
Drugging Hijacked Kinase Pathways in Pediatric Oncology: Opportunities and Current Scenario
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Pediatric Cancer
2. Kinases as Cancer Drivers
3. Protein Kinases in Pediatric Oncology and Their Association with Tumor Prognosis
3.1. Published Evidence of Kinase Dysregulation in Pediatric Oncology
3.1.1. Receptor Tyrosine Kinases (RTK)
3.1.2. PI3K/AKT/mTOR Pathway
3.1.3. MAPK Pathway
3.1.4. Cell Cycle Kinases
Cyclin-Dependent Kinases
Polo-Like Kinases
Aurora Kinases
3.2. In Silico Analysis of Different Kinases Expression and Their Association with Clinical Prognosis
4. Kinases as Druggable Targets—Evidence and Limitations
4.1. RTK Inhibitors
4.2. PI3K/AKT/mTOR Pathway Inhibitors
4.3. MAPK Pathway Inhibitors
4.4. Cyclin-Dependent Kinases Inhibitors
4.5. Polo-Like and Aurora Kinases Inhibitors
5. Kinase Inhibitors in Clinical Trials
5.1. TRK—Tyrosine Receptor Kinases
5.1.1. EGFR and VEGFR
5.1.2. RET Inhibitors
5.1.3. ALK Inhibitors
5.2. PI3K/AKT/mTOR Pathway
5.3. MAPK Pathway
5.4. Cell Cycle Kinases
6. Final Remarks
- (1)
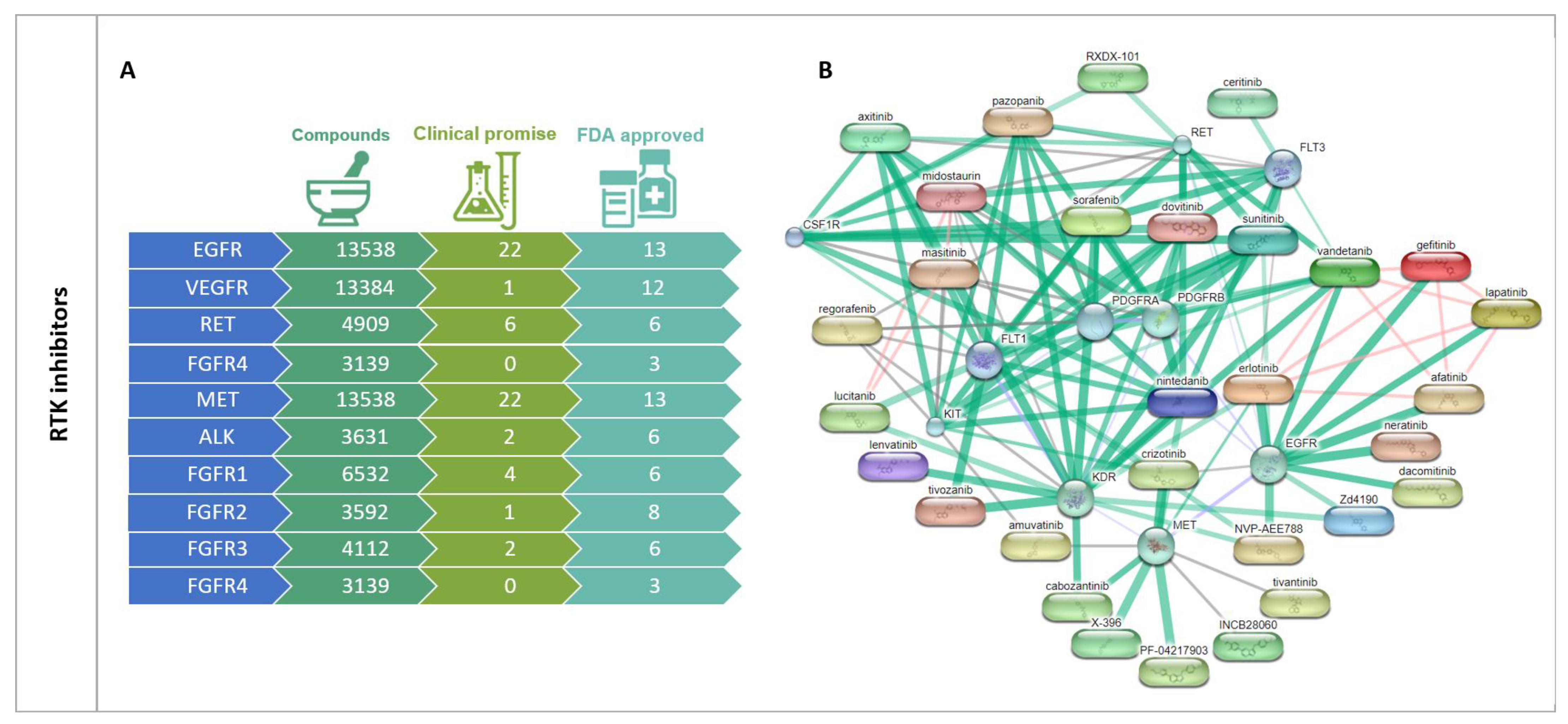
- Selectivity: Most inhibitors developed so far target the ATP-binding site, meaning that they may act on multiple targets simultaneously and open new opportunities for the treatment of different tumor histologies [951,952]. Imatinib, for example, which has led to a significant increase in CML survival rates by selectively targeting the tumor-specific protein BCR/ABL, was included for the treatment of gastrointestinal stromal tumors (GIST), which are characterized by KIT-activating mutations [953]. Nevertheless, most inhibitors discovered to date have faced several adversities limiting their clinical use. First of all, the high sequence similarity in the ATP-binding sites frequently results in poor selectivity (refer to Figure 5 and Supplementary Figure S3) that may lead to undesired side effects. Moreover, these small molecules must compete with high intracellular ATP levels, leading to differences in potency when measured in vivo by biochemical versus cellular assays. In fact, many compounds inhibit their enzymes at nanomolar concentrations when measured biochemically, but only inhibit tumor cell growth under 3-fold higher concentrations [954]. Nevertheless, the increasing number of recognized kinase-specific structural features has allowed the emergence of superior non-ATP competitive kinase inhibitors that target other allosteric sites, which mostly act by inducing a conformational shift in the target enzyme, depleting its function [951,955,956,957].
- (2)
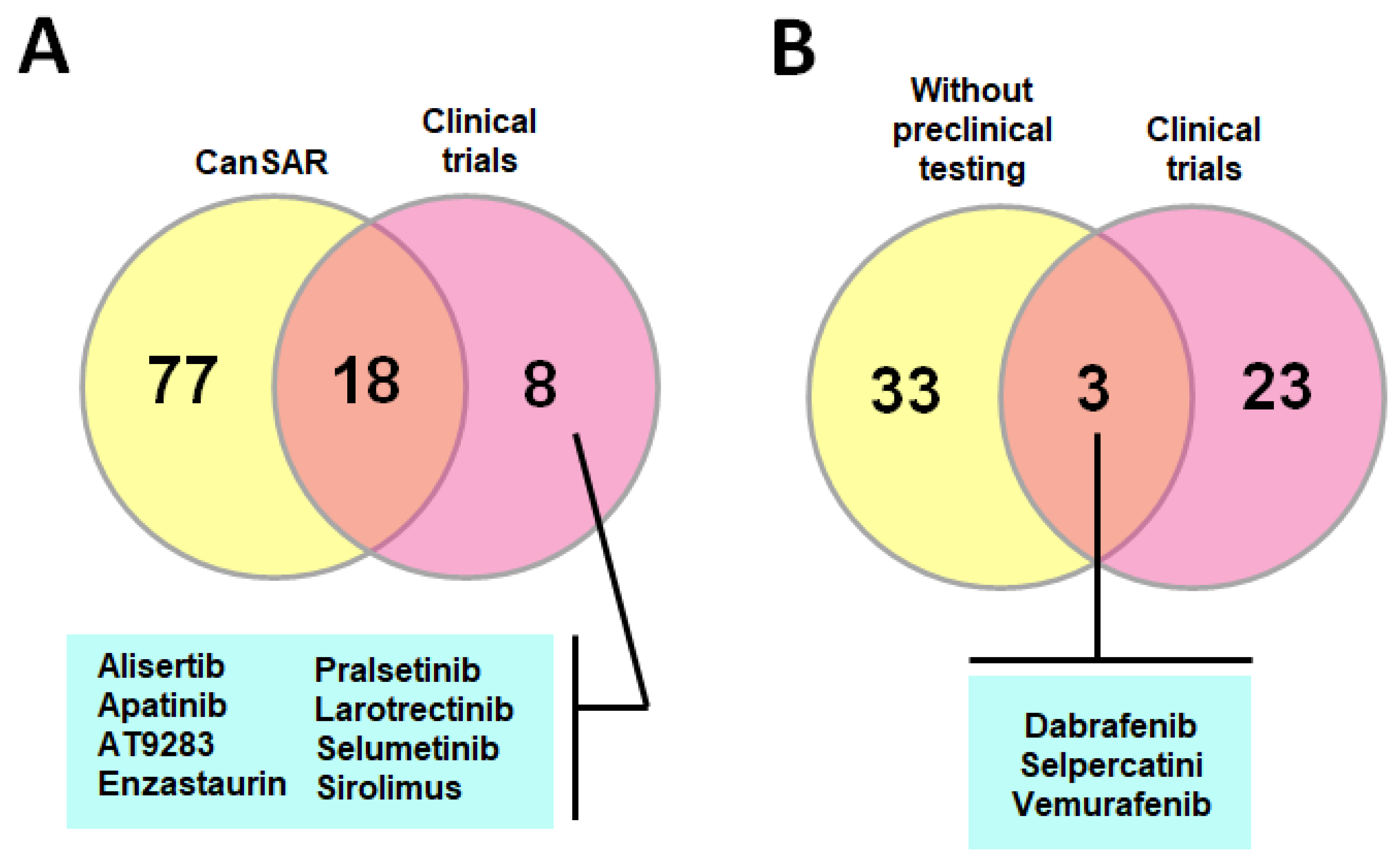
- Adverse effects: Imatinib and dasatinib, for instance, are both licensed for the treatment of children with CML. Despite its undeniable benefits, and with the spectrum of side effects being comparable to what has been reported in adults (i.e., gastrointestinal toxicity, skin rash and muscle cramps), in a growing organism, imatinib treatment impairs longitudinal growth through the disturbance of osseous remodeling and inhibition of growth hormone secretion, which raises concerns about its lifelong use [958]. Moreover, despite the wealth of compounds that emerge on a daily basis, showing selectivity, potency and favorable pharmacological profiles, the probabilities for the translation into effective patient treatment for the great majority of them are extremely low. In this regard, less than 30% of the compounds approved or with clinical promise retrieved from the CanSAR platform have entered clinical trials in the pediatric setting (refer to Figure 6A). PLK1 inhibitors, for instance, despite the robust results obtained in vitro and in vivo, have demonstrated poor applicability due to severe hematological toxicity [959].
- (3)
- Mutational burden and lack of predictive biomarkers: As stated before, the mutational identity may vary between adult and pediatric cancer, a feature that reflects in treatment response. Current treatments targeting ALK mutations in other cancers, for example, have not shown significant efficacy against NB. In this tumor, two hotspot mutations, at positions R1275Q and F1174L, occur in a high proportion of patients; tumors harboring the first are highly sensitive to crizotinib, while tumors bearing the second are resistant [960,961,962]. Moreover, as suggested by Bellantoni and Wagner (2021), childhood solid tumors may have fewer potentially targetable mutations, evinced by the inhibition of RTK for the treatment of OS, where it is necessary to target several relevant RTKs simultaneously to achieve desirable results [821]. Therefore, ground-breaking drugs for adult cancer may not be effective in the pediatric setting.In parallel, inhibitors are not effective if the target is not essential to drive tumor growth or does not represent a prognostic factor, as is the case of ROCK kinases. Even though these proteins have gained popularity and progressively been researched as targets for the development of novel anti-cancer drugs due to their association with metastasis and poorer patient survival in adult tumors, the influence of both isoforms on the prognosis of childhood cancer remains controversial [963].
- (4)
- Intrinsic and acquired resistance: Resistance to targeted therapies is considered a largely inevitable hurdle that has a substantial impact on patients. Refractoriness to chemotherapy due to acquired F1174S ALK mutation in NB has been reported [964]. Likewise, the location of EGFR mutations significantly changes the effectiveness of EGFR; several mutations conferring resistance to EGFR tyrosine kinase inhibitors (such as T790M, L833V, A839T, V851I, A871T and G873E) have been reported [938]. Other examples include inadequate response to imatinib due to BCR-ABL1 kinase domain mutations that impart varying degrees of drug insensitivity, observed as underlying mechanism in 5–10% of adults and children with CML, bypassing pathway activation [965,966]. Moreover, despite an initial benefit of the targeted drug in molecularly well-defined tumors, patients inevitably experience tumor progression due to the development of resistance (i.e., Crizotinib in ALK-rearranged NSCLC population and CNS relapses) [967].
- (5)
- Lack of compounds designed specifically for childhood tumors: In general, few pediatric patients with cancer are enrolled in clinical trials. The perception that adult studies can be generalized to children with similar diseases is a major obstacle. Consequently, most treatments are based on modifications of previously approved regimes for the adult population, and many compounds enter clinical trials without preclinical testing in pediatric oncology (refer to Figure 6B), which is mandatory to obtain a more accurate interpretation of its possible therapeutic potential in a certain cancer entity [968]. Moreover, pediatric cancer is rare, and even among patients with the same cancer type, there is often broad heterogeneity in terms of prognosis, molecular features or pathology. Therefore, few institutions have sufficient patients and the chances of every potential agent or combination being tested are reduced. Even so, priorities for funding are typically assessed according to the “burden of illness” for diseases, which is traditionally determined by disease frequency and mortality rate, leading to reluctance to distribute limited research funding to pediatric trials [969,970,971].
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steliarova-Foucher, E.; Stiller, C.; Lacour, B.; Kaatsch, P. International Classification of Childhood Cancer, third edition. Cancer 2005, 103, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Instituto Nacional de Câncer (Brazil). Coordenação de Prevenção e Vigilância and Sociedade Brasileira de Oncologia Pediátrica, Câncer na Criança e no Adolescente no Brasil: Dados dos Registros de Base Populacional e de Mortalidade; Ministério da Saúde, Instituto Nacional de Câncer–INCA: Brasília, Brazil, 2008. [Google Scholar]
- Downing, J.R.; Wilson, R.K.; Zhang, J.; Mardis, E.R.; Pui, C.-H.; Ding, L.; Ley, T.J.; E Evans, W. The Pediatric Cancer Genome Project. Nat. Genet. 2012, 44, 619–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toren, A.; Rechavi, G.; Ramot, B. Pediatric Cancer: Environmental and Genetic Aspects. Pediatr. Hematol. Oncol. 1996, 13, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Denniston, K.A.; Lin, C.; Lin, C. A Comparison of Pediatric vs. Adult Patients with the Ewing Sarcoma Family of Tumors. Front. Oncol. 2017, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing Adult and Pediatric Rhabdomyosarcoma in the Surveillance, Epidemiology and End Results Program, 1973 to 2005: An Analysis of 2,600 Patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef]
- Spector, L.; Brown, M.B.; Wantman, E.; Letterie, G.S.; Toner, J.P.; Doody, K.; Ginsburg, E.; Williams, M.; Koch, L.; Schymura, M.J.; et al. Association of In Vitro Fertilization With Childhood Cancer in the United States. JAMA Pediatr. 2019, 173, e190392. [Google Scholar] [CrossRef]
- Rahal, Z.; Abdulhai, F.; Kadara, H.; Saab, R. Genomics of adult and pediatric solid tumors. Am. J. Cancer Res. 2018, 8, 1356–1386. [Google Scholar]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Vellichirammal, N.N.; Chaturvedi, N.K.; Joshi, S.S.; Coulter, D.W.; Guda, C. Fusion genes as biomarkers in pediatric cancers: A review of the current state and applicability in diagnostics and personalized therapy. Cancer Lett. 2020, 499, 24–38. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated Molecular Genetic Profiling of Pediatric High-Grade Gliomas Reveals Key Differences With the Adult Disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef] [Green Version]
- Appay, R.; Fina, F.; Macagno, N.; Padovani, L.; Colin, C.; Barets, D.; Ordioni, J.; Scavarda, D.; Giangaspero, F.; Badiali, M.; et al. Duplications of KIAA1549 and BRAF screening by Droplet Digital PCR from formalin-fixed paraffin-embedded DNA is an accurate alternative for KIAA1549-BRAF fusion detection in pilocytic astrocytomas. Mod. Pathol. 2018, 31, 1490–1501. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, K.; on behalf of the Japan Pediatric Molecular Neuro-Oncology Group (JPMNG); Kanemura, Y.; Shofuda, T.; Fukushima, S.; Yamashita, S.; Narushima, D.; Kato, M.; Honda-Kitahara, M.; Ichikawa, H.; et al. Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol. Commun. 2018, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Wachtel, M.; Schäfer, B.W. PAX3-FOXO1: Zooming in on an “undruggable” target. Semin. Cancer Biol. 2018, 50, 115–123. [Google Scholar] [CrossRef]
- Giovannini, M.; Biegel, J.A.; Serra, M.; Wang, J.Y.; Wei, Y.H.; Nycum, L.; Emanuel, B.S.; Evans, G.A. EWS-erg and EWS-Fli1 fusion transcripts in Ewing’s sarcoma and primitive neuroectodermal tumors with variant translocations. J. Clin. Investig. 1994, 94, 489–496. [Google Scholar] [CrossRef]
- Jemal, A.; Murray, T.; Samuels, A.; Ghafoor, A.; Ward, E.; Thun, M.J. Cancer Statistics, 2003. CA A Cancer J. Clin. 2003, 53, 5–26. [Google Scholar] [CrossRef]
- Pui, C.-H. Recent Research Advances in Childhood Acute Lymphoblastic Leukemia. J. Formos. Med. Assoc. 2010, 109, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Pezuk, J.A.; Valera, E.T.; Brassesco, M.S. PLK1 Inhibition: Prospective Role for the Treatment of Pediatric Tumors. Curr. Drug Targets 2016, 17, 1661–1672. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theivendren, P.; Kunjiappan, S.; Hegde, Y.M.; Vellaichamy, S.; Gopal, M.; Dhramalingam, S.R.; Kumar, S. Importance of Protein Kinase and Its Inhibitor: A Review. Protein Kinases-Promis Targets Anticance. Drug Res. IntechOpen Ser. Biochem. 2021, 24, 75–100. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Bruce Alberts, Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Turdo, A.; D’Accardo, C.; Glaviano, A.; Porcelli, G.; Colarossi, C.; Colarossi, L.; Mare, M.; Faldetta, N.; Modica, C.; Pistone, G.; et al. Targeting Phosphatases and Kinases: How to Checkmate Cancer. Front. Cell Dev. Biol. 2021, 9, 690306. [Google Scholar] [CrossRef] [PubMed]
- Giamas, G.; Man, Y.L.; Hirner, H.; Bischof, J.; Kramer, K.; Khan, K.; Ahmed, S.S.L.; Stebbing, J.; Knippschild, U. Kinases as targets in the treatment of solid tumors. Cell Signal. 2010, 22, 984–1002. [Google Scholar] [CrossRef]
- Armstrong, H.; Bording-Jorgensen, M.; Dijk, S.; Wine, E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers 2018, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Bhattacharya, B.; Das, B.; Sinha, B.; Jamatia, T.; Paul, K. Etiologic Role of Kinases in the Progression of Human Cancers and Its Targeting Strategies. Indian J. Surg. Oncol. 2019, 12, 34–45. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R.; Miller, W.T. Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Biol. 2007, 19, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Popovic, N.; Wilson, E. Cell Surface Receptors. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 81–91. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Böhmer, F.-D. Mislocalisation of Activated Receptor Tyrosine Kinases—Challenges for Cancer Therapy. Trends Mol. Med. 2020, 26, 833–847. [Google Scholar] [CrossRef]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR gene amplification in breast cancer: Correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod. Pathol. 2005, 18, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Cappuzzo, F.; Ou, S.-H.I.; Camidge, D.R. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2018, 16, 105–122. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chen, T.; Ding, Z.; Wang, Y.; Wei, Y.; Wei, X. Inhibition of FGF-FGFR and VEGF-VEGFR signalling in cancer treatment. Cell Prolif. 2021, 54, e13009. [Google Scholar] [CrossRef]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Rivera, B.; Gayden, T.; Carrot-Zhang, J.; Nadaf, J.; Boshari, T.; Faury, D.; Zeinieh, M.; Blanc, R.; Burk, D.L.; Fahiminiya, S.; et al. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol. 2016, 131, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Valera, E.T.; McConechy, M.K.; Gayden, T.; Rivera, B.; Jones, D.T.W.; Wittmann, A.; Han, H.; Bareke, E.; Nikbakht, H.; Mikael, L.; et al. Methylome analysis and whole-exome sequencing reveal that brain tumors associated with encephalocraniocutaneous lipomatosis are midline pilocytic astrocytomas. Acta Neuropathol. 2018, 136, 657–660. [Google Scholar] [CrossRef]
- Amary, M.F.; Ye, H.; Berisha, F.; Khatri, B.; Forbes, G.; Lehovsky, K.; Frezza, A.M.; Behjati, S.; Tarpey, P.; Pillay, N.; et al. Fibroblastic growth factor receptor 1 amplification in osteosarcoma is associated with poor response to neo-adjuvant chemotherapy. Cancer Med. 2014, 3, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Berlow, N.E.; Lathara, M.; Bharathy, N.; Martin, L.R.; Purohit, R.; Cleary, M.M.; Liu, Q.; Michalek, J.E.; Srinivasa, G.; et al. Sensitization of osteosarcoma to irradiation by targeting nuclear FGFR1. Biochem. Biophys. Res. Commun. 2022, 621, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Elkrief, A.; Bowman, A.S.; Koche, R.P.; de Stanchina, E.; Benayed, R.; Mauguen, A.; Mattar, M.S.; Khodos, I.; Meyers, P.A.; et al. Prospective Clinical Genomic Profiling of Ewing Sarcoma: ERF and FGFR1 Mutations as Recurrent Secondary Alterations of Potential Biologic and Therapeutic Relevance. JCO Precis. Oncol. 2022, 6, e2200048. [Google Scholar] [CrossRef] [PubMed]
- Agelopoulos, K.; Richter, G.H.; Schmidt, E.; Dirksen, U.; von Heyking, K.; Moser, B.; Klein, H.-U.; Kontny, U.; Dugas, M.; Poos, K.; et al. Deep Sequencing in Conjunction with Expression and Functional Analyses Reveals Activation of FGFR1 in Ewing Sarcoma. Clin. Cancer Res. 2015, 21, 4935–4946. [Google Scholar] [CrossRef] [Green Version]
- Rakheja, D.; Park, J.Y.; Yang, M.S.; Martinez, D.P.; Koduru, P.; Wilson, K.S.; Garcia, R.; Uddin, N. Rhabdomyosarcoma With Epithelioid Features And NSD3::FOXO1 Fusion: Evidence For Reconsideration Of Previously Reported FOXO1::FGFR1 Fusion. Int. J. Surg. Pathol. 2022. [Google Scholar] [CrossRef]
- Goldstein, M.; Meller, I.; Orr-Urtreger, A. FGFR1 over-expression in primary rhabdomyosarcoma tumors is associated with hypomethylation of a 5′ CpG Island and abnormal expression of theAKT1,NOG, andBMP4 genes. Genes Chromosom. Cancer 2007, 46, 1028–1038. [Google Scholar] [CrossRef]
- Gasparini, P.; Fortunato, O.; De Cecco, L.; Casanova, M.; Iannó, M.F.; Carenzo, A.; Centonze, G.; Milione, M.; Collini, P.; Boeri, M.; et al. Age-Related Alterations in Immune Contexture Are Associated with Aggressiveness in Rhabdomyosarcoma. Cancers 2019, 11, 1380. [Google Scholar] [CrossRef] [Green Version]
