1. Introduction
Clinically acquired resistance is an insurmountable dilemma for small-molecule kinase inhibitors to treat cancer [
1]. Nevertheless, locating small-molecule ligands with high affinity and good properties for target proteins in a broad chemical space has been a primary challenge in drug research and development (R&D) [
2]. To date, it cannot be overstated to describe the kinase drugs approved by The U.S. Food and Drug Administration (FDA) to overcome clinical resistance driven by protein kinase “gatekeeper” mutation as “desert oasis”. Lung cancer is the leading cause of cancer-related deaths worldwide, with non-small cell lung cancer (NSCLC) being the most common type of lung cancer. Secondary epidermal growth factor receptor (EGFR) mutations in threonine 790 (T790M) lead to acquired resistance which severely affects patient prognosis. Therefore, strategies or drugs to overcome resistance are urgent to prolong the survival of patients with NSCLC.
Laborious wet labs and high-throughput screening techniques are so time-consuming and challenging that they are unsuitable for screening candidate drugs from a broad range of compound groups in pre-drug R&D. With improvements in machine learning theory and an abundance of pharmacological data available, machine learning provides sufficient power for the development of precision medicine and artificially intelligent drug design (AIDD). Many encouraging scientific achievements have convincingly demonstrated the potential of these approaches. For instance, the knowledge graph (KG) enables to detect of the drivers of tumor resistance and adverse drug reactions in a wider multi-omics space [
3,
4]; reinforcement learning (RL) has been found to be particularly effective in the de novo design and multi-objective optimization of drug molecules [
5,
6,
7]. Deep learning is a powerful data-driven algorithm in machine learning, which offers significant advantages to reveal implicit relationships between drugs, diseases, and genes that are not easily detected, owing to the powerful generalization and representation extraction capability. Some in silico methods that explore potential drug–target associations to advance drug R&D have been developed to narrow the research concentration areas toward the more workable drugs.
Some studies have viewed DTA prediction as a binary classification task, borrowing binary numbers (1/0) to label whether the two are combined [
8,
9,
10], while some others treat it as a regression task and use floating-point numbers to indicate DTAs [
11,
12,
13].
The random forest (RF) algorithm broke the previous methods of relying on multi-parameter scoring functions to infer DTA [
14], which has proven to be convincing for extrapolating drug–target relationships in larger chemical spaces. KronRLS [
15] and SimBoost [
12] were regression-based machine-learning approaches that evaluated similarities between drugs and proteins to determine DTA. Various excellent deep-learning works have been presented. DeepDTA [
8] and Attention-DTA [
16] leveraged the convolutional neural networks (CNNs) to obtain the hidden relationships of atomic and amino acid sequences. DeepCDA incorporated the long- to short-term memory network which aims to alleviate the phenomenon of gradient disappearance and gradient explosion [
17]. MATT-DTI deployed relation-aware self-attention with position embedding to reinforce relative positional associations among atoms [
13]. Transformer-based works have come to the fore in various natural language processing (NLP) tasks. DMIL-PPDTA utilized the transformer encoder to enrich word embeddings of drug and protein sequences, aiming to learn hidden associations from the raw data [
18]. DeepAtom [
19] extrapolated node-level interaction information relevant to binding from the voxelized protein–compound complex structures. Nevertheless, these models rely on known 3D drug–target complexes, and the computational burden of complex 3D convolutional networks to extract the features of massive complexes is expensive. GraphDTA [
11] and MGraphDTA [
20] represented compounds as topological graphs and evaluated several types of Graph Neural Network (GNN) variants, including Graph Convolutional Network (GCN) [
21], Graph Isomorphism Network (GIN) [
22], and the Graph Attention Network (GAT) [
23], with the aim of replacing CNN and achieving excellent performance. Additionally, DGraphDTA encoded both drugs and proteins into the graphs for inferring DTA by GNN [
24]. Among those graph-based methods, they not only effectively avoid the drawbacks of few complex samples and high computational cost, but compensate for the problem of inadequate SMILES (Simplified Molecular Input Line Entry System) [
25] for drug representation, and the molecule graph is closer to the natural description of compounds.
