.5 | 15.675 | 13.875 | 1.8 | 5.425 | 70.975 | 2.925 | 5 |
| 85:15 | 16.35 | 12.75 | 3.6 | 6.85 | 68.95 | 2.85 | 5 |
| 75:25 | 17.25 | 11.25 | 6 | 8.75 | 66.25 | 2.75 | 5 |
| 50:50 | 19.5 | 7.5 | 12 | 13.5 | 59.5 | 2.5 | 5 |
| Group | Type of Group | Formulation Composition | Administration | Total Dose | Average Daily Dose | Week of Administration | Number of Animals |
|---|---|---|---|---|---|---|---|
| A | Treatment | BSA-MSP in CMC solution 1 | 7.28 mg MSP-F in 193 μL CMC solution 1 | 250 μg | 7.1 μg 2 | 0 | 6 |
| B | Treatment | BSA-MSP in CMC solution | 14.6 mg MSP-F in 187 μL CMC solution | 500 μg | 14.3 μg 2 | 0 | 6 |
| C | Treatment | BSA-MSP in CMC solution | 29.2 mg MSP-F in 174 μL CMC solution | 1000 μg | 28.6 μg 2 | 0 | 6 |
| D | Positive control/ treatment | BSA in PBS + BSA-MSP in CMC solution | 500 μg BSA in 100 μL PBS + 14.6 mg MSP-B in 87 μL CMC solution 1 | 1000 μg (500 μg + 500 μg) | 514.3 μg on day 1, 14.3 μg for remaining days 2 | 0 | 6 |
| E | Positive control/placebo | BSA in PBS + placebo MSP in CMC solution | 500 μg BSA in 100 μL PBS + 14.6 mg MSP-F in 87 μL CMC solution | 500 μg | 500 μg on day 1 | 0 | 6 |
| F | Positive control | BSA in PBS (prime-boost) | 500 μg BSA in 200 μL PBS (at 0 and 3 weeks) | 1000 μg (500 μg + 500 μg) | 500 μg on day 1, 500 μg on day 22 | 0 and 3 | 6 |
| G | Positive control | BSA in PBS (prime-boost) | 28.6 μg BSA in 200 μL PBS (at 0 and 3 weeks) | 57.1 μg (28.6 μg + 28.6 μg) | 28.6 μg on day 1, 28.6 μg on day 22 | 0 and 3 | 6 |
| H | Negative control | PBS | 200 μL PBS (at 0 and 3 weeks) | - | - | 0 and 3 | 3 |
| I | Negative control | CMC solution | 200 μL CMC solution | - | - | 0 | 3 |
| Formulation | Ratio Polymer A: Polymer B | d10 (µm) | d50 (µm) | d90 (µm) | CV (%) | Actual Loading (wt-%) | EE (%) |
|---|---|---|---|---|---|---|---|
| A | 100:0 | 30.4 | 38.7 | 50.5 | 21.0 | 4.4 ± 0.1 | 87.4 ± 1.0 |
| B | 92.5:7.5 | 30.8 | 39.9 | 52.4 | 21.8 | 4.5 ± 0.5 | 89.9 ± 10.0 |
| C | 85:15 | 30.6 | 39.5 | 51.5 | 21.7 | 4.9 ± 0.3 | 97.3 ± 5.5 |
| D | 75:25 | 30.5 | 39.2 | 51.4 | 21.9 | 4.9 ± 0.2 | 96.9 ± 4.5 |
| E | 50:50 | 29.9 | 39.0 | 51.6 | 24.5 | 5.1 ± 1.3 | 101.7 ± 26.4 |
| F | 92.5:7.5 | 31.2 | 42.7 | 58.2 | 27.4 | n.a. 2 | n.a. 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Kooij, R.S.; Beukema, M.; Huckriede, A.L.W.; Zuidema, J.; Steendam, R.; Frijlink, H.W.; Hinrichs, W.L.J. A Single Injection with Sustained-Release Microspheres and a Prime-Boost Injection of Bovine Serum Albumin Elicit the Same IgG Antibody Response in Mice. Pharmaceutics 2023, 15, 676. https://doi.org/10.3390/pharmaceutics15020676
van der Kooij RS, Beukema M, Huckriede ALW, Zuidema J, Steendam R, Frijlink HW, Hinrichs WLJ. A Single Injection with Sustained-Release Microspheres and a Prime-Boost Injection of Bovine Serum Albumin Elicit the Same IgG Antibody Response in Mice. Pharmaceutics. 2023; 15(2):676. https://doi.org/10.3390/pharmaceutics15020676
Chicago/Turabian Stylevan der Kooij, Renée S., Martin Beukema, Anke L. W. Huckriede, Johan Zuidema, Rob Steendam, Henderik W. Frijlink, and Wouter L. J. Hinrichs. 2023. "A Single Injection with Sustained-Release Microspheres and a Prime-Boost Injection of Bovine Serum Albumin Elicit the Same IgG Antibody Response in Mice" Pharmaceutics 15, no. 2: 676. https://doi.org/10.3390/pharmaceutics15020676