- Missiaglia, E.; Selfe, J.; Pritchard-Jones, K.; Kool, M.; Shipley, J.; Hamdi, M.; Williamson, D.; Schaaf, G.; Fang, C.; Koster, J.; et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: An approach to identify candidate genes involved in tumor development. Genes Chromosom. Cancer 2009, 48, 455–467. [Google Scholar] [CrossRef]
- Lehtinen, B.; Raita, A.; Kesseli, J.; Annala, M.; Nordfors, K.; Yli-Harja, O.; Zhang, W.; Visakorpi, T.; Nykter, M.; Haapasalo, H.; et al. Clinical association analysis of ependymomas and pilocytic astrocytomas reveals elevated FGFR3 and FGFR1 expression in aggressive ependymomas. BMC Cancer 2017, 17, 310. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, F.; Montella, A.; Tirelli, M.; Avitabile, M.; Lasorsa, V.A.; Visconte, F.; Cantalupo, S.; Maiorino, T.; De Angelis, B.; Morini, M.; et al. FGFR1 is a potential therapeutic target in neuroblastoma. Cancer Cell Int. 2022, 22, 174. [Google Scholar] [CrossRef]
- Schmelz, K.; Toedling, J.; Huska, M.; Cwikla, M.C.; Kruetzfeldt, L.-M.; Proba, J.; Ambros, P.F.; Ambros, I.M.; Boral, S.; Lodrini, M.; et al. Spatial and temporal intratumour heterogeneity has potential consequences for single biopsy-based neuroblastoma treatment decisions. Nat. Commun. 2021, 12, 6804. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, S.; Hirato, J.; Yokoo, H. Molecular genetics of ependymomas and pediatric diffuse gliomas: A short review. Brain Tumor Pathol. 2014, 31, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Won, J.; Kim, S.-I.; Lee, Y.; Park, C.-K.; Kim, S.-K.; Choi, S.-H. Molecular Testing of Brain Tumor. J. Pathol. Transl. Med. 2017, 51, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiou, V.; Gkretsi, V. The role of fibroblast growth factors and their receptors in gliomas: The mutations involved. Rev. Neurosci. 2018, 30, 543–554. [Google Scholar] [CrossRef]
- Vega, J.E.V.; Brat, D.J. Incorporating Advances in Molecular Pathology Into Brain Tumor Diagnostics. Adv. Anat. Pathol. 2018, 25, 143–171. [Google Scholar] [CrossRef]
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast Growth Factor Receptor Functions in Glioblastoma. Cells 2019, 8, 715. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, R.; Matsuda, Y.; Ishiwata, T.; Naito, Z. Downregulation of fibroblast growth factor receptor 2 and its isoforms correlates with a high proliferation rate and poor prognosis in high-grade glioma. Oncol. Rep. 2014, 32, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Li, Z.; Zeng, S.; Wang, X.; Gong, Z.; Xu, Z. FGFR2-mediated phosphorylation of PTEN at tyrosine 240 contributes to the radioresistance of glioma. J. Cell Commun. Signal. 2019, 13, 279–280. [Google Scholar] [CrossRef]
- Salm, F.; Cwiek, P.; Ghosal, A.; Buccarello, A.L.; Largey, F.; Wotzkow, C.; Höland, K.; Styp-Rekowska, B.; Djonov, V.; Zlobec, I.; et al. RNA interference screening identifies a novel role for autocrine fibroblast growth factor signaling in neuroblastoma chemoresistance. Oncogene 2012, 32, 3944–3953. [Google Scholar] [CrossRef]
- Kumar, K.S.; Neve, A.; Stucklin, A.S.G.; Kuzan-Fischer, C.M.; Rushing, E.J.; Taylor, M.D.; Tripolitsioti, D.; Behrmann, L.; Kirschenbaum, D.; Grotzer, M.; et al. TGF-β Determines the Pro-migratory Potential of bFGF Signaling in Medulloblastoma. Cell Rep. 2018, 23, 3798–3812.e8. [Google Scholar] [CrossRef]
- Vignovich, J.; Becker, D. Expression of BFGF and differential expression of FGF receptors in normal human myoblasts and rhabdomyosarcomas. Int. J. Oncol. 1993, 2, 637–642. [Google Scholar] [CrossRef]
- Hirotsu, M.; Setoguchi, T.; Matsunoshita, Y.; Sasaki, H.; Nagao, H.; Gao, H.; Sugimura, K.; Komiya, S. Tumour formation by single fibroblast growth factor receptor 3-positive rhabdomyosarcoma-initiating cells. Br. J. Cancer 2009, 101, 2030–2037. [Google Scholar] [CrossRef] [Green Version]
- Sahu, D.K.; Singh, N.; Das, M.; Rawat, J.; Gupta, D.K. Differential expression profiling of onco and tumor-suppressor genes from major-signaling pathways in Wilms’ tumor. Pediatr. Surg. Int. 2022, 38, 1601–1617. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Holzhauser, S.; Lange, B.; Ohmayer, A.; Andonova, T.; Bersani, C.; Dalianis, T. Analyses of FGFR3 and PIK3CA mutations in neuroblastomas and the effects of the corresponding inhibitors on neuroblastoma cell lines. Int. J. Oncol. 2019, 55, 1372–1384. [Google Scholar] [CrossRef]
- Ahrendsen, J.T.; Sinai, C.; Meredith, D.M.; Malinowski, S.W.; Cooney, T.M.; Bandopadhayay, P.; Ligon, K.L.; Alexandrescu, S. Molecular Alterations in Pediatric Low-Grade Gliomas That Led to Death. J. Neuropathol. Exp. Neurol. 2021, 80, 1052–1059. [Google Scholar] [CrossRef]
- Johnson, A.; Severson, E.; Gay, L.; Vergilio, J.-A.; Elvin, J.; Suh, J.; Daniel, S.; Covert, M.; Frampton, G.M.; Hsu, S.; et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist 2017, 22, 1478–1490. [Google Scholar] [CrossRef] [Green Version]
- Frattini, V.; Pagnotta, S.M.; Tala; Fan, J.J.; Russo, M.V.; Lee, S.B.; Garofano, L.; Zhang, J.; Shi, P.; Lewis, G.; et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018, 553, 222–227. [Google Scholar] [CrossRef]
- Kamura, S.; Matsumoto, Y.; Fukushi, J.-I.; Fujiwara, T.; Iida, K.; Okada, Y.; Iwamoto, Y. Basic fibroblast growth factor in the bone microenvironment enhances cell motility and invasion of Ewing’s sarcoma family of tumours by activating the FGFR1–PI3K–Rac1 pathway. Br. J. Cancer 2010, 103, 370–381. [Google Scholar] [CrossRef]
- Lee, S.; Lim, S.; Cho, D. Personalized genomic analysis based on circulating tumor cells of extra-skeletal Ewing sarcoma of the uterus: A case report of a 16-year-old Korean female. Exp. Ther. Med. 2018, 16, 1343–1349. [Google Scholar] [CrossRef]
- Li, Z.; Dou, P.; Liu, T.; He, S. Application of Long Noncoding RNAs in Osteosarcoma: Biomarkers and Therapeutic Targets. Cell. Physiol. Biochem. 2017, 42, 1407–1419. [Google Scholar] [CrossRef]
- Ren, T.; Qing, Y.; Dai, N.; Li, M.; Qian, C.; Yang, Y.; Cheng, Y.; Li, Z.; Zhang, S.; Zhong, Z.; et al. Apurinic/apyrimidinic endonuclease 1 induced upregulation of fibroblast growth factor 2 and its receptor 3 induces angiogenesis in human osteosarcoma cells. Cancer Sci. 2014, 105, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Jing, Y.; Cao, Y. Overexpression of miR-100 inhibits growth of osteosarcoma through FGFR3. Tumor Biol. 2015, 36, 8405–8411. [Google Scholar] [CrossRef]
- Gabler, L.; Jaunecker, C.N.; Katz, S.; van Schoonhoven, S.; Englinger, B.; Pirker, C.; Mohr, T.; Vician, P.; Stojanovic, M.; Woitzuck, V.; et al. Fibroblast growth factor receptor 4 promotes glioblastoma progression: A central role of integrin-mediated cell invasiveness. Acta Neuropathol. Commun. 2022, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Guo, Z.; Sun, H.; Liu, X.; Zhang, Y.; Zhang, L.; Sun, W.; Tian, Y. Urinary protein biomarkers for pediatric medulloblastoma. J. Proteom. 2020, 225, 103832. [Google Scholar] [CrossRef] [PubMed]
- El Demellawy, D.; McGowan-Jordan, J.; de Nanassy, J.; Chernetsova, E.; Nasr, A. Update on molecular findings in rhabdomyosarcoma. Pathology 2017, 49, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Taylor Vi, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef] [Green Version]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Baird, K.; Davis, S.; Antonescu, C.R.; Harper, U.L.; Walker, R.L.; Chen, Y.; Glatfelter, A.A.; Duray, P.H.; Meltzer, P.S. Gene Expression Profiling of Human Sarcomas: Insights into Sarcoma Biology. Cancer Res 2005, 65, 9226–9235. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-Wide Identification of PAX3-FKHR Binding Sites in Rhabdomyosarcoma Reveals Candidate Target Genes Important for Development and Cancer. Cancer Res 2010, 70, 6497–6508. [Google Scholar] [CrossRef] [Green Version]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Whittle, S.B.; Reyes, S.; Du, M.; Gireud-Goss, M.; Zhang, L.; Woodfield, S.E.; Ittmann, M.; Scheurer, M.; Bean, A.J.; Zage, P.E. A Polymorphism in the FGFR4 Gene Is Associated With Risk of Neuroblastoma and Altered Receptor Degradation. J. Pediatr. Hematol. 2016, 38, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, N.; Varjosalo, M.; Meller, P.; Lohi, J.; Chan, K.M.; Zhou, Z.; Alitalo, K.; Taipale, J.; Keski-Oja, J.; Lehti, K. FGF receptor-4 (FGFR4) polymorphism acts as an activity switch of a membrane type 1 matrix metalloproteinase–FGFR4 complex. Proc. Natl. Acad. Sci. USA 2010, 107, 15786–15791. [Google Scholar] [CrossRef] [Green Version]
- Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Nicholson, R.; Gee, J.; Harper, M. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor α and Poly (ADP-ribose) Polymerase As Potential Therapeutic Targets. J. Clin. Oncol. 2010, 28, 1337–1344. [Google Scholar] [CrossRef]
- Suri, V.; Das, P.; Jain, A.; Sharma, M.C.; Borkar, S.A.; Suri, A.; Gupta, D.; Sarkar, C. Pediatric glioblastomas: A histopathological and molecular genetic study. Neuro-Oncology 2009, 11, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef] [Green Version]
- Kraus, J.A.; Felsberg, J.; Tonn, J.C.; Reifenberger, G.; Pietsch, T. Molecular genetic analysis of the TP53, PTEN, CDKN2A, EGFR, CDK4 and MDM2 tumour-associated genes in supratentorial primitive neuroectodermal tumours and glioblastomas of childhood. Neuropathol. Appl. Neurobiol. 2002, 28, 325–333. [Google Scholar] [CrossRef]
- Pollack, I.F.; Hamilton, R.L.; James, C.D.; Finkelstein, S.D.; Burnham, J.; Yates, A.J.; Holmes, E.J.; Zhou, T.; Finlay, J.L. Rarity ofPTENdeletions andEGFRamplification in malignant gliomas of childhood: Results from the Children’s Cancer Group 945 cohort. J. Neurosurgery Pediatr. 2006, 105, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganigi, P.; Santosh, V.; Anandh, B.; Chandramouli, B.; Kolluri, V.S. Expression of p53, EGFR, pRb and bcl-2 Proteins in Pediatric Glioblastoma Multiforme: A Study of 54 Patients. Pediatr. Neurosurg. 2005, 41, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Hill, D.A. ERBB1 is amplified and overexpressed in high-grade diffusely infiltrative pediatric brain stem glio-ma. Clin. Cancer Res. 2003, 9 Pt 1, 3620–3624. [Google Scholar] [PubMed]
- Korshunov, A.; Golanov, A.; Timirgaz, V. Immunohistochemical markers for intracranial ependymoma recurrence: An analysis of 88 cases. J. Neurol. Sci. 2000, 177, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Mendrzyk, F.; Korshunov, A.; Benner, A.; Toedt, G.; Pfister, S.; Radlwimmer, B.; Lichter, P. Identification of Gains on 1q and Epidermal Growth Factor Receptor Overexpression as Independent Prognostic Markers in Intracranial Ependymoma. Clin. Cancer Res. 2006, 12, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Massimino, M.; Buttarelli, F.R.; Antonelli, M.; Gandola, L.; Modena, P.; Giangaspero, F. Intracranial ependymoma: Factors affecting outcome. Futur. Oncol. 2009, 5, 207–216. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Perry, R.H.; Kelly, P.J.; Pearson, A.D.; Lunec, J. Prognostic significance of HER2 and HER4 coexpres-sion in childhood medulloblastoma. Cancer Res. 1997, 57, 3272–3280. [Google Scholar]
- Bodey, B.; E Kaiser, H.; E Siegel, S. Epidermal growth factor receptor (EGFR) expression in childhood brain tumors. Vivo 2005, 19, 931–941. [Google Scholar]
- Layfield, L.J.; Thompson, J.K.; Ms, R.K.D.; Kerns, B.-J. Prognostic indicators for neuroblastoma: Stage, grade, DNA ploidy, MIB-1-proliferation index, p53, HER-2/neu and EGFr–a survival study. J. Surg. Oncol. 1995, 59, 21–27. [Google Scholar] [CrossRef]
- Izycka-Swieszewska, E.; Wozniak, A.; Drozynska, E.; Kot, J.; Grajkowska, W.; Klepacka, T.; Perek, D.; Koltan, S.; Bien, E.; Limon, J. Expression and significance of HER family receptors in neuroblastic tumors. Clin. Exp. Metastasis 2011, 28, 271–282. [Google Scholar] [CrossRef]
- Izycka-Swieszewska, E.; Wozniak, A.; Kot, J.; Grajkowska, W.; Balcerska, A.; Perek, D.; Dembowska-Baginska, B.; Klepacka, T.; Drozynska, E. Prognostic significance of HER2 expression in neuroblastic tumors. Mod. Pathol. 2010, 23, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Q.; Fu, Z.; Zuo, D.; Hua, Y.; Cai, Z. ErbB Receptors as Prognostic and Therapeutic Drug Targets in Bone and Soft Tissue Sarcomas. Cancer Investig. 2014, 32, 533–542. [Google Scholar] [CrossRef]
- Wen, Y.H.; Koeppen, H.; Garcia, R.; Chiriboga, L.; Tarlow, B.D.; Peters, B.A.; Eigenbrot, C.; Yee, H.; Steiner, G.; Greco, M.A. Epidermal growth factor receptor in osteosarcoma: Expression and mutational analysis. Hum. Pathol. 2007, 38, 1184–1191. [Google Scholar] [CrossRef]
- Liu, L.; Xiao, C.; Sun, Q. MiRNA-375 inhibits retinoblastoma progression through targeting ERBB2 and inhibiting MAPK1/MAPK3 signalling pathway. Cutan. Ocul. Toxicol. 2021, 41, 1–10. [Google Scholar] [CrossRef]
- Vasei, M.; Modjtahedi, H.; Ale-Booyeh, O.; Mosallaei, A.; Kajbafzadeh, A.M.; Shahriari, M.; Ghaderi, A.A.; Soleymanpour, H.; Kosari, F.; Moch, H.; et al. Amplification and expression of EGFR and ERBB2 in Wilms tumor. Cancer Genet. Cytogenet. 2009, 194, 88–95. [Google Scholar] [CrossRef]
- Little, S.E.; Bax, D.A.; Rodriguez-Pinilla, M.; Natrajan, R.; Messahel, B.; Pritchard-Jones, K.; Vujanic, G.M.; Reis-Filho, J.S.; Jones, C. Multifaceted Dysregulation of the Epidermal Growth Factor Receptor Pathway in Clear Cell Sarcoma of the Kidney. Clin. Cancer Res. 2007, 13, 4360–4364. [Google Scholar] [CrossRef] [Green Version]
- Armistead, P.M.; Salganick, J.; Roh, J.S.; Steinert, D.M.; Patel, S.; Munsell, M.; El-Naggar, A.K.; Benjamin, R.S.; Zhang, W.; Trent, J.C. Expression of receptor tyrosine kinases and apoptotic molecules in rhabdomyosarcoma: Correlation with overall survival in 105 patients. Cancer 2007, 110, 2293–2303. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2012, 153, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Sato, M.; Morimoto, Y.; Ohara, K.; Kosugi, K.; Oishi, Y.; Kuranari, Y.; Murase, M.; Yoshida, K.; Toda, M. Quantitative assessment and clinical relevance of VEGFRs-positive tumor cells in refractory brain tumors. Exp. Mol. Pathol. 2020, 114, 104408. [Google Scholar] [CrossRef] [PubMed]
- Slongo, M.L.; Molena, B.; Brunati, A.M.; Frasson, M.; Gardiman, M.; Carli, M.; Perilongo, G.; Rosolen, A.; Onisto, M. Functional VEGF and VEGF receptors are expressed in human medulloblastomas. Neuro-Oncology 2007, 9, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesundheit, B.; Klement, G.; Senger, C.; Kerbel, R.; Kieran, M.; Baruchel, S.; Becker, L. Differences in vasculature between pilocytic and anaplastic astrocytomas of childhood. Med. Pediatr. Oncol. 2003, 41, 516–526. [Google Scholar] [CrossRef]
- Farschtschi, S.; Merker, V.L.; Wolf, D.; Schuhmann, M.; Blakeley, J.; Plotkin, S.R.; Hagel, C.; Mautner, V.F. Bevacizumab treatment for symptomatic spinal ependymomas in neurofibromatosis type 2. Acta Neurol. Scand. 2015, 133, 475–480. [Google Scholar] [CrossRef]
- Virág, J.; Kenessey, I.; Haberler, C.; Piurko, V.; Bálint, K.; Döme, B.; Tímár, J.; Garami, M.; Hegedűs, B. Angiogenesis and Angiogenic Tyrosine Kinase Receptor Expression in Pediatric Brain Tumors. Pathol. Oncol. Res. 2013, 20, 417–426. [Google Scholar] [CrossRef]
- Zhou, Q.; Yan, X.; Zhu, H.; Xin, Z.; Zhao, J.; Shen, W.; Yin, W.; Guo, Y.; Xu, H.; Zhao, M.; et al. Identification of three tumor antigens and immune subtypes for mRNA vaccine development in diffuse glioma. Theranostics 2021, 11, 9775–9790. [Google Scholar] [CrossRef]
- Fakhari, M.; Pullirsch, D.; Abraham, D.; Paya, K.; Hofbauer, R.; Holzfeind, P.; Hofmann, M.; Aharinejad, S. Selective upregulation of vascular endothelial growth factor receptors neuropilin-1 and -2 in human neuroblastoma. Cancer 2001, 94, 258–263. [Google Scholar] [CrossRef]
- Czapiewski, P.; Kunc, M.; Haybaeck, J. Genetic and molecular alterations in olfactory neuroblastoma: Implications for pathogenesis, prognosis and treatment. Oncotarget 2016, 7, 52584–52596. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Joseph, C.G.; Hwang, H.; Jiao, Y.; Wood, L.D.; Kinde, I.; Wu, J.; Mandahl, N.; Luo, J.; Hruban, R.H.; Diaz, L.; et al. Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosom. Cancer 2013, 53, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Negri, G.L.; Grande, B.M.; Delaidelli, A.; El-Naggar, A.; Cochrane, D.; Lau, C.C.; Triche, T.J.; Moore, R.A.; Jones, S.J.M.; Montpetit, A.; et al. Integrative genomic analysis of matched primary and metastatic pediatric osteosarcoma. J. Pathol. 2019, 249, 319–331. [Google Scholar] [CrossRef]
- Subbiah, V.; Cote, G.J. Advances in Targeting RET-Dependent Cancers. Cancer Discov. 2020, 10, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Kawai, K.; Asai, N. Roles of the RET Proto-oncogene in Cancer and Development. JMA J. 2020, 3, 175–181. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Adashek, J.J.; Desai, A.P.; Andreev-Drakhlin, A.Y.; Roszik, J.; Cote, G.J.; Subbiah, V. Hallmarks of RET and Co-occuring Genomic Alterations in RET-aberrant Cancers. Mol. Cancer Ther. 2021, 20, 1769–1776. [Google Scholar] [CrossRef]