Although these methods produce excellent prediction results, they are difficult to generalize to real-world problems. Firstly, the molecular similarity principle [
26] states that molecules with similar structures usually show similar biological activities and physicochemical properties; conversely, there are significant differences. Therefore, the model must discriminate between molecular structures over a wide chemical space. Moreover, modeling underlying complicated mapping patterns between compounds and proteins simply concatenate, which deviates from the non-covalent interaction between the receptor and ligand. More importantly, these approaches have limited interpretability as a result of the “black-box” property of graph neural networks. Considering that the false-positive statistics generated by the binary classification task directly impair the robustness of the model, here, predicting DTA was regarded as a regression problem. We propose a three-channel DoubleSG-DTA theoretical framework based on GINs and multiple attention mechanisms to address the aforementioned problems, which significantly outshines other regression-based SOTA methods on various benchmark datasets. Afterward, we visualize the gradient of atomic contributions in graph representations and compare them with the molecular docking poses to further extend the interpretability of the graph-based model.
This paper presents the main contributions as follows:
DoubleSG-DTA combined graph isomorphism networks and the squeeze-and-excitation networks to extract multimodal representations of drugs in parallel, aiming to enhance the model to discriminate between compound structures and selectively suppress redundant information to disturb model decisions.
The design of cross-multi-head attention mechanisms to model the reality-based non-covalent molecular docking behavior of drug substructures and subsequences with target proteins, respectively;
Application of the DoubleSG-DTA to screen promising hit compounds of the NSCLC harboring mutation from natural products, which have been consistent with reported laboratory studies.
2. Double Sequence and Graph to Predict Drug–Target Affinity (DoubleSG-DTA)
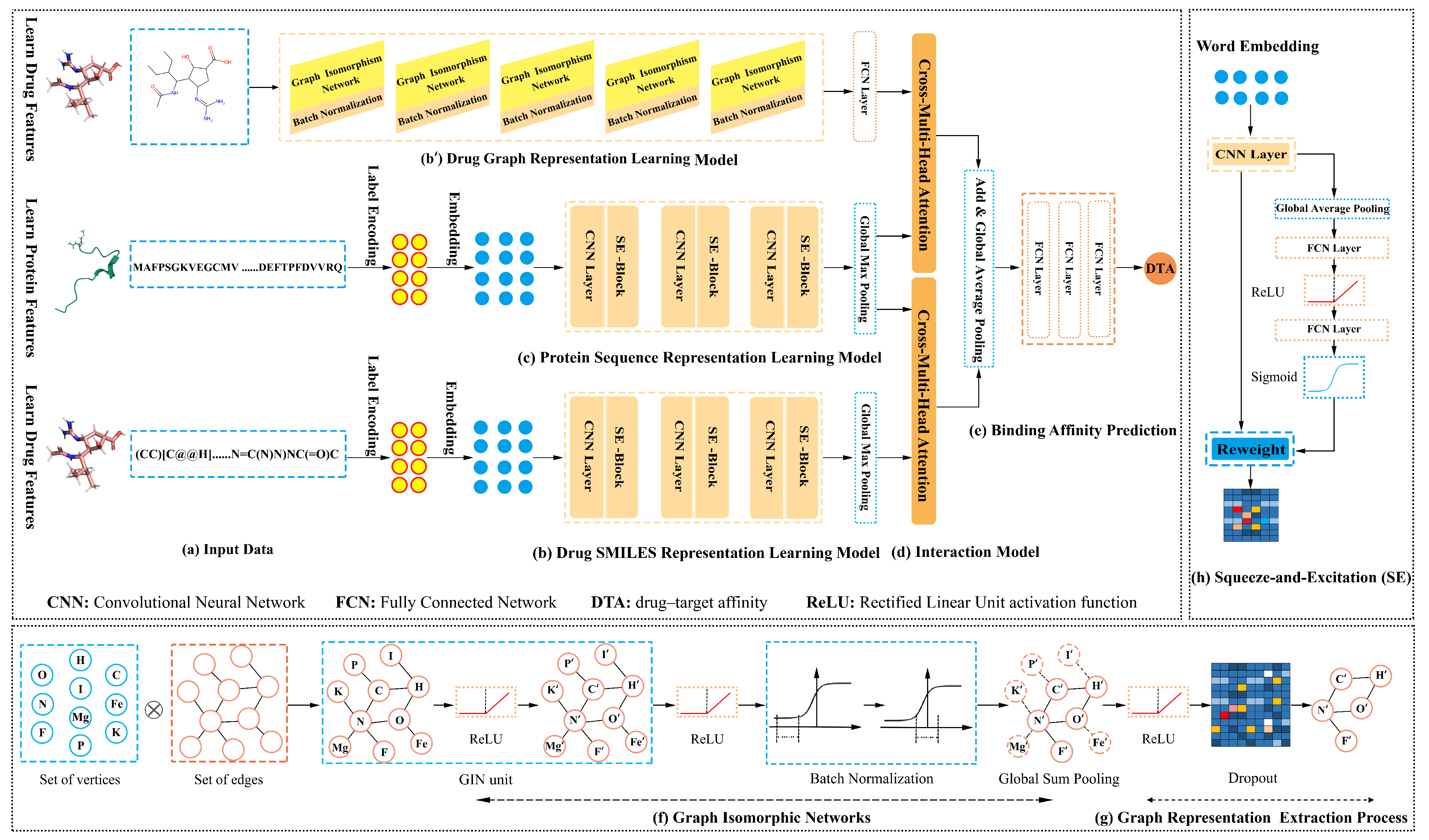