- Elisei, R.; Romei, C.; Vorontsova, T.; Cosci, B.; Veremeychik, V.; Kuchinskaya, E.; Basolo, F.; Demidchik, E.P.; Miccoli, P.; Pinchera, A.; et al. RET/PTC Rearrangements in Thyroid Nodules: Studies in Irradiated and Not Irradiated, Malignant and Benign Thyroid Lesions in Children and Adults1. J. Clin. Endocrinol. Metab. 2001, 86, 3211–3216. [Google Scholar] [CrossRef] [Green Version]
- Rabes, H.M.; Klugbauer, S. Molecular genetics of childhood papillary thyroid carcinomas after irradiation: High prevalence of RET rearrangement. Genes Environ. Cancer 1998, 154, 248–264. [Google Scholar] [CrossRef]
- Zimmerman, D. Thyroid neoplasia in children. Curr. Opin. Pediatr. 1997, 9, 413–418. [Google Scholar] [CrossRef]
- Ortiz, M.V.; Gerdemann, U.; Raju, S.G.; Henry, D.; Smith, S.; Rothenberg, S.M.; Cox, M.C.; Proust, S.; Bender, J.G.; Frazier, A.L.; et al. Activity of the Highly Specific RET Inhibitor Selpercatinib (LOXO-292) in Pediatric Patients With Tumors Harboring RET Gene Alterations. JCO Precis. Oncol. 2020, 4, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Kovac, M.; Woolley, C.; Ribi, S.; Blattmann, C.; Roth, E.; Morini, M.; Kovacova, M.; Ameline, B.; Kulozik, A.; Bielack, S.; et al. Germline RET variants underlie a subset of paediatric osteosarcoma. J. Med. Genet. 2020, 58, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, E.M.; Collier, C.D.; Getty, P.J. Receptor Tyrosine Kinases in Osteosarcoma: 2019 Update. Adv. Exp. Med. Biol. 2020, 1258, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xia, Y.; Yin, Y.; Luo, J.; Liu, M.; Zhang, C.; Zhao, Y.; Yang, L.; Kong, L. ATF4 destabilizes RET through nonclassical GRP78 inhibition to enhance chemosensitivity to bortezomib in human osteosarcoma. Theranostics 2019, 9, 6334–6353. [Google Scholar] [CrossRef] [PubMed]
- Rettew, A.N.; Young, E.D.; Lev, D.C.; Kleinerman, E.S.; Abdulkarim, F.W.; Getty, P.J.; Greenfield, E.M. Multiple receptor tyrosine kinases promote the in vitro phenotype of metastatic human osteosarcoma cell lines. Oncogenesis 2012, 1, e34. [Google Scholar] [CrossRef] [Green Version]
- Rettew, A.N.; Getty, P.J.; Greenfield, E.M. Receptor Tyrosine Kinases in Osteosarcoma: Not Just the Usual Suspects. Curr. Adv. Osteosarcoma 2014, 804, 47–66. [Google Scholar] [CrossRef]
- Dabir, S.; Babakoohi, S.; Kluge, A.; Morrow, J.J.; Kresak, A.; Yang, M.; MacPherson, D.; Wildey, G.; Dowlati, A. RET Mutation and Expression in Small-Cell Lung Cancer. J. Thorac. Oncol. 2014, 9, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Cockburn, J.G.; Richardson, D.S.; Gujral, T.S.; Mulligan, L.M. RET-Mediated Cell Adhesion and Migration Require Multiple Integrin Subunits. J. Clin. Endocrinol. Metab. 2010, 95, E342–E346. [Google Scholar] [CrossRef] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Park, K.C.; Richardson, D.R. The c-MET oncoprotein: Function, mechanisms of degradation and its targeting by novel anti-cancer agents. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129650. [Google Scholar] [CrossRef]
- Marona, P.; Górka, J.; Kotlinowski, J.; Majka, M.; Jura, J.; Miekus, K. C-Met as a Key Factor Responsible for Sustaining Undifferentiated Phenotype and Therapy Resistance in Renal Carcinomas. Cells 2019, 8, 272. [Google Scholar] [CrossRef] [Green Version]
- Zambelli, A.; Biamonti, G.; Amato, A. HGF/c-Met Signalling in the Tumor Microenvironment. TumorMicroenviron. Signal. Pathw. Part B 2020, 1270, 31–44. [Google Scholar] [CrossRef]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Grundy, M.; Narendran, A. The hepatocyte growth factor/mesenchymal epithelial transition factor axis in high-risk pediatric solid tumors and the anti-tumor activity of targeted therapeutic agents. Front. Pediatr. 2022, 10, 910268. [Google Scholar] [CrossRef]
- Stucklin, A.S.G.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, P.; Müller, S.; Schirmacher, P.; Stavrou, D.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Expression and localization of scatter factor/hepatocyte growth factor in human astrocytomas. Neuro-Oncology 2001, 3, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lal, B.; Kwon, S.; Fan, X.; Saldanha, U.; Reznik, T.E.; Kuchner, E.B.; Eberhart, C.; Laterra, J.; Abounader, R. The Scatter Factor/Hepatocyte Growth Factor: C-Met Pathway in Human Embryonal Central Nervous System Tumor Malignancy. Cancer Res 2005, 65, 9355–9362. [Google Scholar] [CrossRef] [Green Version]
- Provençal, M.; Labbé, D.; Veitch, R.; Boivin, D.; Rivard, G.; Sartelet, H.; Robitaille, Y.; Gingras, D.; Béliveau, R. c-Met activation in medulloblastoma induces tissue factor expression and activity: Effects on cell migration. Carcinog. 2009, 30, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- E Crosswell, H.; Dasgupta, A.; Alvarado, C.S.; Watt, T.; Christensen, J.G.; De, P.; Durden, D.L.; Findley, H.W. PHA665752, a small-molecule inhibitor of c-Met, inhibits hepatocyte growth factor-stimulated migration and proliferation of c-Met-positive neuroblastoma cells. BMC Cancer 2009, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Hecht, M.; Papoutsi, M.; Tran, H.D.; Wilting, J.; Schweigerer, L. Hepatocyte Growth Factor/c-Met Signaling Promotes the Progression of Experimental Human Neuroblastomas. Cancer Res 2004, 64, 6109–6118. [Google Scholar] [CrossRef] [Green Version]
- Alami, J.; Williams, B.R.G.; Yeger, H. Expression and localization of HGF and met in Wilms’ tumours. J. Pathol. 2001, 196, 76–84. [Google Scholar] [CrossRef]
- Cao, X.; Liu, D.-H.; Zhou, Y.; Yan, X.-M.; Yuan, L.-Q.; Pan, J.; Fu, M.-C.; Zhang, T.; Wang, J. Histone deacetylase 5 promotes Wilms’ tumor cell proliferation through the upregulation of c-Met. Mol. Med. Rep. 2016, 13, 2745–2750. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.M.; Prabhu, V.; Manukonda, R.; Mishra, D.K.; Kaliki, S.; Vemuganti, G.K. Overexpression of metastasis-associated in colon cancer 1 in retinoblastoma. Tumor Biol. 2020, 42, 1010428320975973. [Google Scholar] [CrossRef]
- Patanè, S.; Avnet, S.; Coltella, N.; Costa, B.; Sponza, S.; Olivero, M.; Vigna, E.; Naldini, L.; Baldini, N.; Ferracini, R.; et al. MET Overexpression Turns Human Primary Osteoblasts into Osteosarcomas. Cancer Res 2006, 66, 4750–4757. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wu, S.; Huang, Y.; Zhang, T.; Dong, H.; Zheng, X.; Chen, T.; Gong, X.; Liu, G.; Zhao, X. A c-Met Inhibitor Suppresses Osteosarcoma Progression via the ERK1/2 Pathway in Human Osteosarcoma Cells. OncoTargets Ther. 2021, 14, 4791–4804. [Google Scholar] [CrossRef]
- Entz-Werle, N.; Lavaux, T.; Metzger, N.; Stoetzel, C.; Lasthaus, C.; Marec, P.; Kalita, C.; Brugieres, L.; Pacquement, H.; Schmitt, C.; et al. Involvement of MET/TWIST/APC Combination or the Potential Role of Ossification Factors in Pediatric High-Grade Osteosarcoma Oncogenesis. Neoplasia 2007, 9, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Fleuren, E.D.; Roeffen, M.H.; Leenders, W.P.; Flucke, U.E.; Vlenterie, M.; Schreuder, H.W.; Boerman, O.C.; van der Graaf, W.T.; Versleijen-Jonkers, Y.M. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int. J. Cancer 2013, 133, 427–436. [Google Scholar] [CrossRef]
- Yan, D.; Da Dong, X.; Chen, X.; Wang, L.; Lu, C.; Wang, J.; Qu, J.; Tu, L. MicroRNA-1/206 Targets c-Met and Inhibits Rhabdomyosarcoma Development. J. Biol. Chem. 2009, 284, 29596–29604. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Wang, Y.; Meng, L.; Liu, Y.; Pang, Y.; Cui, W.; Zhang, L.; Li, Z.; Liu, Q.; Shang, H.; et al. c-MET expression potentially contributes to the poor prognosis of rhabdomyosarcoma. Int. J. Clin. Exp. Pathol. 2018, 11, 4083–4092. [Google Scholar]
- Otabe, O.; Kikuchi, K.; Tsuchiya, K.; Katsumi, Y.; Yagyu, S.; Miyachi, M.; Iehara, T.; Hosoi, H. MET/ERK2 pathway regulates the motility of human alveolar rhabdomyosarcoma cells. Oncol. Rep. 2016, 37, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Taulli, R.; Scuoppo, C.; Bersani, F.; Accornero, P.; Forni, P.E.; Miretti, S.; Grinza, A.; Allegra, P.; Schmitt-Ney, M.; Crepaldi, T.; et al. Validation of Met as a Therapeutic Target in Alveolar and Embryonal Rhabdomyosarcoma. Cancer Res 2006, 66, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NPM, in Non-Hodgkin’s Lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef]
- Takita, J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017, 108, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Peron, M.; Lovisa, F.; Poli, E.; Basso, G.; Bonvini, P. Understanding the Interplay between Expression, Mutation and Activity of ALK Receptor in Rhabdomyosarcoma Cells for Clinical Application of Small-Molecule Inhibitors. PLoS ONE 2015, 10, e0132330. [Google Scholar] [CrossRef] [Green Version]
- Felkai, L.; Bánusz, R.; Kovalszky, I.; Sápi, Z.; Garami, M.; Papp, G.; Karászi, K.; Varga, E.; Csóka, M. The Presence of ALK Alterations and Clinical Relevance of Crizotinib Treatment in Pediatric Solid Tumors. Pathol. Oncol. Res. 2017, 25, 217–224. [Google Scholar] [CrossRef]
- Aygun, N. Biological and Genetic Features of Neuroblastoma and Their Clinical Importance. Curr. Pediatr. Rev. 2018, 14, 73–90. [Google Scholar] [CrossRef]
- Pastor, E.R.; Mousa, S.A. Current management of neuroblastoma and future direction. Crit. Rev. Oncol. 2019, 138, 38–43. [Google Scholar] [CrossRef]
- Pacenta, H.L.; E Macy, M. Entrectinib and other ALK/TRK inhibitors for the treatment of neuroblastoma. Drug Des. Dev. Ther. 2018, 12, 3549–3561. [Google Scholar] [CrossRef] [Green Version]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALKF1174L Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Valera, E.T.; Neder, L.; Queiroz, R.G.; Santos, A.C.; Sousa, G.R.; Oliveira, R.S.; Santos, M.V.; Machado, H.R.; Tone, L.G. Perinatal complex low- and high-grade glial tumor harboring a novel GIGYF2-ALK fusion. Pediatr. Blood Cancer 2019, 67, e28015. [Google Scholar] [CrossRef]
- Argani, P.; Lian, D.W.; Agaimy, A.; Metzler, M.; Wobker, S.E.; Matoso, A.; Epstein, J.I.; Sung, Y.-S.; Zhang, L.; Antonescu, C.R. Pediatric Mesothelioma With ALK Fusions. Am. J. Surg. Pathol. 2021, 45, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.K.; Panagopoulos, I.; Meling, T.R.; Micci, F.; Gorunova, L.; Thorsen, J.; Due-Tønnessen, B.; Scheie, D.; Lund-Iversen, M.; Krossnes, B.; et al. Fusion genes withALKas recurrent partner in ependymoma-like gliomas: A new brain tumor entity? Neuro-Oncology 2015, 17, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łastowska, M.; Trubicka, J.; Niemira, M.; Paczkowska-Abdulsalam, M.; Karkucińska-Więckowska, A.; Kaleta, M.; Drogosiewicz, M.; Tarasińska, M.; Perek-Polnik, M.; Krętowski, A.; et al. ALK Expression Is a Novel Marker for the WNT-activated Type of Pediatric Medulloblastoma and an Indicator of Good Prognosis for Patients. Am. J. Surg. Pathol. 2017, 41, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More than Just another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Barrett, D.; Brown, V.I.; Grupp, S.A.; Teachey, D.T. Targeting the PI3K/AKT/mTOR Signaling Axis in Children with Hematologic Malignancies. Pediatr. Drugs 2012, 14, 299–316. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Fujishita, T. Oncogenic Roles of the PI3K/AKT/mTOR Axis. Curr. Top. Microbiol. Immunol. 2017, 407, 153–189. [Google Scholar] [CrossRef]
- Shorning, B.; Dass, M.; Smalley, M.; Pearson, H. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/ Akt/ mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef]
- Fattahi, S.; Amjadi-Moheb, F.; Tabaripour, R.; Ashrafi, G.H.; Akhavan-Niaki, H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. 2020, 262, 118513. [Google Scholar] [CrossRef]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Gann, C.-N.; Morsli, N.; Chen, X.; Barrueco, J. Response to ‘Dai W et al. Am J Cancer Res 2015;5(10):3270-3275′ from the makers of nintedanib. Am. J. Cancer Res. 2016, 6, 1547–1548. [Google Scholar]
- Remke, M.; Pfister, S.; Kox, C.; Toedt, G.; Becker, N.; Benner, A.; Werft, W.; Breit, S.; Liu, S.; Engel, F.; et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-β and PI3K-AKT pathways and deletions at 6q15-16.1 as a genomic marker for unfavorable early treatment response. Blood 2009, 114, 1053–1062. [Google Scholar] [CrossRef] [Green Version]
- Armengol, G.; Canellas, A.; Álvarez, Y.; Bastida, P.; De Toledo, J.S.; Pérez-Iribarne, M.D.M.; Camós, M.; Tuset, E.; Estella, J.; Coll, M.D.; et al. Genetic changes including gene copy number alterations and their relation to prognosis in childhood acute myeloid leukemia. Leuk. Lymphoma 2009, 51, 114–124. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, G. Genetic Alterations and Aberrant Expression of Genes Related to the Phosphatidyl-lnositol-3′-Kinase/Protein Kinase B (Akt) Signal Transduction Pathway in Glioblastomas. Brain Pathol. 2006, 13, 507–518. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular Function of Phosphoinositide 3-Kinases: Implications for Development, Immunity, Homeostasis, and Cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110 of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; He, T.; Liu, S.; Zheng, Y.; Xiang, L.; Pei, X.; Wang, Z.; Yang, H. The PIK3CA E542K and E545K mutations promote glycolysis and proliferation via induction of the β-catenin/SIRT3 signaling pathway in cervical cancer. J. Hematol. Oncol. 2018, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional Analysis of PIK3CA Gene Mutations in Human Colorectal Cancer. Cancer Res 2005, 65, 4562–4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murat, C.; Braga, P.; Fortes, M.; Bronstein, M.; Correa-Giannella, M.L.; Giorgi, R. Mutation and genomic amplification of the PIK3CA proto-oncogene in pituitary adenomas. Braz. J. Med. Biol. Res. 2012, 45, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Yao, D.; Liu, W.; Wang, N.; Lv, H.; Zhang, G.; Ji, M.; Xu, L.; He, N.; Shi, B.; et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer 2012, 12, 50. [Google Scholar] [CrossRef] [Green Version]
- Holst, F.; Werner, H.M.; Mjøs, S.; Hoivik, E.A.; Kusonmano, K.; Wik, E.; Berg, A.; Birkeland, E.; Gibson, W.J.; Halle, M.K.; et al. PIK3CA Amplification Associates with Aggressive Phenotype but Not Markers of AKT-MTOR Signaling in Endometrial Carcinoma. Clin. Cancer Res. 2019, 25, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Huw, L.-Y.; O’Brien, C.; Pandita, A.; Mohan, S.; Spoerke, J.M.; Lu, S.; Wang, Y.; Hampton, G.M.; Wilson, T.R.; Lackner, M.R. Acquired PIK3CA amplification causes resistance to selective phosphoinositide 3-kinase inhibitors in breast cancer. Oncogenesis 2013, 2, e83. [Google Scholar] [CrossRef] [Green Version]
- Salm, F.; Dimitrova, V.; Von Bueren, A.O.; Ćwiek, P.; Rehrauer, H.; Djonov, V.; Anderle, P.; Arcaro, A. The Phosphoinositide 3-Kinase p110α Isoform Regulates Leukemia Inhibitory Factor Receptor Expression via c-Myc and miR-125b to Promote Cell Proliferation in Medulloblastoma. PLoS ONE 2015, 10, e0123958. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, A.S.; Fattet, S.; Fischer, B.; Shalaby, T.; Jackson, S.P.; Schoenwaelder, S.M.; Grotzer, M.A.; Delattre, O.; Arcaro, A. Targeting the PI3K p110α Isoform Inhibits Medulloblastoma Proliferation, Chemoresistance, and Migration. Clin. Cancer Res. 2008, 14, 6761–6769. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, A.S.; Fattet, S.; Kulesza, D.W.; Atamer, A.; Elsing, A.N.; Shalaby, T.; Jackson, S.P.; Schoenwaelder, S.M.; Grotzer, M.A.; Delattre, O.; et al. A Sensitized RNA Interference Screen Identifies a Novel Role for the PI3K p110γ Isoform in Medulloblastoma Cell Proliferation and Chemoresistance. Mol. Cancer Res. 2011, 9, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Luk, S.K.; Piekorz, R.P.; Nürnberg, B.; To, S.-S.T. The catalytic phosphoinositol 3-kinase isoform p110δ is required for glioma cell migration and invasion. Eur. J. Cancer 2012, 48, 149–157. [Google Scholar] [CrossRef]
- Boller, D.; Schramm, A.; Doepfner, K.T.; Shalaby, T.; von Bueren, A.O.; Eggert, A.; Grotzer, M.A.; Arcaro, A. Targeting the Phosphoinositide 3-Kinase Isoform p110δ Impairs Growth and Survival in Neuroblastoma Cells. Clin. Cancer Res. 2008, 14, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Fransson, S.; Martinsson, T.; Ejeskär, K. Neuroblastoma tumors with favorable and unfavorable outcomes: Significant differences in mRNA expression of genes mapped at 1p36.2. Genes Chromosom. Cancer 2006, 46, 45–52. [Google Scholar] [CrossRef]
- Wang, Q.; Diskin, S.; Rappaport, E.; Attiyeh, E.; Mosse, Y.; Shue, D.; Seiser, E.; Jagannathan, J.; Shusterman, S.; Bansal, M.; et al. Integrative Genomics Identifies Distinct Molecular Classes of Neuroblastoma and Shows That Multiple Genes Are Targeted by Regional Alterations in DNA Copy Number. Cancer Res 2006, 66, 6050–6062. [Google Scholar] [CrossRef] [Green Version]