This work developed the DoubleSG-DTA model with three-channel multimodal representations, four-channel interaction, and one-channel output for DTA prediction, which deployed multilayer GINs and multiple attention blocks, as shown in
Figure 1. Primarily, we took the drug graphs and SMILES as inputs into the drug representation learning models. Multilayer GINs [
22] and squeeze-and-excitation networks (SENets) [
27] are jointly used as feature extractors for drugs. Additionally, the protein representation learning model captures the dominant feature of the over-redundant protein sequences that are highly dependent on stacked SENets. Moreover, to further encode the drug–target mutual interaction information, we designed cross-multi-head attention to model the reality-based non-covalent molecular docking behavior of drug substructures and subsequences with target proteins, respectively. Ultimately, we decoupled the attention coefficients into the Multilayer Perceptrons (MLPs) to predict DTA. This section presents the building blocks of our framework in order.
2.1. Word Embedding and Graph Encoding
Initially, we utilized high-dimensional word embeddings to uniquely encode drug and protein sequences. To this aim, we built label/integer dictionaries for drug SMILES and protein FASTA sequences, which consist of 64 and 22 key-value pairs, respectively. For example, the SMILES of Propylene glycol “CC(O)CO” and the
[
28] protein subsequence “NWCVQIA” are encoded as
and
according to the SMILES dictionary {‘C’:22, ‘N’:34, ‘O’:33, ‘(’:4, ‘)’:3} and the protein dictionary {‘A’:1, ‘N’:14, ‘C’:2, ‘Q’:15, ‘I’:8, ‘V’:22, ‘W’:21}. We then map each integer vector into word embeddings
and
by embedding layers. Where
and
denote the size of the SMILES and protein FASTA sequence,
represents the embedding dimensions.
We convert SMILES to their corresponding molecular graphs
and extract atom features by RDKit [
29], where E and V are the sets of edges and atoms, respectively. Each atom node in a drug is represented by a multi-dimension vector of 10 molecular descriptors (atom symbol, atom number, hybridization, number of adjacent atoms, chirality, formal charge, aromaticity, number of bonded hydrogens, and explicit and implicit valence).
2.2. Drug and Protein Sequence Representation Learning Model
The CNNs construct text features by fusing spatial correlations between features that benefit from the convolutional kernel’s local receptive field but are likewise limited by it. In computer vision, the squeeze-and-excitation (SE) block with channel attention was integrated into existing architectures, which adaptively rescales channel-wise feature weights by explicitly modeling non-mutually-exclusive relationships between channels [
27]. The research has confirmed that the SENets achieved superior performance for image classification with a slight increase in computational cost [
27]. Accordingly, we stacked multilayer SENets designed to selectively enhance effective statistics and suppress noise to disturb model decisions. Given
as the feature matrix of the convolution layer output, we routed it to the SE block, where
.
SE module makes use of squeeze, excitation, and reweighting operators. The squeeze operator intrinsically aims to transform the dimensions of the feature matrix
U and obtain channel-wise statistics
by applying the global average pooling operation.
The excitation module leverages two learnable FCNs with the gating mechanism to learn inter-channel non-linear interaction and filter non-dominant features.
where the
is the Rectified Linear Unit (ReLU) activation function, and
is the sigmoid function, and
and
are the two learnable weight matrices. The reduction ratio was set to r = 16 to reconcile the balance between performance and complexity [
27].
The reweighting representation
was computed by applying the channel-wise multiplication operation to the channel attention weight
and the feature map
.
where
,
.
The word embeddings
and
are directly fed into the convolutional layers, then delivered to the SE block accompanied by a global max pooling operation to calculate desired feature information. Hence, the drug and protein sequence representations can be expressed as:
2.3. Drug Graph Representation Learning Model
Drug molecules are non-Euclidean chemical structures that consist of entities (atoms) and relations (bonds) with rich semantic information and complex spatial structures. This is essential for accurately discriminating between drug molecules and precisely predicting the binding affinity of different compound molecules with proteins. Nevertheless, that is beyond the reach of traditional GNNs.
Meanwhile, we take into account that drugs with similar substructures may react pharmacologically with target proteins with the same or similar protein binding pockets. Interestingly, graph isomorphism networks [
22] with injectivity broadly follow a flexible message-passing scheme that enables atoms to recursively update semantic information through aggregating near and far neighboring atomic features. A sufficient number of iterations allows the GIN to be perfectly equipped with the most powerful ability to “read-out” drug graph representations and identify drug molecules.
GIN updates atom feature vectors via the MLPs, ensuring that GIN still satisfies injectivity after K-iterations of aggregation. The graph representation is obtained by summing all of the atom feature vectors in the drug. Formally, the kernel function of GINs updates atom feature vector
, and the drug graph representation
is:
where
is a set of nodes adjacent to atom
i. The
function is a graph-level pooling function. We made
a learnable parameter.
The successful construction of deep GINs is highly dependent on the ReLU activation function and batch normalization, while batch normalization can effectively alleviate the vanishing gradient and over-smoothing problems.
where
denotes node-level batch normalization.
2.4. Drug Molecule and Target Protein Interaction Model
Drug molecules binding to target proteins is actually an identification relationship similar to the “lock and key” model. Inspired by previous attention-based methods [
13,
17,
30], we constructed two cross-multi-head attention modules to model non-covalent molecular docking behavior between compounds and proteins, instead of simply connecting drug and protein representations that inherently generates more intrusive information. Concretely, we observed the associations among molecules’ substructures, subsequences, and residues from multiple independent perspectives. The cross-multi-head attention blocks take the drug and protein sequences feature matrices
and
of SENets, and the drug graph-level representation
of the GIN as inputs, respectively.
In the following paragraphs, we construct learnable linear transition layers so that each head can fully learn from the high-dimensional features. Afterward, we combine
,
with
by adopting the cross-multi-head attention mechanism.
where
,
, and
,
are the learnable weights and bias terms, respectively.
Q,
K, and
V represent queries, keys, and values vectors. An individual scaled dot–product attention module was expressed as mapping the
Q with
K-
V pairs to the similarity matrix. Multi-head attention jointly concerned different representation subspaces at distinct positions by concatenating
h individual attention units [
31].