- Yoon, C.; Lu, J.; Ryeom, S.W.; Simon, M.C.; Yoon, S.S. PIK3R3, part of the regulatory domain of PI3K, is upregulated in sarcoma stem-like cells and promotes invasion, migration, and chemotherapy resistance. Cell Death Dis. 2021, 12, 749. [Google Scholar] [CrossRef]
- Staff, T.P.O. Correction: Variable expression of PIK3R3 and PTEN in Ewing sarcoma impacts oncogenic phenotypes. PLoS ONE 2015, 10, e0120830. [Google Scholar] [CrossRef]
- Bellacosa, A.; Franke, T.F.; E Gonzalez-Portal, M.; Datta, K.; Taguchi, T.; Gardner, J.; Cheng, J.Q.; Testa, J.R.; Tsichlis, P.N. Structure, expression and chromosomal mapping of c-akt: Relationship to v-akt and its implications. Oncogene 1993, 8, 745–754. [Google Scholar]
- Basu, A.; Lambring, C. Akt Isoforms: A Family Affair in Breast Cancer. Cancers 2021, 13, 3445. [Google Scholar] [CrossRef]
- Meier, R.; Alessi, D.R.; Cron, P.; Andjelković, M.; Hemmings, B.A. Mitogenic Activation, Phosphorylation, and Nuclear Translocation of Protein Kinase Bβ. J. Biol. Chem. 1997, 272, 30491–30497. [Google Scholar] [CrossRef] [Green Version]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K AKT pathway in the pathogenesis of prostate cancer. Front. Biosci. 2016, 21, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Schüller, U.; Ruiter, M.; Herms, J.; Kretzschmar, H.A.; Grasbon-Frodl, E. Absence of mutations in the AKT1 oncogene in glioblastomas and medulloblastomas. Acta Neuropathol. 2008, 115, 367–368. [Google Scholar] [CrossRef]
- Hartmann, W.; Digon-Söntgerath, B.; Koch, A.; Waha, A.; Endl, E.; Dani, I.; Denkhaus, D.; Goodyer, C.G.; Sörensen, N.; Wiestler, O.D.; et al. Phosphatidylinositol 3′-Kinase/AKT Signaling Is Activated in Medulloblastoma Cell Proliferation and Is Associated with Reduced Expression of PTEN. Clin. Cancer Res. 2006, 12, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Granados, V.A.; Avirneni-Vadlamudi, U.; Dalal, P.; Scarborough, S.R.; Galindo, K.A.; Mahajan, P.; Galindo, R.L. Selective Targeting of Myoblast Fusogenic Signaling and Differentiation-Arrest Antagonizes Rhabdomyosarcoma Cells. Cancer Res 2019, 79, 4585–4591. [Google Scholar] [CrossRef] [PubMed]
- Hotfilder, M.; Sondermann, P.; Senß, A.; van Valen, F.; Jürgens, H.; Vormoor, J. PI3K/AKT is involved in mediating survival signals that rescue Ewing tumour cells from fibroblast growth factor 2-induced cell death. Br. J. Cancer 2005, 92, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Pan, R.; Hou, L.; Wu, H.; Sun, J.; Zhang, W.; Tian, X.; Chen, H. Suppression of CLEC3A inhibits osteosarcoma cell proliferation and promotes their chemosensitivity through the AKT1/mTOR/HIF1α signaling pathway. Mol. Med. Rep. 2020, 21, 1739–1748. [Google Scholar] [CrossRef] [Green Version]
- Kuijjer, M.L.; Akker, B.E.W.M.V.D.; Hilhorst, R.; Mommersteeg, M.; Buddingh, E.; Serra, M.; Bürger, H.; Hogendoorn, P.C.W.; Cleton-Jansen, A.-M. Kinome and mRNA expression profiling of high-grade osteosarcoma cell lines implies Akt signaling as possible target for therapy. BMC Med. Genom. 2014, 7, 4. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, J.; Ji, Y.; Yu, B. Elevated expression of AKT2 correlates with disease severity and poor prognosis in human osteosarcoma. Mol. Med. Rep. 2014, 10, 737–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhou, Z.; Zhang, Q.; Rong, Y.; Li, L.; Luo, Y.; Wang, J.; Yin, G.; Lv, C.; Cai, W. Overexpression of miR-1258 inhibits cell proliferation by targeting AKT3 in osteosarcoma. Biochem. Biophys. Res. Commun. 2019, 510, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Lee, S.; Paul, P.; Qiao, L.; Taylor, C.J.; Schlegel, C.; Colon, N.C.; Chung, D.H. Akt2 Regulates Metastatic Potential in Neuroblastoma. PLoS ONE 2013, 8, e56382. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976, Erratum in Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Zarogoulidis, P.; Lampaki, S.; Turner, J.F.; Huang, H.; Kakolyris, S.; Syrigos, K.; Zarogoulidis, K. mTOR pathway: A current, up-to-date mini-review (Review). Oncol. Lett. 2014, 8, 2367–2370. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A Diverse Array of Cancer-Associated MTOR Mutations Are Hyperactivating and Can Predict Rapamycin Sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Culjkovic, B.; Topisirovic, I.; Skrabanek, L.; Ruiz-Gutierrez, M.; Borden, K.L. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J. Cell Biol. 2006, 175, 415–426. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; Rong, L.; Yoshida, K.; Ler, L.W.; Sonenberg, N. eIF4E—From translation to transformation. Oncogene 2004, 23, 3172–3179. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Jia, Y.; Ye, Y.; Kang, E.; Chen, H.; Wang, J.; He, X. Identification of key methylation differentially expressed genes in posterior fossa ependymoma based on epigenomic and transcriptome analysis. J. Transl. Med. 2021, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Machado, L.E.; Alvarenga, A.W.; Da Silva, F.F.; Roffé, M.; Begnami, M.D.; Torres, L.F.B.; Da Cunha, I.W.; Martins, V.R.; Hajj, G.N.M. Overexpression of mTOR and p(240–244)S6 in IDH1 Wild-Type Human Glioblastomas Is Predictive of Low Survival. J. Histochem. Cytochem. 2018, 66, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhang, P.; Su, H.; Cai, L.; Zhao, L.; Zhou, H. Bioinformatics Analysis of Neuroblastoma miRNA Based on GEO Data. Pharmacogenomics Pers. Med. 2021, 14, 849–858. [Google Scholar] [CrossRef]
- Pócza, T.; Sebestyén, A.; Turányi, E.; Krènacs, T.; Márk, A.; Sticz, T.B.; Jakab, Z.; Hauser, P. mTOR Pathway As a Potential Target In a Subset of Human Medulloblastoma. Pathol. Oncol. Res. 2014, 20, 893–900. [Google Scholar] [CrossRef]
- Kaid, C.; Assoni, A.; Marçola, M.; Semedo-Kuriki, P.; Bortolin, R.H.; Carvalho, V.M.; Okamoto, O.K. Proteome and miRNome profiling of microvesicles derived from medulloblastoma cell lines with stem-like properties reveals biomarkers of poor prognosis. Brain Res. 2020, 1730, 146646. [Google Scholar] [CrossRef]
- Chakraborty, S.; Khare, S.; Dorairaj, S.K.; Prabhakaran, V.C.; Prakash, D.R.; Kumar, A. Identification of genes associated with tumorigenesis of retinoblastoma by microarray analysis. Genomics 2007, 90, 344–353. [Google Scholar] [CrossRef]
- Subbiah, V.; Brown, R.E.; Jiang, Y.; Buryanek, J.; Hayes-Jordan, A.; Kurzrock, R.; Anderson, P.M. Morphoproteomic Profiling of the Mammalian Target of Rapamycin (mTOR) Signaling Pathway in Desmoplastic Small Round Cell Tumor (EWS/WT1), Ewing’s Sarcoma (EWS/FLI1) and Wilms’ Tumor(WT1). PLoS ONE 2013, 8, e68985. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Sherman, A.K.; Pawel, B.R. Expression of therapeutic targets in Ewing sarcoma family tumors. Hum. Pathol. 2012, 43, 1077–1083. [Google Scholar] [CrossRef]
- Dobashi, Y.; Suzuki, S.; Sato, E.; Hamada, Y.; Yanagawa, T.; Ooi, A. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod. Pathol. 2009, 22, 1328–1340. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, K.; Bruce, B.; Hewitt, S.; Thomas, D.; Khanna, C.; Helman, L.J. Ezrin mediates growth and survival in Ewing’s sarcoma through the AKT/mTOR, but not the MAPK, signaling pathway. Clin. Exp. Metastasis 2006, 23, 227–236. [Google Scholar] [CrossRef]
- Hu, K.; Dai, H.-B.; Qiu, Z.-L. mTOR signaling in osteosarcoma: Oncogenesis and therapeutic aspects (Review). Oncol. Rep. 2016, 36, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egas-Bejar, D.; Anderson, P.M.; Agarwal, R.; Corrales-Medina, F.; Devarajan, E.; Huh, W.W.; Brown, R.E.; Subbiah, V. Theranostic profiling for actionable aberrations in advanced high risk osteosarcoma with aggressive biology reveals high molecular diversity: The human fingerprint hypothesis. Oncoscience 2014, 1, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petricoin, E.F.; Espina, V.; Araujo, R.P.; Midura, B.; Yeung, C.; Wan, X.; Eichler, G.S.; Johann, D.J.; Qualman, S.; Tsokos, M.; et al. Phosphoprotein Pathway Mapping: Akt/Mammalian Target of Rapamycin Activation Is Negatively Associated with Childhood Rhabdomyosarcoma Survival. Cancer Res 2007, 67, 3431–3440. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Kockeritz, L.; Doble, B.; Patel, S.; Woodgett, J. Glycogen Synthase Kinase-3—An Overview of An Over-Achieving Protein Kinase. Curr. Drug Targets 2006, 7, 1377–1388. [Google Scholar] [CrossRef]
- Sutherland, C. What Are the bona fideGSK3 Substrates? Int. J. Alzheimer’s Dis. 2011, 2011, 505607. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Davis, N.M.; Abrams, S.L.; Montalto, G.; Cervello, M.; Basecke, J.; Libra, M.; Nicoletti, F.; Cocco, L.; Martelli, A.M.; et al. Diverse roles of GSK-3: Tumor promoter–tumor suppressor, target in cancer therapy. Adv. Biol. Regul. 2014, 54, 176–196. [Google Scholar] [CrossRef]
- Patel, S.; Woodgett, J. Glycogen Synthase Kinase-3 and Cancer: Good Cop, Bad Cop? Cancer Cell 2008, 14, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Luo, J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2008, 273, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Investig. 2012, 122, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farago, M.; Dominguez, I.; Landesman-Bollag, E.; Xu, X.; Rosner, A.; Cardiff, R.D.; Seldin, D.C. Kinase-Inactive Glycogen Synthase Kinase 3β Promotes Wnt Signaling and Mammary Tumorigenesis. Cancer Res 2005, 65, 5792–5801. [Google Scholar] [CrossRef] [Green Version]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen Synthase Kinase-3β Participates in Nuclear Factor κB–Mediated Gene Transcription and Cell Survival in Pancreatic Cancer Cells. Cancer Res 2005, 65, 2076–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.P.; Cleary, M.L. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brassesco, M.S.; Valera, E.T.; Meyer, C.; Marschalek, R.; Lopes, B.A.; Queiroz, R.G.D.P.; Calado, R.D.T.; Scrideli, C.; Tone, L.G. A new complex rearrangement in infant ALL: T(X;11;17)(p11.2;q23;q12). Cancer Genet. 2018, 228–229, 110–114. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Ocasio, J.K.; Bates, R.D.P.; Rapp, C.D.; Gershon, T.R. GSK-3 modulates SHH-driven proliferation in postnatal cerebellar neurogenesis and medulloblastoma. Development 2019, 146, dev177550. [Google Scholar] [CrossRef] [Green Version]
- Silva-Evangelista, C.; Barret, E.; Ménez, V.; Merlevede, J.; Kergrohen, T.; Saccasyn, A.; Oberlin, E.; Puget, S.; Beccaria, K.; Grill, J.; et al. A kinome-wide shRNA screen uncovers vaccinia-related kinase 3 (VRK3) as an essential gene for diffuse intrinsic pontine glioma survival. Oncogene 2019, 38, 6479–6490. [Google Scholar] [CrossRef]
- Lenz, J.E.; Riester, R.; Schleicher, S.B.; Handgretinger, R.; Boehme, K.A.; Traub, F. Interaction of arsenic trioxide and etoposide in Ewing sarcoma cell lines. Oncol. Rep. 2019, 43, 337–345. [Google Scholar] [CrossRef]
- Machado, I.; López-Guerrero, J.A.; Navarro, S.; Alberghini, M.; Scotlandi, K.; Picci, P.; Llombart-Bosch, A. Epithelial cell adhesion molecules and epithelial mesenchymal transition (EMT) markers in Ewing’s sarcoma family of tumors (ESFTs). Do they offer any prognostic significance? Virchows Arch. 2012, 461, 333–337. [Google Scholar] [CrossRef]
- Ma, C.; Bower, K.A.; Chen, G.; Shi, X.; Ke, Z.-J.; Luo, J. Interaction between ERK and GSK3β Mediates Basic Fibroblast Growth Factor-induced Apoptosis in SK-N-MC Neuroblastoma Cells. J. Biol. Chem. 2008, 283, 9248–9256. [Google Scholar] [CrossRef] [Green Version]
- Woodgett, J.R. Can a two-faced kinase be exploited for osteosarcoma? Gynecol. Oncol. 2012, 104, 722–723. [Google Scholar] [CrossRef] [Green Version]
- Le Guellec, S.; Moyal, E.C.-J.; Filleron, T.; Delisle, M.-B.; Chevreau, C.; Rubie, H.; Castex, M.-P.; De Gauzy, J.S.; Bonnevialle, P.; Gomez-Brouchet, A. The β5/focal adhesion kinase/glycogen synthase kinase 3β integrin pathway in high-grade osteosarcoma: A protein expression profile predictive of response to neoadjuvant chemotherapy. Hum. Pathol. 2013, 44, 2149–2158. [Google Scholar] [CrossRef]
- Zeng, F.-Y.; Dong, H.; Cui, J.; Liu, L.; Chen, T. Glycogen synthase kinase 3 regulates PAX3–FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2010, 391, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Dionyssiou, M.G.; Ehyai, S.; Avrutin, E.; Connor, M.K.; McDermott, J.C. Glycogen synthase kinase 3β represses MYOGENIN function in alveolar rhabdomyosarcoma. Cell Death Dis. 2014, 5, e1094. [Google Scholar] [CrossRef] [Green Version]
- Belyea, B.; Kephart, J.G.; Blum, J.; Kirsch, D.G.; Linardic, C.M. Embryonic Signaling Pathways and Rhabdomyosarcoma: Contributions to Cancer Development and Opportunities for Therapeutic Targeting. Sarcoma 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small-molecule glycogen synthase kinase-3 inhibitor, is active in neuroblastoma. Anti-Cancer Drugs 2018, 29, 717–724. [Google Scholar] [CrossRef]
- Li, Z.; Tan, F.; Thiele, C.J. Inactivation of glycogen synthase kinase-3β contributes to brain-derived neutrophic factor/TrkB-induced resistance to chemotherapy in neuroblastoma cells. Mol. Cancer Ther. 2007, 6, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.J.; Krstic, A.; Schwarzl, T.; Higgins, D.G.; Kolch, W. GSK3 Inhibitors Regulate MYCN mRNA Levels and Reduce Neuroblastoma Cell Viability through Multiple Mechanisms, Including p53 and Wnt Signaling. Mol. Cancer Ther. 2014, 13, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Dickey, A.; Schleicher, S.; Leahy, K.; Hu, R.; Hallahan, D.; Thotala, D.K. GSK-3β inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J. Neuro-Oncology 2010, 104, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Katoh, M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, K.; Trojanek, J.; Del Valle, L.; Eldeen, M.B.; Hofmann, F.; Garcia-Echeverria, C.; Khalili, K.; Reiss, K. Inhibition of IGF-I receptor in anchorage-independence attenuates GSK-3β constitutive phosphorylation and compromises growth and survival of medulloblastoma cell lines. Oncogene 2006, 26, 2308–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, R.; Stylli, S.; Luwor, R.; Kaye, A.; Hovens, C. Glycogen synthase kinase-3β (GSK-3β) and its dysregulation in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3β is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. 2013, 22, 1698–1710. [Google Scholar] [CrossRef]
- Schaeffer, H.J.; Weber, M.J. Mitogen-Activated Protein Kinases: Specific Messages from Ubiquitous Messengers. Mol. Cell. Biol. 1999, 19, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Zalyte, E.; Rimkus, A.; Dapkus, D.; Noreika, R.; Urbonavicius, S. JNK, p38, ERK, and SGK1 Inhibitors in Cancer. Cancers 2017, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Mishima, K.; Inoue, K.; Hayashi, Y. Overexpression of extracellular-signal regulated kinases on oral squamous cell carcinoma. Oral Oncol. 2002, 38, 468–474. [Google Scholar] [CrossRef]
- Kudaravalli, S.; Hollander, P.D.; Mani, S.A. Role of p38 MAP kinase in cancer stem cells and metastasis. Oncogene 2022, 41, 3177–3185. [Google Scholar] [CrossRef]
- Tournier, C. The 2 Faces of JNK Signaling in Cancer. Genes Cancer 2013, 4, 397–400. [Google Scholar] [CrossRef]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.; Raingeaud, J.; Kaneki, M.; Weichselbaum, R.; Davis, R.J.; Kufe, D.; Kharbanda, S. Activation of p38 Mitogen-activated Protein Kinase by c-Abl-dependent and -independent Mechanisms. J. Biol. Chem. 1996, 271, 23775–23779. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38α MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Garinet, S.; Pasmant, E. RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget 2015, 7, 38892–38907. [Google Scholar] [CrossRef] [Green Version]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF–MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, E.; Bister, K. MYC and RAF: Key Effectors in Cellular Signaling and Major Drivers in Human Cancer. Poxviruses 2017, 407, 117–151. [Google Scholar] [CrossRef]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer–roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Pfister, S.; Janzarik, W.G.; Remke, M.; Ernst, A.; Werft, W.; Becker, N.; Toedt, G.; Wittmann, A.; Kratz, C.; Olbrich, H.; et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J. Clin. Investig. 2008, 118, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Bar, E.E.; Lin, A.; Tihan, T.; Burger, P.C.; Eberhart, C.G. Frequent Gains at Chromosome 7q34 Involving BRAF in Pilocytic Astrocytoma. J. Neuropathol. Exp. Neurol. 2008, 67, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Reis, G.F.; Bloomer, M.M.; Perry, A.; Phillips, J.J.; Grenert, J.P.; Karnezis, A.N.; Tihan, T. Pilocytic astrocytomas of the optic nerve and their relation to pilocytic astrocytomas elsewhere in the central nervous system. Mod. Pathol. 2013, 26, 1279–1287. [Google Scholar] [CrossRef] [Green Version]
- Anagnostopoulos, A.K.; Dimas, K.S.; Papathanassiou, C.; Braoudaki, M.; Anastasiadou, E.; Vougas, K.; Karamolegou, K.; Kontos, H.; Prodromou, N.; Tzortzatou-Stathopoulou, F.; et al. Proteomics Studies of Childhood Pilocytic Astrocytoma. J. Proteome Res. 2011, 10, 2555–2565. [Google Scholar] [CrossRef]
- MacDonald, T.J.; Brown, K.M.; LaFleur, B.; Peterson, K.; Lawlor, C.; Chen, Y.; Packer, R.J.; Cogen, P.; Stephan, D.A. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat. Genet. 2001, 29, 143–152. [Google Scholar] [CrossRef]
- Badodi, S.; Pomella, N.; Lim, Y.M.; Brandner, S.; Morrison, G.; Pollard, S.M.; Zhang, X.; Zabet, N.R.; Marino, S. Combination of BMI1 and MAPK/ERK inhibitors is effective in medulloblastoma. Neuro-Oncology 2022, 24, 1273–1285. [Google Scholar] [CrossRef]
- Tsumura, H.; Yoshida, T.; Saito, H.; Imanaka-Yoshida, K.; Suzuki, N. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene 2006, 25, 7673–7679. [Google Scholar] [CrossRef]
- Na, K.Y.; Kim, Y.W.; Park, Y.-K. Mitogen-activated protein kinase pathway in osteosarcoma. Pathology 2012, 44, 540–546. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, C.; Chen, L. MiR-511 mimic transfection inhibits the proliferation, invasion of osteosarcoma cells and reduces metastatic osteosarcoma tumor burden in nude mice via targeting MAPK1. Cancer Biomarkers 2019, 26, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhang, H.-Y.; Wang, S.-L.; Ye, L.; Yang, G.-H.; Bu, H. Smad4 and ERK2 stimulated by transforming growth factor beta1 in rhabdomyosarcoma. Chin. Med. J. 2007, 120, 515–521. [Google Scholar] [CrossRef]
- Lynch, J.; Fay, J.; Meehan, M.; Bryan, K.; Watters, K.M.; Murphy, D.M.; Stallings, R.L. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-β signalling pathway. Carcinog. 2012, 33, 976–985. [Google Scholar] [CrossRef]
- Tabatabaei, S.N.; Derbali, R.M.; Yang, C.; Superstein, R.; Hamel, P.; Chain, J.L.; Hardy, P. Co-delivery of miR-181a and melphalan by lipid nanoparticles for treatment of seeded retinoblastoma. J. Control. Release 2019, 298, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.Y.C. Cell cycle control: A system of interlinking oscillators. Methods Mol. Biol. 2016, 1342, 3–19. [Google Scholar] [PubMed]
- Saka, Y.; Giuraniuc, C.V.; Ohkura, H. Accurate chromosome segregation by probabilistic self-organisation. BMC Biol. 2015, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.-W.; Liu, F. Novel insights into cell cycle regulation of cell fate determination. J. Zhejiang Univ. B 2019, 20, 467–475. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [Green Version]
- Pines, J. The cell cycle kinases. Semin. Cancer Biol. 1994, 5, 305–313. [Google Scholar]
- Bruyère, C.; Meijer, L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr. Opin. Cell Biol. 2013, 25, 772–779. [Google Scholar] [CrossRef]
- Chilà, R.; Guffanti, F.; Damia, G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat. Rev. 2016, 50, 83–88. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer Cell Cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.; Peters, G. Genetic Alterations of Cyclins, Cyclin-Dependent Kinases, and Cdk Inhibitors in Human Cancer. Adv.Cancer Res. 1996, 68, 67–108. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclins and related kinases in cancer cells. J. BUON. 2007, 12 (Suppl. S1), S45–S52. [Google Scholar]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Niwa, T.; Akaike, Y.; Watanabe, K.; Chibazakura, T. Hyperactivation of cyclin A-CDK induces centrosome overduplication and chromosome tetraploidization in mouse cells. Biochem. Biophys. Res. Commun. 2021, 549, 91–97. [Google Scholar] [CrossRef]
- Viotto, D.; Russo, F.; Anania, I.; Segatto, I.; Vinciguerra, G.L.R.; Dall’Acqua, A.; Bomben, R.; Perin, T.; Cusan, M.; Schiappacassi, M.; et al. CDKN1B mutation and copy number variation are associated with tumor aggressiveness in luminal breast cancer. J. Pathol. 2020, 253, 234–245. [Google Scholar] [CrossRef]
- Lu, Y.; Leow, A.; O Madu, C. The Role of Cyclin-Dependent Kinases on the Metastasis of Breast Cancer. Nov. Approaches Cancer Study 2020, 4, 377–385. [Google Scholar] [CrossRef]
- Lam, Y.; E. di Tomaso, H.-K.; Ng, J.C.S.; Pang, M.F.; Roussel, N.M.; Hjelm, P. Expression of p19 INK4d, CDK4, CDK6 in glioblastoma multiforme. Br. J. Neurosurg. 2000, 14, 28–32. [Google Scholar] [CrossRef]
- Richardson, S.; Hill, R.M.; Kui, C.; Lindsey, J.C.; Grabovksa, Y.; Keeling, C.; Pease, L.; Bashton, M.; Crosier, S.; Vinci, M.; et al. Emergence and maintenance of actionable genetic drivers at medulloblastoma relapse. Neuro-Oncology 2021, 24, 153–165. [Google Scholar] [CrossRef]
- Wood, A.C.; Krytska, K.; Ryles, H.T.; Infarinato, N.R.; Sano, R.; Hansel, T.D.; Hart, L.S.; King, F.J.; Smith, T.R.; Ainscow, E.; et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin. Cancer Res. 2017, 23, 2856–2868. [Google Scholar] [CrossRef] [Green Version]
- Iolascon, A.; Faienza, M.F.; Coppola, B.; Rosolen, A.; Basso, G.; Ragione, F.D.; Schettini, F. Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, andCDKN2C) in childhood rhabdomyosarcoma. Genes Chromosomes Cancer 1996, 15, 217–222. [Google Scholar] [CrossRef]
- Komuro, H.; Valentine, M.B.; Rubnitz, J.E.; Saito, M.; Raimondi, S.C.; Carroll, A.J.; Yi, T.; Sherr, C.J.; Look, A.T. p27KIP1 Deletions in Childhood Acute Lymphoblastic Leukemia. Neoplasia 1999, 1, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation. Cancer Cell 2018, 34, 626–642.e8, Erratum in 2019, 35, P705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, H.; Liu, Z. An Integrative Human Pan-Cancer Analysis of Cyclin-Dependent Kinase 1 (CDK1). Cancers 2022, 14, 2658. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Che, X.; Wang, J.; Zou, G.; Yu, Q.; Zhang, X. CDK1 serves as a novel therapeutic target for endometrioid endometrial cancer. J. Cancer 2021, 12, 2206–2215. [Google Scholar] [CrossRef]
- Li, M.; He, F.; Zhang, Z.; Xiang, Z.; Hu, D. CDK1 serves as a potential prognostic biomarker and target for lung cancer. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Nakayama, S.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Matsushima, T.; Torikoshi, Y.; Tanaka, S.; Yoshida, T.; Ishihara, H.; et al. Determination of the specific activity of CDK1 and CDK2 as a novel prognostic indicator for early breast cancer. Ann. Oncol. 2007, 19, 68–72. [Google Scholar] [CrossRef]
- Wang, C.; Xie, X.; Li, W.; Jiang, D. Expression of KIF2A, NDC80, CDK1, and CCNB1 in breast cancer patients: Their interaction and linkage with tumor features and prognosis. J. Clin. Lab. Anal. 2022, 36, e24647. [Google Scholar] [CrossRef]
- Xing, Z.; Wang, X.; Liu, J.; Zhang, M.; Feng, K.; Wang, X. Expression and prognostic value of CDK1, CCNA2, and CCNB1 gene clusters in human breast cancer. J. Int. Med. Res. 2021, 49. [Google Scholar] [CrossRef]
- Huang, Z.; Shen, G.; Gao, J. CDK1 promotes the stemness of lung cancer cells through interacting with Sox2. Clin. Transl. Oncol. 2021, 23, 1743–1751. [Google Scholar] [CrossRef]
- Ravindran Menon, D.; Luo, Y.; Arcaroli, J.J.; Liu, S.; Krishnankutty, L.N.; Osborne, D.G.; Li, Y.; Samson, J.M.; Bagby, S.; Tan, A.C.; et al. CDK1 Interacts with Sox2 and Promotes Tumor Initiation in Human Melanoma. Cancer Res. 2018, 78, 6561–6574. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Yan, Q.; Ge, J.; Dou, G.; Zhao, G. Identification of driver genes and key pathways of ependymoma. Turk. Neurosurg. 2018. [Google Scholar] [CrossRef]
- Pérez-Ramírez, M.; Hernández-Jiménez, A.J.; Guerrero-Guerrero, A.; Benadón-Darszon, E.; Pérezpeña-Díazconti, M.; Siordia-Reyes, A.G.; García-Méndez, A.; de León, F.C.-P.; Salamanca-Gómez, F.A.; García-Hernández, N. Genomics and epigenetics: A study of ependymomas in pediatric patients. Clin. Neurol. Neurosurg. 2016, 144, 53–58. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, L.; Jiang, J.; Zhang, Y.; Wang, X.; Zhang, Q.; Wang, Y.; Liu, C.; Li, F. CDK1 and CCNB1 as potential diagnostic markers of rhabdomyosarcoma: Validation following bioinformatics analysis. BMC Med. Genom. 2019, 12, 198. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Sun, C.; Chen, H.; Zhang, C.; Li, W.; Wu, L.; Zhu, J.; Sun, F.; Huang, J.; Wang, J.; et al. Bioinformatics Analysis and Validation Identify CDK1 and MAD2L1 as Prognostic Markers of Rhabdomyosarcoma. Cancer Manag. Res. 2020, 12, 12123–12136. [Google Scholar] [CrossRef]
- Schwermer, M.; Lee, S.; Köster, J.; van Maerken, T.; Stephan, H.; Eggert, A.; Morik, K.; Schulte, J.H.; Schramm, A. Sensitivity to cdk1-inhibition is modulated by p53 status in preclinical models of embryonal tumors. Oncotarget 2015, 6, 15425–15435. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Zhu, X.; Wu, J.; Chen, Y.; Zhang, J.; Sun, X. Centromere protein E as a novel biomarker and potential therapeutic target for retinoblastoma. Bioengineered 2021, 12, 5950–5970. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Xie, X.; Wang, Z.; Lei, Q. Identification of potential crucial genes and key pathways in osteosarcoma. Hereditas 2020, 157, 29. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Y.; Reiter, R.J. Melatonin inhibits the proliferation of human osteosarcoma cell line MG-63. Bone 2013, 55, 432–438. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, X.; Li, H.; Li, B.; Sun, L.; Xie, T.; Zhu, T.; Zhou, H.; Ye, Z. Piperine inhibits proliferation of human osteosarcoma cells via G2/M phase arrest and metastasis by suppressing MMP-2/-9 expression. Int. Immunopharmacol. 2015, 24, 50–58. [Google Scholar] [CrossRef]
- Cai, D.; Latham, V.M.; Zhang, X.; Shapiro, G.I. Combined Depletion of Cell Cycle and Transcriptional Cyclin-Dependent Kinase Activities Induces Apoptosis in Cancer Cells. Cancer Res 2006, 66, 9270–9280. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Huang, Q.; Zhai, D.; Dong, J.; Wang, A.; Lan, Q. CDK1 expression and effects of CDK1 silencing on the malig-nant phenotype of glioma cells. Zhonghua Zhong Liu Za Zhi 2007, 29, 484–488. [Google Scholar]
- Zhou, Y.; Yang, L.; Zhang, X.; Chen, R.; Chen, X.; Tang, W.; Zhang, M. Identification of Potential Biomarkers in Glioblastoma through Bioinformatic Analysis and Evaluating Their Prognostic Value. BioMed Res. Int. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCurdy, S.R.; Pacal, M.; Ahmad, M.; Bremner, R. A CDK2 activity signature predicts outcome in CDK2-low cancers. Oncogene 2016, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2018, 62, 4233–4251. [Google Scholar] [CrossRef]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin E Deregulation Promotes Loss of Specific Genomic Regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2019, 25, 406–413. [Google Scholar] [CrossRef]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjö, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nature 2009, 12, 54–59. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Pang, J.; Yu, Y.; Bieerkehazhi, S.; Lu, J.; Hu, T.; Zhao, Y.; Xu, X.; Zhang, H.; et al. Multiple CDK inhibitor dinaciclib suppresses neuroblastoma growth via inhibiting CDK2 and CDK9 activity. Sci. Rep. 2016, 6, 29090. [Google Scholar] [CrossRef] [Green Version]
- Bo, L.; Wei, B.; Wang, Z.; Kong, D.; Gao, Z.; Miao, Z. Bioinformatics analysis of the CDK2 functions in neuroblastoma. Mol. Med. Rep. 2017, 17, 3951–3959. [Google Scholar] [CrossRef]
- Bolin, S.; Borgenvik, A.; Persson, C.; Rosén, G.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; et al. Abstract 2473: Combined BET-bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Cancer Res 2016, 76, 2473. [Google Scholar] [CrossRef]
- Liu, H.; Weng, J. A comprehensive bioinformatic analysis of cyclin-dependent kinase 2 (CDK2) in glioma. Gene 2022, 822, 146325. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, C.; Zhu, X.; Zhu, X.; Xian, H.; Huang, Z. Suppression of microRNA-125a-5p upregulates the TAZ-EGFR signaling pathway and promotes retinoblastoma proliferation. Cell Signal. 2016, 28, 850–860. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, C.; Cui, H.; Huang, Z. High expression of TAZ indicates a poor prognosis in retinoblastoma. Diagn. Pathol. 2015, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Pazzagli, C.; Born, T.L.; Bertolaet, B.L.; Knudsen, K.; Arden, K.C.; Henry, R.R.; Feramisco, J.R. Elevated cyclins and cyclin-dependent kinase activity in the rhabdomyosarcoma cell line RD. Cancer Res 1998, 58, 2042–2049. [Google Scholar]
- Fu, W.; Ma, L.; Chu, B.; Wang, X.; Bagui, T.K.; Bui, M.M.; Gemmer, J.; Altiok, S.; Letson, D.G.; Pledger, W.J. Abstract 3596: SCH727965, a cyclin-dependent kinases inhibitor, induces apoptosis in sarcoma cells through caspase 3- dependent pathway. Cancer Res 2011, 71, 3596. [Google Scholar] [CrossRef]
- Musa, J.; Cidre-Aranaz, F.; Aynaud, M.-M.; Orth, M.F.; Knott, M.M.L.; Mirabeau, O.; Mazor, G.; Varon, M.; Hölting, T.L.B.; Grossetête, S.; et al. Cooperation of cancer drivers with regulatory germline variants shapes clinical outcomes. Nat. Commun. 2019, 10, 4128. [Google Scholar] [CrossRef] [Green Version]
- Ohali, A.; Avigad, S.; Zaizov, R.; Ophir, R.; Horn-Saban, S.; Cohen, I.J.; Meller, I.; Kollender, Y.; Issakov, J.; Yaniv, I. Prediction of high risk Ewing’s sarcoma by gene expression profiling. Oncogene 2004, 23, 8997–9006. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Leone, G.W.; Wang, H. Cyclin D-CDK4/6 functions in cancer. Adv. Cancer Res. 2020, 148, 147–169. [Google Scholar]
- Dobashi, Y.; Goto, A.; Fukayama, M.; Abe, A.; Ooi, A. Overexpression of cdk4/cyclin D1, a possible mediator of apoptosis and an indicator of prognosis in human primary lung carcinoma. Int. J. Cancer 2004, 110, 532–541. [Google Scholar] [CrossRef]
- Nadal, A.; Jares, P.; Pinyol, M.; Conde, L.; Romeu, C.; Fernández, P.L.; Campo, E.; Cardesa, A. Association of CDK4 and CCND1 mRNA overexpression in laryngeal squamous cell carcinomas occurs without CDK4 amplification. Virchows Arch. 2006, 450, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Kaku-Ito, Y.; Oda, Y.; Ito, T. CDK4: A Novel Therapeutic Target for Extramammary Paget’s Disease. Front. Oncol. 2021, 11, 710378. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sui, L.; Sugimoto, K.; Tai, Y.; Tokuda, M. Cyclin D1-CDK4 complex, a possible critical factor for cell prolifera-tion and prognosis in laryngeal squamous cell carcinomas. Int. J. Cancer 2001, 95, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-J.; Lee, S.-W.; Lin, L.-C.; Lin, C.-Y.; Chang, K.-Y.; Li, C.-F. Cyclin-dependent kinase 4 overexpression is mostly independent of gene amplification and constitutes an independent prognosticator for nasopharyngeal carcinoma. Tumor Biol. 2014, 35, 7209–7216. [Google Scholar] [CrossRef]
- Wu, A.; Wu, B.; Guo, J.; Luo, W.; Wu, D.; Yang, H.; Zhen, Y.; Yu, X.; Wang, H.; Zhou, Y.; et al. Elevated expression of CDK4 in lung cancer. J. Transl. Med. 2011, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.-W.; Lin, Y.-M.; Chang, J.-G.; Yeh, K.-T.; Chen, R.-M.; Tsai, J.J.P.; Su, W.-W.; Hu, R.-M. Clinical implications of deregulated CDK4 and Cyclin D1 expression in patients with human hepatocellular carcinoma. Med. Oncol. 2013, 30, 379. [Google Scholar] [CrossRef]
- An, H.-X.; Beckmann, M.W.; Reifenberger, G.; Bender, H.G.; Niederacher, D. Gene Amplification and Overexpression of CDK4 in Sporadic Breast Carcinomas Is Associated with High Tumor Cell Proliferation. Am. J. Pathol. 1999, 154, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2016, 36, 2255–2264. [Google Scholar] [CrossRef] [Green Version]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Büschenfelde, K.-H.M.Z.; Beach, D. A p16 INK4a -Insensitive CDK4 Mutant Targeted by Cytolytic T Lymphocytes in a Human Melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Kim, H.; Ham, E.K.; Kim, Y.I.; Chi, J.G.; Lee, H.S.; Park, S.H.; Jung, Y.M.; Myung, N.K.; Lee, M.J.; Jang, J.-J. Overexpression of cyclin D1 and cdk4 in tumorigenesis of sporadic hepatoblastomas. Cancer Lett. 1998, 131, 177–183. [Google Scholar] [CrossRef]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, 147, 2988–2995. [Google Scholar] [CrossRef]
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; A Pathak, A.; Brown, N.; A Deshpande, A.; et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia 2015, 30, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, K.; Sexl, V. CDK6 and p16INK4A in lymphoid malignancies. Oncotarget 2013, 4, 1858–1859. [Google Scholar] [CrossRef]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Van der Linden, M.; Willekes, M.; van Roon, E.; Seslija, L.; Schneider, P.; Pieters, R.; Stam, R. MLL fusion-driven activation ofCDK6potentiates proliferation inMLL-rearranged infant ALL. Cell Cycle 2014, 13, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Faussillon, M.; Monnier, L.; Junien, C.; Jeanpierre, C. Frequent overexpression of cyclin D2/cyclin-dependent kinase 4 in Wilms’ tumor. Cancer Lett. 2005, 221, 67–75. [Google Scholar] [CrossRef]