We obtained one of the cross-multi-head attention weight
as follows:
where
,
,
, and
are parameter matrices for learning linear projections. Next, another cross-multi-head attention coefficient
was computed as:
Afterward, we decoupled the attention weight
to obtain drug attention weight
and protein attention weight
by applying row-wise sum and column-wise sum operations. We updated the drug representation
and protein representation
.
where ⊙ is an element-wise product. The drug–target interaction weight
can be interpreted as modeling the significant semantic correlations between target proteins and compound features.
where
is the global average pooling operation.
2.5. Drug and Target Protein Binding Affinity Prediction
Finally, interaction information
was fed directly into MLPs to map the drug–target affinity score. Here, this MLPs consists of four layers, each followed by a ReLU and dropout layer, which are applied to alleviate the model from over-fitting.
5. Case Study on the NSCLC with Mutation
According to the statistics of cancer data in 2021 [
37], lung cancer mortality increased to around 46% of total cancer mortality, among which NSCLC accounted for approximately 85% of lung malignancies. Patients with NSCLC are normally accompanied by epidermal growth factor receptor (EGFR) mutations [
38], which brings great challenges to the treatment of NSCLC. In recent years, the remarkable achievements of small-molecule EGFR tyrosine kinase inhibitors (EGFR-TKIs) in targeted therapy have brought light to NSCLC patients. First-generation EGFR-TKIs (Gefitinib and Erlotinib) and second-generation EGFR-TKI (Afatinib) significantly improved the prognosis of advanced NSCLC patients compared to platinum-based chemotherapy. Unfortunately, the majority of patients develop
mutation, resulting in severe resistance symptoms [
39]. Inevitably, despite the high selectivity of the third-generation EGFR-TKI (Osimertinib) targeting NSCLC harboring
mutation, patients develop secondary resistance [
40].
Natural products continue to be a precious source of templates with structural complexity and numerous pharmacophores in drug R&D, especially effective in cancer. For instance, paclitaxel [
41] and vincristine [
42] have been widely invested in the clinical treatment of tumors. In this section, we preferred to screen high-affinity and good properties targeted inhibitors of NSCLC with
mutation from natural products. We hope our results may provide clues for medical scientists to develop highly selective natural drugs.
For the above purpose, we acquired the FASTA sequence of mutant protein
(PDB ID:2JIT [
28]) from the Protein Data Bank [
43] and collected 2645 natural compounds from Selleck Chemicals
https://www.selleck.cn/ (accessed on 4 January 2023), which are easily optimized for good human oral bioavailability (OB > 40%) and drug-likeness (DL > 0.18) [
44,
45].
Table 8 provides information on the top 10 natural products predicted by DoubleSG-DTA, which have the highest affinity to the
mutant protein.
Then, we carried out a comprehensive literature survey on the top 10 natural products. Based on the study [
46], gossypol not only significantly increased the sensitivity to EGFR-TKIs in H1975 cells carrying
, but inhibited cell proliferation and induced apoptosis. The Gö6976 is derived from Staurosporine, experimental confirmation that Gö6976 (at 500 nanomolar) exhibits significant binding affinity for
mutants, while it shows a significantly lower affinity for wild-type EGFR [
47]. The research results indicate that Shikonin has selective cytotoxic effects on gefitinib-resistant NSCLC cell lines carrying
mutation, while relatively safe to normal lung cells [
48]. Gossypol acetic acid significantly enhances sensitized lung cancer cells carrying
mutation to gefitinib and overcomes EGFR-TKIs resistance [
49,
50]. According to the above-mentioned report, such natural products may be promising strategies to combat resistance in NSCLC harboring
mutation.
Table 8.
Docking information of the top 10 natural products with the highest affinity.
Table 8.
Docking information of the top 10 natural products with the highest affinity.
| Natural Products | MF | MW | H-Bonds | Binding-Energy (KJ/mol) |
|---|
| Gossypol [46] | C30H30O8 | 518.60 | 4 | −12.636 |
| Gossypol acetic acid [50] | C32H34O10 | 578.60 | 3 | −14.644 |
| Staurosporine [47] | C28H26N4O3 | 466.50 | 3 | −18.744 |
| Emodin | C15H10O5 | 270.24 | 4 | −13.933 |
| Physcion | C16H12O5 | 284.26 | 3 | −16.862 |
| Aurantio-obtusin | C17H14O7 | 330.29 | 4 | −17.531 |
| Shikonin [48] | C16H16O5 | 288.29 | 3 | −13.180 |
| Rhein | C15H8O6 | 284.22 | 6 | −16.192 |
| Obtusifolin | C16H12O5 | 284.26 | 3 | −15.104 |
| Chrysophanol | C15H10O4 | 254.24 | 5 | −16.025 |
6. Molecular Docking and Biological Interpretation
To further validate such new interactions, computational docking was performed via AutoDock [
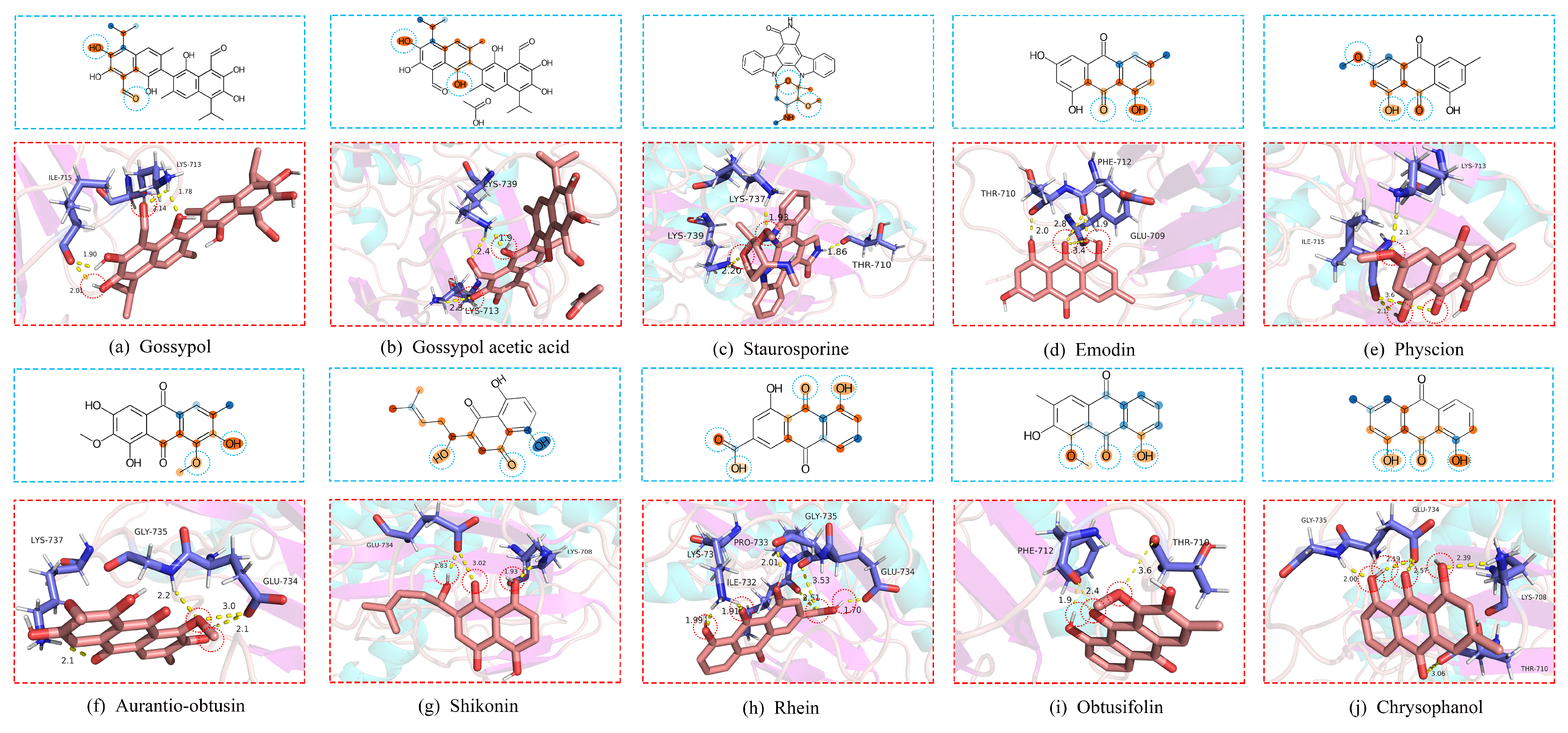
51]. As shown in
Figure 4, we employed the most efficient, reliable, and successful Lamarckian genetic algorithm in Autodock to perform an adaptive global–local search for the lowest-energy ligand–receptor docked conformation, and predicted the binding free energy via an empirical binding free energy force field [
52]. The ligand–receptor binding energy includes electrostatic interactions, hydrogen bonding, van der Waals forces and hydrophobic interactions, and so forth, and the structural stability is negatively correlated with the binding energy value. Furthermore, an acceptable molecular docking conformation that has a binding energy of less than −5.0208 KJ/mol. Drug molecule ligands interact stably with target proteins in the above manner, aiming to exert a variety of biological activities such as anti-inflammatory and anti-tumor activities of the drug molecules, and to stimulate the physiological and pharmacological functions of the protein. As shown in
Figure 4 and
Table 8, the docking indicates that the top 10 natural compounds can be stably docked to the
protein by generating multiple hydrogen bonds.