- Haruta, M.; Arai, Y.; Okita, H.; Tanaka, Y.; Takimoto, T.; Sugino, R.P.; Yamada, Y.; Kamijo, T.; Oue, T.; Fukuzawa, M.; et al. Combined Genetic and Chromosomal Characterization of Wilms Tumors Identifies Chromosome 12 Gain as a Potential New Marker Predicting a Favorable Outcome. Neoplasia 2018, 21, 117–131. [Google Scholar] [CrossRef]
- Schubert, N.A.; Chen, C.Y.; Rodríguez, A.; Koster, J.; Dowless, M.; Pfister, S.M.; Shields, D.J.; Stancato, L.F.; Vassal, G.; Caron, H.N.; et al. Target actionability review to evaluate CDK4/6 as a therapeutic target in paediatric solid and brain tumours. Eur. J. Cancer 2022, 170, 196–208. [Google Scholar] [CrossRef]
- Iwata, S.; Tatsumi, Y.; Yonemoto, T.; Araki, A.; Itami, M.; Kamoda, H.; Tsukanishi, T.; Hagiwara, Y.; Kinoshita, H.; Ishii, T.; et al. CDK4 overexpression is a predictive biomarker for resistance to conventional chemotherapy in patients with osteosarcoma. Oncol. Rep. 2021, 46, 1–11. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, J.K.; Yu, Z.; Hornicek, F.J.; Kan, Q.; Duan, Z. Expression and therapeutic implications of cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 1573–1582. [Google Scholar] [CrossRef]
- Wunder, J.S.; Eppert, K.; Burrow, S.R.; Gogkoz, N.; Bell, R.S.; Andrulis, I.L. Co-amplification and overexpression of CDK4, SAS and MDM2 occurs frequently in human parosteal osteosarcomas. Oncogene 1999, 18, 783–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suehara, Y.; Alex, D.; Bowman, A.; Middha, S.; Zehir, A.; Chakravarty, D.; Wang, L.; Jour, G.; Nafa, K.; Hayashi, T.; et al. Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin. Cancer Res. 2019, 25, 6346–6356. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, G.; Tesser-Gamba, F.; Petrilli, A.; Donato-Macedo, C.; Alves, M.; de Lima, F.; Garcia-Filho, R.; Oliveira, R.; Toledo, S. Molecular profiling of osteosarcoma in children and adolescents from different age groups using a next-generation sequencing panel. Cancer Genet. 2021, 258–259, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Hettmer, S.; Linardic, C.M.; Kelsey, A.; Rudzinski, E.R.; Vokuhl, C.; Selfe, J.; Ruhen, O.; Shern, J.F.; Khan, J.; Kovach, A.R.; et al. Molecular testing of rhabdomyosarcoma in clinical trials to improve risk stratification and outcome: A consensus view from European paediatric Soft tissue sarcoma Study Group, Children’s Oncology Group and Cooperative Weichteilsarkom-Studiengruppe. Eur. J. Cancer 2022, 172, 367–386. [Google Scholar] [CrossRef]
- Barr, F.G.; Duan, F.; Smith, L.M.; Gustafson, D.; Pitts, M.; Hammond, S.; Gastier-Foster, J.M. Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: A report from the Children’s Oncology Group. Genes Chromosom. Cancer 2009, 48, 661–672. [Google Scholar] [CrossRef] [Green Version]
- de Andrade, C.R.; Jr, A.T.; Nishimoto, I.N.; Kowalski, L.P.; Lopes, M.A. Rhabdomyosarcoma of the head and neck: A clinicopathological and immunohistochemical analysis of 29 cases. Braz. Dent. J. 2010, 21, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Ragazzini, P.; Gamberi, G.; Pazzaglia, L.; Serra, M.; Magagnoli, G. Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyosarcoma. Histol. Histopathol. 2004, 401–411. [Google Scholar] [CrossRef]
- Saab, R.; Bills, J.L.; Miceli, A.P.; Anderson, C.M.; Khoury, J.D.; Fry, D.W.; Navid, F.; Houghton, P.J.; Skapek, S.X. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol. Cancer Ther. 2006, 5, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Barghi, F.; Shannon, H.E.; Saadatzadeh, M.R.; Bailey, B.J.; Riyahi, N.; Bijangi-Vishehsaraei, K.; Just, M.; Ferguson, M.J.; Pandya, P.H.; Pollok, K.E. Precision Medicine Highlights Dysregulation of the CDK4/6 Cell Cycle Regulatory Pathway in Pediatric, Adolescents and Young Adult Sarcomas. Cancers 2022, 14, 3611. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, J.J.; Ebus, M.E.; Koster, J.; van Sluis, P.; van Noesel, C.J.; Versteeg, R.; Caron, H.N. Cyclin D1 and CDK4 Activity Contribute to the Undifferentiated Phenotype in Neuroblastoma. Cancer Res 2008, 68, 2599–2609. [Google Scholar] [CrossRef] [Green Version]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, L.; Ognibene, M.; Morini, M.; Conte, M.; Di Cataldo, A.; Tondo, A.; D’Angelo, P.; Castellano, A.; Garaventa, A.; Lasorsa, V.A.; et al. Genomic coamplification of CDK4/MDM2/FRS2 is associated with very poor prognosis and atypical clinical features in neuroblastoma patients. Genes Chromosom. Cancer 2019, 59, 277–285. [Google Scholar] [CrossRef]
- Huang, W.; Hao, Z.; Mao, F.; Guo, D. Small Molecule Inhibitors in Adult High-Grade Glioma: From the Past to the Future. Front. Oncol. 2022, 12, 911876. [Google Scholar] [CrossRef]
- Liu, A.; Zhao, H.; Sun, B.; Han, X.; Zhou, D.; Cui, Z.; Ma, X.; Zhang, J.; Yuan, L. A predictive analysis approach for paediatric and adult high-grade glioma: miRNAs and network insight. Ann. Transl. Med. 2020, 8, 242. [Google Scholar] [CrossRef]
- Rallis, K.S.; George, A.M.; Wozniak, A.M.; Bigogno, C.M.; Chow, B.; Hanrahan, J.G.; Sideris, M. Molecular Genetics and Targeted Therapies for Paediatric High-grade Glioma. Cancer Genom.-Proteom. 2022, 19, 390–414. [Google Scholar] [CrossRef]
- Sepúlveda-Sánchez, J.M.; Gil-Gil, M.; Alonso-García, M.; Salgado, M.V.; Vicente, E.; Barroso, C.M.; Sánchez, R.; Durán, G.; Peñas, R.D.L.; Muñoz-Langa, J.; et al. Phase II Trial of Palbociclib in Recurrent Retinoblastoma-Positive Anaplastic Oligodendroglioma: A Study from the Spanish Group for Research in Neuro-Oncology (GEINO). Target. Oncol. 2020, 15, 613–622. [Google Scholar] [CrossRef]
- Zangen, I.L.; Kneitz, S.; Monoranu, C.-M.; Rutkowski, S.; Hinkes, B.; Vince, G.H.; Huang, B.; Roggendorf, W. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. 2007, 113, 325–337. [Google Scholar] [CrossRef]
- Liang, M.-L.; Chen, C.-H.; Liu, Y.-R.; Huang, M.-H.; Lin, Y.-C.; Wong, T.-T.; Lin, S.-E.; Chu, S.-S.; Ding, Y.-H.; Hsieh, T.-H. Abemaciclib, A Selective CDK4/6 Inhibitor, Restricts the Growth of Pediatric Ependymomas. Cancers 2020, 12, 3597. [Google Scholar] [CrossRef]
- Magalhães, T.D.A.; Cruzeiro, G.A.V.; de Sousa, G.R.; da Silva, K.R.; Lira, R.C.P.; Scrideli, C.A.; Tone, L.G.; Valera, E.T.; Borges, K.S. Notch pathway in ependymoma RELA-fused subgroup: Upregulation and association with cancer stem cells markers expression. Cancer Gene Ther. 2019, 27, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Lummus, S.C.; Donson, A.M.; Gowan, K.; Jones, K.L.; Vibhakar, R.; Foreman, N.K.; Kleinschmidt-DeMasters, B.K. p16Loss and E2F/cell cycle deregulation in infant posterior fossa ependymoma. Pediatr. Blood Cancer 2017, 64, e26656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shupp, A.; Casimiro, M.C.; Pestell, R.G. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget 2017, 8, 17373–17382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Sicinski, P. A kinase of many talents: Non-neuronal functions of CDK5 in development and disease. Open Biol. 2020, 10, 190287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.; Rossie, S. Tale of the Good and the Bad Cdk5: Remodeling of the Actin Cytoskeleton in the Brain. Mol. Neurobiol. 2017, 55, 3426–3438. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Song, Y.-S.; Li, Y.; Jia, Y.-H.; Zhao, H.-D. Cdk5 links with DNA damage response and cancer. Mol. Cancer 2017, 16, 60. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.-T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 53, 4328–4342. [Google Scholar] [CrossRef]
- Liang, Q.; Li, L.; Zhang, J.; Lei, Y.; Wang, L.; Liu, D.-X.; Feng, J.; Hou, P.; Yao, R.; Zhang, Y.; et al. CDK5 is essential for TGF-β1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013, 3, 2932. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Xie, S.; Liu, Y.; Shen, C.; Song, X.; Zhou, G.-L.; Wang, C. CDK5 Functions as a Tumor Promoter in Human Lung Cancer. J. Cancer 2018, 9, 3950–3961. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Lin, T.-Y.; Juang, J.-L. Abl deregulates Cdk5 kinase activity and subcellular localization in Drosophila neurodegeneration. Cell Death Differ. 2006, 14, 607–615. [Google Scholar] [CrossRef]
- de Porras, V.R.; Bystrup, S.; Heras, S.C.-D.L.; Musulén, E.; Palomero, L.; Alonso, M.H.; Nieto, R.; Arango, D.; Moreno, V.; Queralt, C.; et al. Tumor Expression of Cyclin-Dependent Kinase 5 (Cdk5) Is a Prognostic Biomarker and Predicts Outcome of Oxaliplatin-Treated Metastatic Colorectal Cancer Patients. Cancers 2019, 11, 1540. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, X.; Lv, P.; Yu, H.; Jiang, X. CDK5 Knockdown inhibits proliferation and induces apoptosis and Cell Cycle Arrest in Human Glioblastoma. J. Cancer 2021, 12, 3958–3966. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.-C.; Chiu, C.-Y.; Song, Y.-M.; Lin, S.-Y. Cdk5 Regulates STAT3 Activation and Cell Proliferation in Medullary Thyroid Carcinoma Cells. J. Biol. Chem. 2007, 282, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- Oner, M.; Lin, E.; Chen, M.-C.; Hsu, F.-N.; Prince, G.M.S.H.; Chiu, K.-Y.; Teng, C.-L.J.; Yang, T.-Y.; Wang, H.-Y.; Yue, C.-H.; et al. Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications. Int. J. Mol. Sci. 2019, 20, 3881. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.-H.; Chen, M.-C.; Peng, Y.-T.; Kao, W.-H.; Chang, C.-H.; Wang, Y.-C.; Lai, C.-H.; Hsieh, J.-T.; Wang, J.-H.; Lee, Y.-T.; et al. Cdk5 Directly Targets Nuclear p21CIP1 and Promotes Cancer Cell Growth. Cancer Res 2016, 76, 6888–6900. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-Q.; Xie, J.-W.; Xie, H.-T.; Chen, P.-C.; Zhang, X.-L.; Zheng, C.-H.; Li, P.; Wang, J.-B.; Lin, J.-X.; Cao, L.-L.; et al. Expression of CRM1 and CDK5 shows high prognostic accuracy for gastric cancer. World J. Gastroenterol. 2017, 23, 2012–2022. [Google Scholar] [CrossRef]
- Kour, S.; Rana, S.; Contreras, J.I.; King, H.M.; Robb, C.M.; Sonawane, Y.A.; Bendjennat, M.; Crawford, A.J.; Barger, C.J.; Kizhake, S.; et al. CDK5 Inhibitor Downregulates Mcl-1 and Sensitizes Pancreatic Cancer Cell Lines to Navitoclax. Mol. Pharmacol. 2019, 96, 419–429. [Google Scholar] [CrossRef]
- Mukherjee, S.; Tucker-Burden, C.; Kaissi, E.; Newsam, A.; Duggireddy, H.; Chau, M.; Zhang, C.; Diwedi, B.; Rupji, M.; Seby, S.; et al. CDK5 Inhibition Resolves PKA/cAMP-Independent Activation of CREB1 Signaling in Glioma Stem Cells. Cell Rep. 2018, 23, 1651–1664. [Google Scholar] [CrossRef] [Green Version]
- Do, P.A.; Lee, C.H. The Role of CDK5 in Tumours and Tumour Microenvironments. Cancers 2020, 13, 101. [Google Scholar] [CrossRef]
- Gao, G.-B.; Sun, Y.; Fang, R.-D.; Wang, Y.; Wang, Y.; He, Q.-Y. Post-translational modifications of CDK5 and their biological roles in cancer. Mol. Biomed. 2021, 2, 1–15. [Google Scholar] [CrossRef]
- Peyressatre, M.; Laure, A.; Pellerano, M.; Boukhaddaoui, H.; Soussi, I.; Morris, M.C. Fluorescent Biosensor of CDK5 Kinase Activity in Glioblastoma Cell Extracts and Living Cells. Biotechnol. J. 2020, 15, e1900474. [Google Scholar] [CrossRef]
- Binlateh, T.; Reudhabibadh, R.; Prommeenate, P.; Hutamekalin, P. Investigation of mechanisms underlying the inhibitory effects of metformin against proliferation and growth of neuroblastoma SH-SY5Y cells. Toxicol. Vitr. 2022, 83, 105410. [Google Scholar] [CrossRef] [PubMed]
- Yushan, R.; Wenjie, C.; Suning, H.; Yiwu, D.; Tengfei, Z.; Madushi, W.M.; Feifei, L.; Changwen, Z.; Xin, W.; Roodrajeetsing, G.; et al. Insights into the clinical value of cyclin-dependent kinase 5 in glioma: A retrospective study. World J. Surg. Oncol. 2015, 13, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catania, A.; Urban, S.; Yan, E.; Hao, C.; Barron, G.; Allalunis-Turner, J. Expression and localization of cyclin-dependent kinase 5 in apoptotic human glioma cells. Neuro-Oncology 2001, 3, 89–98. [Google Scholar] [CrossRef] [PubMed]
- de Nigris, F.; Mancini, F.P.; Schiano, C.; Infante, T.; Zullo, A.; Minucci, P.B.; Al-Omran, M.; Giordano, A.; Napoli, C. Osteosarcoma cells induce endothelial cell proliferation during neo-angiogenesis. J. Cell. Physiol. 2012, 228, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.-X.; Bi, Q.; Han, Y.; Zhao, C.; Zou, H. Potential mechanisms underlying CDK5 related Osteosarcoma progression. Expert Opin. Ther. Targets 2017, 21, 455–460. [Google Scholar] [CrossRef]
- Fu, H.; Zhao, H.; Yang, Y.; Duan, K.; Guo, T. CDK5 Inhibitor Seliciclib Promotes Osteoblastic Differentiation of MSCs and Suppresses the Migration of MG-63 Osteosarcoma Cells. BioRixv 2020. [Google Scholar] [CrossRef]
- Saidak, Z.; Le Henaff, C.; Azzi, S.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Wnt/β-Catenin Signaling Mediates Osteoblast Differentiation Triggered by Peptide-induced α5β1 Integrin Priming in Mesenchymal Skeletal Cells. J. Biol. Chem. 2015, 290, 6903–6912. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Schachter, M.M.; Fisher, R.P. The CDK-activating kinase Cdk7: Taking yes for an answer. Cell Cycle 2013, 12, 3239–3240. [Google Scholar] [CrossRef] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.P. Cdk7: A kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2018, 10, 47–56. [Google Scholar] [CrossRef]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2018, 10, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Abduljabbar, R.; Lai, C.-F.; Periyasamy, M.; Harrod, A.; Gemma, C.; Steel, J.H.; Patel, N.; Busonero, C.; Jerjees, D.; et al. Expression of CDK7, Cyclin H, and MAT1 Is Elevated in Breast Cancer and Is Prognostic in Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2016, 22, 5929–5938. [Google Scholar] [CrossRef] [Green Version]
- Naseh, G.; Mohammadifard, M. Upregulation of cyclin-dependent kinase 7 and matrix metalloproteinase-14 expression contribute to metastatic properties of gastric cancer. IUBMB Life 2016, 68, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Jagomast, T.; Idel, C.; Klapper, L.; Kuppler, P.; Offermann, A.; Dreyer, E.; Bruchhage, K.-L.; Ribbat-Idel, J.; Perner, S. CDK7 Predicts Worse Outcome in Head and Neck Squamous-Cell Cancer. Cancers 2022, 14, 492. [Google Scholar] [CrossRef]
- Kim, J.; Cho, Y.-J.; Ryu, J.-Y.; Hwang, I.; Han, H.D.; Ahn, H.J.; Kim, W.Y.; Cho, H.; Chung, J.-Y.; Hewitt, S.M.; et al. CDK7 is a reliable prognostic factor and novel therapeutic target in epithelial ovarian cancer. Gynecol. Oncol. 2020, 156, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Zhu, C.; Jin, J.; Wang, X.; Yu, L.; Guan, X. Expression of CDK7 correlates with molecular subtypes and predicts clinical outcomes in breast cancer. Transl. Cancer Res. 2021, 10, 669–680. [Google Scholar] [CrossRef]
- Kretz, A.-L.; Schaum, M.; Richter, J.; Kitzig, E.F.; Engler, C.C.; Leithäuser, F.; Henne-Bruns, D.; Knippschild, U.; Lemke, J. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Liu, S.; Luo, Q.; Tan, X. Expression of CDK9 in endometrial cancer tissues and its effect on the proliferation of HEC-1B. Open Life Sci. 2021, 16, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Rasool, R.U.; Natesan, R.; Deng, Q.; Aras, S.; Lal, P.; Effron, S.S.; Mitchell-Velasquez, E.; Posimo, J.M.; Carskadon, S.; Baca, S.C.; et al. CDK7 Inhibition Suppresses Castration-Resistant Prostate Cancer through MED1 Inactivation. Cancer Discov. 2019, 9, 1538–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Liang, J.; Wang, L.; Zhang, Z.; Yuan, P.; Wang, J.; Gao, Y.; Ma, F.; Calagua, C.; Ye, H.; et al. Phosphorylation of the androgen receptor at Ser81 is co-sustained by CDK1 and CDK9 and leads to AR-mediated transactivation in prostate cancer. Mol. Oncol. 2021, 15, 1901–1920. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Dean, D.C.; Wei, R.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 7 (CDK7) is an emerging prognostic biomarker and therapeutic target in osteosarcoma. Ther. Adv. Musculoskelet. Dis. 2021, 13. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Zou, C.; Zhao, Z.; Lai, Y.; Shi, Z.; Xie, X.; Huang, G.; Wang, Y.; Zhang, X.; et al. Targeting Super-Enhancer–Associated Oncogenes in Osteosarcoma with THZ2, a Covalent CDK7 Inhibitor. Clin. Cancer Res. 2020, 26, 2681–2692. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Seebacher, N.A.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. Ebiomedicine 2018, 39, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.-J. Is CDK9 a promising target for both primary and metastatic osteosarcoma? Ebiomedicine 2019, 40, 27–28. [Google Scholar] [CrossRef] [Green Version]
- Iniguez, A.B.; Stolte, B.; Wang, E.J.; Conway, A.S.; Alexe, G.; Dharia, N.V.; Kwiatkowski, N.; Zhang, T.; Abraham, B.J.; Mora, J.; et al. EWS/FLI Confers Tumor Cell Synthetic Lethality to CDK12 Inhibition in Ewing Sarcoma. Cancer Cell 2018, 33, 202–216.e6. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.; Ma, X.; Long, C.; Mao, Y.; Kuang, X.; Huang, Z.; Fan, Y.; Zhang, H.; Xia, Q.; Wang, R.; et al. Anti-tumor Drug THZ1 Suppresses TGFβ2-mediated EMT in Lens Epithelial Cells via Notch and TGFβ/Smad Signaling Pathway. J. Cancer 2019, 10, 3778–3788. [Google Scholar] [CrossRef] [Green Version]
- Cassandri, M.; Fioravanti, R.; Pomella, S.; Valente, S.; Rotili, D.; Del Baldo, G.; De Angelis, B.; Rota, R.; Mai, A. CDK9 as a Valuable Target in Cancer: From Natural Compounds Inhibitors to Current Treatment in Pediatric Soft Tissue Sarcomas. Front. Pharmacol. 2020, 11, 1230. [Google Scholar] [CrossRef]
- Simone, C.; Giordano, A. Abrogation of signal-dependent activation of the cdk9/cyclin T2a complex in human RD rhabdomyosarcoma cells. Cell Death Differ. 2006, 14, 192–195. [Google Scholar] [CrossRef] [Green Version]
- Richter, G.H.; Hensel, T.; Schmidt, O.; Saratov, V.; von Heyking, K.; Becker-Dettling, F.; Prexler, C.; Yen, H.-Y.; Steiger, K.; Fulda, S.; et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers 2020, 12, 304. [Google Scholar] [CrossRef] [Green Version]
- De Falco, G.; Bellan, C.; D’Amuri, A.; Angeloni, G.; Leucci, E.; Giordano, A.; Leoncini, L. Cdk9 regulates neural differentiation and its expression correlates with the differentiation grade of neuroblastoma and PNET tumors. Cancer Biol. Ther. 2005, 4, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Poon, E.; Liang, T.; Jamin, Y.; Walz, S.; Kwok, C.; Hakkert, A.; Barker, K.; Urban, Z.; Thway, K.; Zeid, R.; et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J. Clin. Investig. 2020, 130, 5875–5892. [Google Scholar] [CrossRef]
- Hamanaka, R.; Maloid, S.; Smith, M.R.; O’Connell, C.D.; Longo, D.L.; Ferris, D.K. Cloning and characterization of hu-man and murine homologues of the Drosophila polo serine-threonine kinase. Cell Growth Differ. 1994, 5, 249–257. [Google Scholar]
- Johnson, E.F.; Stewart, K.; Woods, K.W.; Giranda, V.L.; Luo, Y. Pharmacological and Functional Comparison of the Polo-like Kinase Family: Insight into Inhibitor and Substrate Specificity. Biochemistry 2007, 46, 9551–9563. [Google Scholar] [CrossRef]
- Cizmecioglu, O.; Warnke, S.; Arnold, M.; Duensing, S.; Hoffmann, I. Plk2 regulated centriole duplication is dependent on its localization to the centrosome and a functional polo-box domain. Cell Cycle 2008, 7, 3548–3555. [Google Scholar] [CrossRef]
- Archambault, V.; Glover, D. Polo-like kinases: Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009, 10, 265–275. [Google Scholar] [CrossRef]
- Glover, D.M.; Hagan, I.M.; Tavares, A. Polo-like kinases: A team that plays throughout mitosis. Genes Dev. 1998, 12, 3777–3787. [Google Scholar] [CrossRef] [Green Version]
- Barr, F.; Silljé, H.H.W.; Nigg, E. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–441. [Google Scholar] [CrossRef] [PubMed]
- A Winkles, J.; Alberts, G.F. Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 2005, 24, 260–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747. [Google Scholar] [CrossRef] [PubMed]
- Raab, C.A.; Raab, M.; Becker, S.; Strebhardt, K. Non-mitotic functions of polo-like kinases in cancer cells. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1875, 188467. [Google Scholar] [CrossRef]
- Cholewa, B.D.; Liu, X.; Ahmad, N. The Role of Polo-like Kinase 1 in Carcinogenesis: Cause or Consequence? Cancer Res 2013, 73, 6848–6855. [Google Scholar] [CrossRef] [Green Version]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Pellegrino, R.; Calvisi, D.F.; Ladu, S.; Ehemann, V.; Staniscia, T.; Evert, M.; Dombrowski, F.; Schirmacher, P.; Longerich, T. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology 2009, 51, 857–868. [Google Scholar] [CrossRef]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like Kinase 1 (Plk1) Inhibits p53 Function by Physical Interaction and Phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, H.; Cao, Y.; Geng, H.; Ren, F.; Li, K.; Dai, C.; Li, N. Integrated bioinformatics analysis reveals CDK1 and PLK1 as potential therapeutic targets of lung adenocarcinoma. Medicine 2021, 100, e26474. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Sun, Z.; Guo, Q.; Shi, H.; Jia, Y. The clinical and prognostic value of polo-like kinase 1 in lung squamous cell carcinoma patients: Immunohistochemical analysis. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Ramani, P.; Nash, R.; Sowa-Avugrah, E.; A Rogers, C. High levels of polo-like kinase 1 and phosphorylated translationally controlled tumor protein indicate poor prognosis in neuroblastomas. J. Neuro-Oncology 2015, 125, 103–111. [Google Scholar] [CrossRef]
- Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Donson, A.M.; Knipstein, J.; Dubuc, A.; Taylor, M.D.; Handler, M.H.; Foreman, N.K.; et al. Polo-like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells. BMC Cancer 2012, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Triscott, J.; Lee, C.; Foster, C.; Manoranjan, B.; Pambid, M.R.; Berns, R.; Fotovati, A.; Venugopal, C.; O’Halloran, K.; Narendran, A.; et al. Personalizing the Treatment of Pediatric Medulloblastoma: Polo-like Kinase 1 as a Molecular Target in High-Risk Children. Cancer Res 2013, 73, 6734–6744. [Google Scholar] [CrossRef] [Green Version]
- Pezuk, J.A.; Brassesco, M.S.; de Oliveira, R.S.; Machado, H.R.; Neder, L.; Scrideli, C.A.; Tone, L.G. PLK1-associated microRNAs are correlated with pediatric medulloblastoma prognosis. Child’s Nerv. Syst. 2017, 33, 609–615. [Google Scholar] [CrossRef]
- Ma, H.; Nie, C.; Chen, Y.; Li, J.; Xie, Y.; Tang, Z.; Gao, Y.; Ai, S.; Mao, Y.; Sun, Q.; et al. Therapeutic Targeting PLK1 by ON-01910.Na Is Effective in Local Treatment of Retinoblastoma. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2021, 28, 745–761. [Google Scholar] [CrossRef]
- Singh, L.; Pushker, N.; Sen, S.; Singh, M.K.; A Chauhan, F.; Kashyap, S. Prognostic significance of polo-like kinases in retinoblastoma: Correlation with patient outcome, clinical and histopathological parameters. Clin. Exp. Ophthalmol. 2015, 43, 550–557. [Google Scholar] [CrossRef]
- Ackermann, S.; Goeser, F.; Schulte, J.H.; Schramm, A.; Ehemann, V.; Hero, B.; Eggert, A.; Berthold, F.; Fischer, M. Polo-Like Kinase 1 is a Therapeutic Target in High-Risk Neuroblastoma. Clin. Cancer Res. 2011, 17, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Mo, H.; He, J.; Yuan, Z.; Wu, Z.; Liu, B.; Lin, X.; Guan, J. PLK1 contributes to autophagy by regulating MYC stabilization in osteosarcoma cells. OncoTargets Ther. 2019, 12, 7527–7536. [Google Scholar] [CrossRef] [Green Version]
- Mountzios, G.; Terpos, E.; Dimopoulos, M. Aurora kinases as targets for cancer therapy. Cancer Treat. Rev. 2008, 34, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2014, 34, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toji, S.; Yabuta, N.; Hosomi, T.; Nishihara, S.; Kobayashi, T.; Suzuki, S.; Tamai, K.; Nojima, H. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells 2004, 9, 383–397. [Google Scholar] [CrossRef]
- Naso, F.D.; Boi, D.; Ascanelli, C.; Pamfil, G.; Lindon, C.; Paiardini, A.; Guarguaglini, G. Nuclear localisation of Aurora-A: Its regulation and significance for Aurora-A functions in cancer. Oncogene 2021, 40, 3917–3928. [Google Scholar] [CrossRef] [PubMed]
- Marumoto, T.; Honda, S.; Hara, T.; Nitta, M.; Hirota, T.; Kohmura, E.; Saya, H. Aurora-A Kinase Maintains the Fidelity of Early and Late Mitotic Events in HeLa Cells. J. Biol. Chem. 2003, 278, 51786–51795. [Google Scholar] [CrossRef] [Green Version]
- Crane, R.; Gadea, B.; Littlepage, L.; Wu, H.; Ruderman, J.V. Aurora A, Meiosis and Mitosis. Biol. Cell 2004, 96, 215–229. [Google Scholar] [CrossRef]
- Musacchio, A.; Hardwick, K.G. The spindle checkpoint: Structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 731–741. [Google Scholar] [CrossRef]
- Werner, M.; Mattis, A.; Aubele, M.; Cummings, M.; Zitzelsberger, H.; Hutzler, P.; Höfler, H. 20q13.2 Amplification in intraductal hyperplasia adjacent to in situ and invasive ductal carcinoma of the breast. Virchows Arch. 1999, 435, 469–472. [Google Scholar] [CrossRef]
- Bui, V.-M.; Mettling, C.; Jou, J.; Sun, H.S. Genomic amplification of chromosome 20q13.33 is the early biomarker for the development of sporadic colorectal carcinoma. BMC Med. Genom. 2020, 13, 149. [Google Scholar] [CrossRef]
- Tanner, M.M.; Tirkkonen, M.; Kallioniemi, A.; Holli, K.; Collins, C.; Kowbel, D.; Gray, J.W.; Kallioniemi, O.; Isola, J. Amplification of chromosomal region 20q13 in invasive breast cancer: Prognostic implications. Clin. Cancer Res. 1995, 1, 1455–1461. [Google Scholar]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.; Inamdar, K.V.; Wilcox, R.A. The role of aurora A and polo-like kinases in high-risk lymphomas. Blood Adv. 2019, 3, 1778–1787. [Google Scholar] [CrossRef] [Green Version]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics 2020, 10, 6928–6945. [Google Scholar] [CrossRef]
- Wan, X.-B.; Long, Z.-J.; Yan, M.; Xu, J.; Xia, L.-P.; Liu, L.; Zhao, Y.; Huang, X.-F.; Wang, X.-R.; Zhu, X.-F.; et al. Inhibition of Aurora-A suppresses epithelial–mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinog. 2008, 29, 1930–1937. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.B.; Silva, F.N.M.; Golemis, E.A. Aurora Kinases as Therapeutic Targets in Head and Neck Cancer. Cancer J. 2022, 28, 387–400. [Google Scholar] [CrossRef]
- Do, T.-V.; Xiao, F.; E Bickel, L.; Klein-Szanto, A.J.; Pathak, H.B.; Hua, X.; Howe, C.; O’Brien, S.W.; Maglaty, M.; A Ecsedy, J.; et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene 2013, 33, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Wang, J.; Treekitkarnmongkol, W.; Kawai, H.; Sasai, K.; Zhang, H.; Wang, H.; Adams, H.P.; Jiang, S.; Chakraborty, S.N.; et al. Aurora Kinase-A Inactivates DNA Damage-Induced Apoptosis and Spindle Assembly Checkpoint Response Functions of p73. Cancer Cell 2012, 21, 196–211. [Google Scholar] [CrossRef] [Green Version]
- Anand, S.; Penrhyn-Lowe, S.; Venkitaraman, A.R. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 2003, 3, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Cirak, Y.; Furuncuoglu, Y.; Yapicier, O.; Aksu, A.; Cubukcu, E. Aurora A overexpression in breast cancer patients induces taxane resistance and results in worse prognosis. J. BUON. 2016, 20, 1414–1419. [Google Scholar]
- Reiter, R.; Gais, P.; Jütting, U.; Steuer-Vogt, M.K.; Pickhard, A.; Bink, K.; Rauser, S.; Lassmann, S.; Höfler, H.; Werner, M.; et al. Aurora Kinase A Messenger RNA Overexpression Is Correlated with Tumor Progression and Shortened Survival in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2006, 12, 5136–5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, E.-M.; Lee, Y.-R.; Hong, O.-Y.; Jung, S.H.; Youn, H.J.; Kim, J.-S. Aurora kinases are essential for PKC-induced invasion and matrix metalloproteinase-9 expression in MCF-7 breast cancer cells. Oncol. Rep. 2015, 34, 803–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N.; Lin, Y.G.; Immaneni, A.; Deavers, M.T.; Merritt, W.M.; Spannuth, W.A.; Bodurka, D.C.; Gershenson, D.M.; Brinkley, W.R.; Sood, A.K. Overexpression of the Centrosomal Protein Aurora-A Kinase is Associated with Poor Prognosis in Epithelial Ovarian Cancer Patients. Clin. Cancer Res. 2007, 13, 4098–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuncel, H.; Shimamoto, F.; Qi, H.K.; Aoki, E.; Jikihara, H.; Nakai, S.; Takata, T.; Tatsuka, M. Nuclear Aurora B and cytoplasmic Survivin expression is involved in lymph node metastasis of colorectal cancer. Oncol. Lett. 2012, 3, 1109–1114. [Google Scholar] [CrossRef] [Green Version]
- Rannou, Y.; Troadec, M.-B.; Petretti, C.; Hans, F.; Dutertre, S.; Dimitrov, S.; Prigent, C. Localization of aurora A and aurora B kinases during interphase: Role of the N-terminal domain. Cell Cycle 2008, 7, 3012–3020. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Wei, P.; Zhang, H.; Ding, Z.; Yang, L.; Huang, Z.; Zhang, N. AURKA Governs Self-Renewal Capacity in Glioma-Initiating Cells via Stabilization/Activation of β-catenin/Wnt Signaling. Mol. Cancer Res. 2013, 11, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Li, L.; Zhu, J.; Liu, J.; Lin, A.; Fu, W.; Liu, G.; Xia, H.; Zhang, T.; He, J. AURKA rs8173 G>C Polymorphism Decreases Wilms Tumor Risk in Chinese Children. J. Oncol. 2019, 2019, 9074908–7. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M. Unholy Matrimony: Aurora A and N-Myc as Malignant Partners in Neuroblastoma. Cancer Cell 2009, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Ommer, J.; Selfe, J.L.; Wachtel, M.; O’Brien, E.M.; Laubscher, D.; Roemmele, M.; Kasper, S.; Delattre, O.; Surdez, D.; Petts, G.; et al. Aurora A Kinase Inhibition Destabilizes PAX3-FOXO1 and MYCN and Synergizes with Navitoclax to Induce Rhabdomyosarcoma Cell Death. Cancer Res 2020, 80, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Meller, I.; Issakov, J.; Orr-Urtreger, A. Novel Genes Implicated in Embryonal, Alveolar, and Pleomorphic Rhabdomyosarcoma: A Cytogenetic and Molecular Analysis of Primary Tumors. Neoplasia 2006, 8, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Li, Z.; Huang, Y.; Xiong, C.; Zhang, C.; Liang, H.; Xu, J.; Luo, X. A Novel Ferroptosis-Related Gene Signature for Prognosis Prediction in Ewing Sarcoma. Anal. Cell Pathol. 2022, 2022, 1–22. [Google Scholar] [CrossRef]
- Liao, H.; Xie, X.; Xu, Y.; Huang, G. Identification of driver genes associated with chemotherapy resistance of Ewing’s sarcoma. OncoTargets Ther. 2018, 11, 6947–6956. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-T.; Liu, A.-G.; Cai, K.-T.; He, R.-Q.; Li, Z.; Wei, Q.-J.; Chen, M.-Y.; Huang, J.-Y.; Yan, W.-Y.; Zhou, H.; et al. Exploration and validation of downregulated microRNA-199a-3p, downstream messenger RNA targets and transcriptional regulation in osteosarcoma. Am. J. Transl. Res. 2019, 11, 7538–7554. [Google Scholar]
- Zhu, X.; Mei, J.; Wang, Z. Aurora-A kinase: Potential tumor marker of osteosarcoma. J. Cancer Res. Ther. 2014, 10, 102–107. [Google Scholar] [CrossRef]
- Yang, W.; Jiang, X.; Zhao, X.; Mao, P. Treatment of RB -deficient retinoblastoma with Aurora-A kinase inhibitor. Kaohsiung J. Med. Sci. 2021, 38, 244–252. [Google Scholar] [CrossRef]
- Lehman, N.L.; O’Donnell, J.P.; Whiteley, L.J.; Stapp, R.T.; Lehman, T.D.; Roszka, K.M.; Schultz, L.R.; Williams, C.J.; Mikkelsen, T.; Brown, S.L.; et al. Aurora A is differentially expressed in gliomas, is associated with patient survival in glioblastoma and is a potential chemotherapeutic target in gliomas. Cell Cycle 2012, 11, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Zhou, Y.; Jiao, J.; Xu, L.; Yan, Y.; Wu, Q.; Tong, X.; Yan, H. Integrated Analysis of Transcriptome Data Revealed AURKA and KIF20A as Critical Genes in Medulloblastoma Progression. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Vader, G.; Medema, R.; Lens, S.M. The chromosomal passenger complex: Guiding Aurora-B through mitosis. J. Cell Biol. 2006, 173, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Vagnarelli, P.; Earnshaw, W.C. Chromosomal passengers: The four-dimensional regulation of mitotic events. Chromosoma 2004, 113, 211–222. [Google Scholar] [CrossRef]
- Adams, R.R.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001, 11, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, Y.; Kawashima, T.; Hirose, K.; Tonozuka, Y.; Kawajiri, A.; Bao, Y.C.; Deng, X.; Tatsuka, M.; Narumiya, S.; May, W.; et al. Phosphorylation by Aurora B Converts MgcRacGAP to a RhoGAP during Cytokinesis. Dev. Cell 2003, 4, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-Y.; Sun, Z.-W.; Li, X.; Reuben, M.; Tatchell, K.; Bishop, D.K.; Grushcow, J.M.; Brame, C.J.; A Caldwell, J.; Hunt, D.F.; et al. Mitotic Phosphorylation of Histone H3 Is Governed by Ipl1/aurora Kinase and Glc7/PP1 Phosphatase in Budding Yeast and Nematodes. Cell 2000, 102, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murnion, M.E.; Adams, R.R.; Callister, D.M.; Allis, C.D.; Earnshaw, W.C.; Swedlow, J.R. Chromatin-associated Protein Phosphatase 1 Regulates Aurora-B and Histone H3 Phosphorylation. J. Biol. Chem. 2001, 276, 26656–26665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, W.; Zhang, X.; Kline-Smith, S.L.; E Rosasco, S.; A Barrett-Wilt, G.; Shabanowitz, J.; Hunt, D.F.; E Walczak, C.; Stukenberg, T. Aurora B Phosphorylates Centromeric MCAK and Regulates Its Localization and Microtubule Depolymerization Activity. Curr. Biol. 2004, 14, 273–286. [Google Scholar] [CrossRef]
- Ma, H.T.; Poon, R.Y. Aurora kinases and DNA damage response. Mutat. Res. Mol. Mech. Mutagen. 2020, 821, 111716. [Google Scholar] [CrossRef]
- González-Loyola, A.; Fernández-Miranda, G.; Trakala, M.; Partida, D.; Samejima, K.; Ogawa, H.; Cañamero, M.; de Martino, A.; Martínez-Ramírez, A.; de Cárcer, G.; et al. Aurora B Overexpression Causes Aneuploidy and p21 Cip1 Repression during Tumor Development. Mol. Cell. Biol. 2015, 35, 3566–3578. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, M.; Koga, T.; Takayama, K.; Ijichi, K.; Yano, T.; Maehara, Y.; Nakanishi, Y.; Sueishi, K. Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung Cancer 2013, 80, 85–90. [Google Scholar] [CrossRef]