Graph neural networks have always been criticized because of their poor interpretability, and these models are commonly thought of as “black boxes”. In this work, inspired by Grad-AAM [
20] and Grad-CAM [
53], which employed the gradient-weighted class activation mapping method, the regions of graph structure that contribute most to the prediction results are visualized as heatmaps, enhancing the interpretability of deep learning-based network models processing graph data.
Since the last layer of the GINs of DoubleSG-DTA incorporates the richest high-level semantic information, the drug graph representations are visualized to produce heatmaps depicting the atoms and functional groups that contribute most prominently to predicting DTA. We denote the feature map of the last graph convolution layer as
F. In order to obtain the probability map
P of atomic node
v for a given drug molecule, we calculate the gradient of the predicted affinity
of the molecule binding to the target protein at the
c-th channel of the feature map
F and atomic node
v. The gradient
has been calculated as follows.
Next, a weighted combination of the data for each channel of the feature map F was performed, followed by the ReLU activation function.
Finally, the gradient weights were scaled to the range of 0 to 1 using min–max normalization to obtain a probability map P of the weighted distribution of the drug molecules, which was further rendered into a heatmap.
As shown in
Figure 4, the active structures in the heatmaps overlap with molecular docking sites by more than 77.14%, and the mathematical calculation formulation is given as Equation (
23).
Figure 4 explains that describing the drug molecules as graphs and learning the topological pattern structures of the drug molecules with an appropriate depth of GIN can accurately discriminate between drug molecular active structures.
where
N denotes the number of drugs,
stands for the number of molecular docking sites, and
is the number of atoms and functional groups that contributes the most and is identical to the molecular docking site.
7. Conclusions
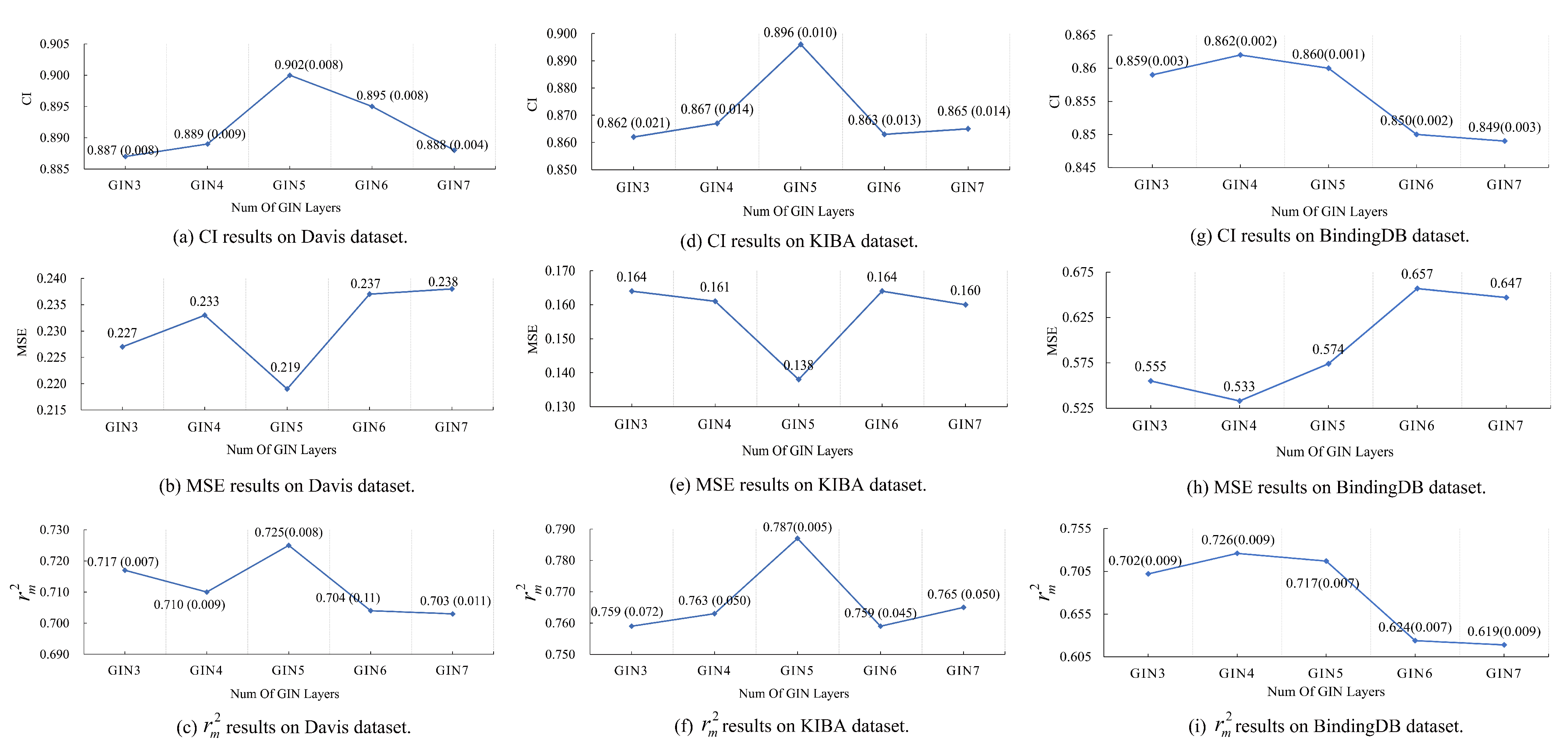
This investigation presented an interpretable deep learning-based computational model to project the affinity of drug–target pairs for aiding in drug discovery. The experimental results indicated that the simple yet powerful graph isomorphism networks coupled with the lightweight squeeze-and-excitation networks made the DoubleSG-DTA perform exceptionally well with the support of cross-multi-head attention compared with all previously reported works. Extensive experiments have revealed that (i) the most appropriate number of graph isomorphism network layers for extracting drug graph representations and discriminating between molecular structures is , (ii) the SE block with the soft attention mechanism selectively emphasized information features by expanding the perceptual field, significantly boosting the model’s decision making, and (iii) fully modeling the interaction between compounds and proteins facilitates further performance in predicting drug–target binding affinity. Ultimately, the well-established DoubleSG-DTA was applied to screen promising high-affinity compounds of Non-Small Cell Lung Cancer with mutation from natural products to provide some clues for medical scientists. In addition, drug graph representations were visualized as heatmaps, in which the active structures that contributed the most covered almost all molecular docking sites, which may provide biological interpretation and entry points for later molecular optimization. Overall, DoubleSG-DTA may be an effective in silico drug discovery tool for medical challenges and urgent public health emergencies.



{kind=link}
{kind=link}
{kind=link}
{kind=link}