- Porcelli, L.; Guida, G.; E Quatrale, A.; Cocco, T.; Sidella, L.; Maida, I.; Iacobazzi, R.M.; Ferretta, A.; A Stolfa, D.; Strippoli, S.; et al. Aurora kinase B inhibition reduces the proliferation of metastatic melanoma cells and enhances the response to chemotherapy. J. Transl. Med. 2015, 13, 26. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, J.; Cao, W.; Sun, L.; Sun, H.; Liu, Y. Aurora-B and HDAC synergistically regulate survival and proliferation of lymphoma cell via AKT, mTOR and Notch pathways. Eur. J. Pharmacol. 2016, 779, 1–7. [Google Scholar] [CrossRef]
- Wan, B.; Huang, Y.; Liu, B.; Lu, L.; Lv, C. AURKB: A promising biomarker in clear cell renal cell carcinoma. Peerj 2019, 7, e7718. [Google Scholar] [CrossRef] [Green Version]
- Twu, N.-F.; Yuan, C.-C.; Yen, M.-S.; Lai, C.-R.; Chao, K.-C.; Wang, P.-H.; Wu, H.-H.; Chen, Y.-J. Expression of Aurora kinase A and B in normal and malignant cervical tissue: High Aurora A kinase expression in squamous cervical cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 142, 57–63. [Google Scholar] [CrossRef]
- Pannone, G.; Hindi, S.; Santoro, A.; Sanguedolce, F.; Rubini, C.; Cincione, R.; De Maria, S.; Tortorella, S.; Rocchetti, R.; Cagiano, S.; et al. Aurora B Expression as a Prognostic Indicator and Possibile Therapeutic Target in Oral Squamous Cell Carcinoma. Int. J. Immunopathol. Pharmacol. 2011, 24, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Alafate, W.; Zuo, J.; Deng, Z.; Guo, X.; Wu, W.; Zhang, W.; Xie, W.; Wang, M.; Wang, J. Combined elevation of AURKB and UBE2C predicts severe outcomes and therapy resistance in glioma. Pathol.-Res. Pract. 2019, 215, 152557. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, Y.; Zhang, C.; Zhang, Q. Role of aurora kinase B in regulating resistance to paclitaxel in breast cancer cells. Hum. Cell 2022, 35, 678–693. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, Z.; Wang, G.-H.; Zhou, Y.-M.; Deng, J.-P.; Feng, Y.; Chen, J.-Q.; Tian, L. AURKB Promotes the Metastasis of Gastric Cancer, Possibly by Inducing EMT. Cancer Manag. Res. 2020, 12, 6947–6958. [Google Scholar] [CrossRef]
- Nie, M.; Wang, Y.; Yu, Z.; Li, X.; Deng, Y.; Wang, Y.; Yang, D.; Li, Q.; Zeng, X.; Ju, J.; et al. AURKB promotes gastric cancer progression via activation of CCND1 expression. Aging 2020, 12, 1304–1321. [Google Scholar] [CrossRef]
- Yuan, K.; Wu, M.; Lyu, S.; Li, Y. Identification of prognostic genes for early basal-like breast cancer with weighted gene co-expression network analysis. Medicine 2022, 101, e30581. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, A.; Shen, Y.; Lu, H.M.; Chen, R. Expression and clinical significance of AURKB gene in lung adenocarcinoma. Medicine 2021, 100, e26439. [Google Scholar] [CrossRef]
- Yang, Y.; Sheng, Y.; Sun, D.; Sun, J.; Li, L.; Sun, L. AURKB promotes tumorigenesis and carboplatin resistance by regulating the ERK pathway in neuroblastoma cells. Int. J. Neurosci. 2021, 1–11. [Google Scholar] [CrossRef]
- Bogen, D.; Wei, J.S.; Azorsa, D.O.; Ormanoglu, P.; Buehler, E.; Guha, R.; Keller, J.M.; Griner, L.A.M.; Ferrer, M.; Song, Y.K.; et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget 2015, 6, 35247–35262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartsink-Segers, S.; Zwaan, C.; Exalto, C.; Luijendijk, M.W.J.; Calvert, V.; Petricoin, E.F.; Evans, W.; Reinhardt, D.; De Haas, V.; Hedtjärn, M.; et al. Aurora kinases in childhood acute leukemia: The promise of aurora B as therapeutic target. Leukemia 2012, 27, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saletta, F.; Wadham, C.; Ziegler, D.; Marshall, G.M.; Haber, M.; McCowage, G.; Norris, M.D.; Byrne, J.A. Molecular profiling of childhood cancer: Biomarkers and novel therapies. BBA Clin. 2014, 1, 59–77. [Google Scholar] [CrossRef] [Green Version]
- Gibson, S.E.; Zeng, W.F.; Weil, R.J.; Prayson, R.A. Aurora B Kinase Expression in Ependymal Neoplasms. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 274–278. [Google Scholar] [CrossRef]
- Wang, S.; Hwang, E.E.; Guha, R.; O’Neill, A.F.; Melong, N.; Veinotte, C.J.; Saur, A.C.; Wuerthele, K.; Shen, M.; McKnight, C.; et al. High-throughput Chemical Screening Identifies Focal Adhesion Kinase and Aurora Kinase B Inhibition as a Synergistic Treatment Combination in Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 4552–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Jin, G.; Yao, K.; Liu, K.; Liu, F.; Chen, H.; Wang, K.; Gorja, D.R.; Reddy, K.; Bode, A.M.; et al. Aurora B kinase as a novel molecular target for inhibition the growth of osteosarcoma. Mol. Carcinog. 2019, 58, 1056–1067. [Google Scholar] [CrossRef]
- Borah, N.A.; Sradhanjali, S.; Barik, M.R.; Jha, A.; Tripathy, D.; Kaliki, S.; Rath, S.; Raghav, S.K.; Patnaik, S.; Mittal, R.; et al. Aurora Kinase B Expression, Its Regulation and Therapeutic Targeting in Human Retinoblastoma. Investig. Opthalmology Vis. Sci. 2021, 62, 16. [Google Scholar] [CrossRef]
- Kimura, M.; Matsuda, Y.; Yoshioka, T.; Okano, Y. Cell Cycle-dependent Expression and Centrosome Localization of a Third Human Aurora/Ipl1-related Protein Kinase, AIK3. J. Biol. Chem. 1999, 274, 7334–7340. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Srivastava, V.; Hegde, A.; Kondo, Y.; Shen, L.; Hoshino, K.; Gonzalez, Y.; Wang, J.; Sasai, K.; Ma, X.; et al. Regulation of AURKC expression by CpG island methylation in human cancer cells. Tumor Biol. 2015, 36, 8147–8158. [Google Scholar] [CrossRef]
- Tseng, T.-C.; Chen, S.-H.; Hsu, Y.-P.P.; Tang, T.K. Protein Kinase Profile of Sperm and Eggs: Cloning and Characterization of Two Novel Testis-Specific Protein Kinases (AIE1, AIE2) Related to Yeast and Fly Chromosome Segregation Regulators. DNA Cell Biol. 1998, 17, 823–833. [Google Scholar] [CrossRef]
- Santos, M.A.; van de Werken, C.; de Vries, M.; Jahr, H.; Vromans, M.J.; Laven, J.S.; Fauser, B.C.; Kops, G.J.; Lens, S.M.; Baart, E.B. A role for Aurora C in the chromosomal passenger complex during human preimplantation embryo development. Hum. Reprod. 2011, 26, 1868–1881. [Google Scholar] [CrossRef] [Green Version]
- Khan, J.; Ezan, F.; Crémet, J.-Y.; Fautrel, A.; Gilot, D.; Lambert, M.; Benaud, C.; Troadec, M.-B.; Prigent, C. Overexpression of Active Aurora-C Kinase Results in Cell Transformation and Tumour Formation. PLoS ONE 2011, 6, e26512. [Google Scholar] [CrossRef]
- Yan, X.; Cao, L.; Li, Q.; Wu, Y.; Zhang, H.; Saiyin, H.; Liu, X.; Zhang, X.; Shi, Q.; Yu, L. Aurora C is directly associated with Survivin and required for cytokinesis. Genes Cells 2005, 10, 617–626. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kiyomura, H. The theoretical analysis on the tooth movement (II). Nihon Kyosei Shika Gakkai zasshi = J. Jpn. Orthod. Soc. 1982, 41, 716–722. [Google Scholar]
- Tsou, J.-H.; Chang, K.-C.; Chang-Liao, P.-Y.; Yang, S.-T.; Lee, C.-T.; Chen, Y.-P.; Lee, Y.-C.; Lin, B.-W.; Lee, J.-C.; Shen, M.-R.; et al. Aberrantly expressed AURKC enhances the transformation and tumourigenicity of epithelial cells. J. Pathol. 2011, 225, 243–254. [Google Scholar] [CrossRef]
- Zekri, A.; Lesan, V.; Ghaffari, S.H.; Tabrizi, M.H.; Modarressi, M.H. Gene Amplification and Overexpression of Aurora-C in Breast and Prostate Cancer Cell Lines. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2012, 20, 241–250. [Google Scholar] [CrossRef]
- Seuánez, H.N.; Pereira, H.S.; Lima, S.C.S.; de Faria, P.S.; Cardoso, L.C.D.A. i RPS6KA4 i i MIR1237 i and i AURKC i promoter regions are differentially methylated in Wilms rsquo tumor. Front. Biosci. 2018, 10, 143–154. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Cheung, C.H.Y.; Liu, Y.-L.; Hou, C.-L.; Hsu, C.-L.; Huang, C.-T.; Yang, T.-S.; Chen, S.-F.; Chen, C.-N.; Hsu, W.-M.; et al. Quantitative Proteomics of Th-MYCN Transgenic Mice Reveals Aurora Kinase Inhibitor Altered Metabolic Pathways and Enhanced ACADM To Suppress Neuroblastoma Progression. J. Proteome Res. 2019, 18, 3850–3866. [Google Scholar] [CrossRef]
- Bejar, J.F.; DiSanza, Z.; Quartuccio, S.M. The oncogenic role of meiosis-specific Aurora kinase C in mitotic cells. Exp. Cell Res. 2021, 407, 112803. [Google Scholar] [CrossRef]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar]
- Seabright, M. A rapid banding technique for human chromosomes. Lancet 1971, 298, 971–972. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Heisterkamp, N.; Stephenson, J.R.; Groffen, J.; Hansen, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the c-abl oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 1983, 306, 239–242. [Google Scholar] [CrossRef] [PubMed]
- de Klein, A.; van Kessel, A.G.; Grosveld, G.; Bartram, C.R.; Hagemeijer, A.; Bootsma, D.; Spurr, N.K.; Heisterkamp, N.; Groffen, J.; Stephenson, J.R. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1982, 300, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; De Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Koschmieder, S.; Göttgens, B.; Zhang, P. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a trans-genic model of BCR-ABL leukemogenesis. Blood 2005, 105, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Festuccia, C.; Gravina, G.L.; Biordi, L.; D’Ascenzo, S.; Dolo, V.; Ficorella, C.; Ricevuto, E.; Tombolini, V. Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate cancer cells in vitro. Prostate 2009, 69, 1529–1537. [Google Scholar] [CrossRef]
- Abdelgalil, A.A.; Al-Kahtani, H.M.; Al-Jenoobi, F.I. Erlotinib. Profiles DrugSubst. Excip. Relat. Methodol. 2019, 45, 93–117. [Google Scholar] [CrossRef]
- Ms, K.N.R.; Zweidler-McKay, P.A.; Van Roy, N.; Speleman, F.; Trevino, J.; Zage, P.E.; Hughes, D.P.M. Signaling of ERBB receptor tyrosine kinases promotes neuroblastoma growth in vitro and in vivo. Cancer 2010, 116, 3233–3243. [Google Scholar] [CrossRef]
- Ji, X.L.; He, M. Sodium cantharidate targets STAT3 and abrogates EGFR inhibitor resistance in osteosarcoma. Aging 2019, 11, 5848–5863. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Favours, E.; Phelps, D.A.; Del Pozo, V.; Ghilu, S.; Kurmashev, D.; Michalek, J.; Trevino, A.; Guttridge, D.; London, C.; et al. Evaluation of patritumab with or without erlotinib in combination with standard cytotoxic agents against pediatric sarcoma xenograft models. Pediatr. Blood Cancer 2017, 65, e26870. [Google Scholar] [CrossRef]
- Abraham, J.; Nelon, L.D.; Kubicek, C.B.; Kilcoyne, A.; Hampton, S.T.; Zarzabal, L.A.; Giles, F.J.; Michalek, J.E.; Rubin, B.P.; Keller, C. Preclinical Testing of Erlotinib in a Transgenic Alveolar Rhabdomyosarcoma Mouse Model. Sarcoma 2011, 2011, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Hernan, R.; Fasheh, R.; Calabrese, C.; Frank, A.J.; MacLean, K.H.; Allard, D.; Barraclough, R.; Gilbertson, R.J. ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res 2003, 63, 140–148. [Google Scholar]
- Guan, S.; Shen, R.; Lafortune, T.; Tiao, N.; Houghton, P.; Yung, W.A.; Koul, D. Establishment and characterization of clinically relevant models of ependymoma: A true challenge for targeted therapy. Neuro-Oncology 2011, 13, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Yu, Y.; Zong, R.; Quyang, L.; He, H.; Zhou, Q.; Pei, C. Erlotinib has tumor inhibitory effect in human retinoblastoma cells. Biomed. Pharmacother. 2017, 85, 479–485. [Google Scholar] [CrossRef]
- Jakacki, R.I.; Hamilton, M.; Gilbertson, R.J.; Blaney, S.M.; Tersak, J.; Krailo, M.D.; Ingle, A.M.; Voss, S.D.; Dancey, J.E.; Adamson, P.C. Pediatric Phase I and Pharmacokinetic Study of Erlotinib Followed by the Combination of Erlotinib and Temozolomide: A Children’s Oncology Group Phase I Consortium Study. J. Clin. Oncol. 2008, 26, 4921–4927. [Google Scholar] [CrossRef]
- Geoerger, B.; Hargrave, D.; Thomas, F.; Ndiaye, A.; Frappaz, D.; Andreiuolo, F.; Varlet, P.; Aerts, I.; Riccardi, R.; Jaspan, T.; et al. Innovative Therapies for Children with Cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro-Oncology 2010, 13, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Frampton, J.E. Vandetanib. Drugs 2012, 72, 1423–1436. [Google Scholar] [CrossRef]
- Karras, S.; Anagnostis, P.; E Krassas, G. Vandetanib for the treatment of thyroid cancer: An update. Expert Opin. Drug Metab. Toxicol. 2014, 10, 469–481. [Google Scholar] [CrossRef]
- Hatem, R.; Labiod, D.; Château-Joubert, S.; de Plater, L.; El Botty, R.; Vacher, S.; Bonin, F.; Servely, J.-L.; Dieras, V.; Bièche, I.; et al. Vandetanib as a potential new treatment for estrogen receptor-negative breast cancers. Int. J. Cancer 2016, 138, 2510–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerio, L.; Bottici, V.; Matrone, A.; Piaggi, P.; Viola, D.; Cappagli, V.; Agate, L.; Molinaro, E.; Ciampi, R.; Tacito, A.; et al. Medullary thyroid cancer treated with vandetanib: Predictors of a longer and durable response. Endocrine-Related Cancer 2020, 27, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Xiang, L.; Wang, N.; Zhao, Z.; Jin, X.; Sun, Y.; Duan, W.; Wang, S.; Jin, X. Vandetanib-induced inhibition of neuroblastoma cell migration and invasion is associated with downregulation of the SDF-1/CXCR4 axis and matrix metalloproteinase 14. Oncol. Rep. 2013, 31, 1165–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, C.-W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.-C.; Hsu, J.-M.; Dong, Q.; et al. MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1. Gastroenterology 2019, 156, 1849–1861.e7. [Google Scholar] [CrossRef]
- Beaudry, P.; Nilsson, M.; Rioth, M.; Prox, D.; Poon, D.; Xu, L.; Zweidler-Mckay, P.; Ryan, A.; Folkman, J.; Ryeom, S.; et al. Potent antitumor effects of ZD6474 on neuroblastoma via dual targeting of tumor cells and tumor endothelium. Mol. Cancer Ther. 2008, 7, 418–424. [Google Scholar] [CrossRef] [Green Version]
- Cazes, A.; Lopez-Delisle, L.; Tsarovina, K.; Pierre-Eugène, C.; De Preter, K.; Peuchmaur, M.; Nicolas, A.; Provost, C.; Louis-Brennetot, C.; Daveau, R.; et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 2688–2702. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yang, C.; Wei, G. Vandetanib inhibits cisplatin-resistant neuroblastoma tumor growth and invasion. Oncol. Rep. 2018, 39, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Zage, P.E.; Zeng, L.; Palla, S.; Fang, W.; Nilsson, M.B.; Heymach, J.V.; Zweidler-McKay, P.A. A novel therapeutic combination for neuroblastoma. Cancer 2010, 116, 2465–2475. [Google Scholar] [CrossRef]
- Craveiro, R.B.; Ehrhardt, M.; Velz, J.; Olschewski, M.; Goetz, B.; Pietsch, T.; Dilloo, D. The anti-neoplastic activity of Vandetanib against high-risk medulloblastoma variants is profoundly enhanced by additional PI3K inhibition. Oncotarget 2017, 8, 46915–46927. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, J.; Zhou, L.; Pan, C.; Zhou, Y.; Du, W.; Chen, J.-M.; Zhu, X.; Shen, J.; Chen, S.; et al. ZD6474, a new treatment strategy for human osteosarcoma, and its potential synergistic effect with celecoxib. Oncotarget 2015, 6, 21341–21352. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.K.; Åman, P. Proliferation of Ewing sarcoma cell lines is suppressed by the receptor tyrosine kinase inhibitors gefitinib and vandetanib. Cancer Cell Int. 2008, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Maloney, C.; Kallis, M.P.; Edelman, M.; Tzanavaris, C.; Lesser, M.; Soffer, S.Z.; Symons, M.; Steinberg, B.M. Gefitinib Inhibits Invasion and Metastasis of Osteosarcoma via Inhibition of Macrophage Receptor Interacting Serine-Threonine Kinase 2. Mol. Cancer Ther. 2020, 19, 1340–1350. [Google Scholar] [CrossRef]
- Wakeling, A.E.; Guy, S.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Barker, A.J.; Gibson, K.H. ZD1839 (Iressa): An orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002, 62, 5749–5754. [Google Scholar]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Pomatico, G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res 2000, 6, 2053–2063. [Google Scholar]
- Foreman, N.K.; Gore, L.; Wells, D.; Bs, J.S.; Heideman, R.; Donson, A.M. Gefitinib is effective against juvenile pilocytic astrocytoma in vitro. Pediatr. Blood Cancer 2005, 47, 293–298. [Google Scholar] [CrossRef]
- Schaiquevich, P.; Panetta, J.C.; Throm, S.; Daw, N.C.; Geyer, J.R.; Furman, W.L.; Stewart, C.F. Population pharmacokinetic (PK) analysis of gefitinib in pediatric cancer patients. J. Clin. Oncol. 2008, 26, 2523. [Google Scholar] [CrossRef]
- Daudigeos-Dubus, E.; Le Dret, L.; Lanvers-Kaminsky, C.; Bawa, O.; Opolon, P.; Vievard, A.; Villa, I.; Pagès, M.; Bosq, J.; Vassal, G.; et al. Regorafenib: Antitumor Activity upon Mono and Combination Therapy in Preclinical Pediatric Malignancy Models. PLoS ONE 2015, 10, e0142612. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. In Recent Results in Cancer Research; Springer LLC: New York, NY, USA, 2018; Volume 211, pp. 45–56. [Google Scholar] [CrossRef]
- Subramonian, D.; Phanhthilath, N.; Rinehardt, H.; Flynn, S.; Huo, Y.; Zhang, J.; Messer, K.; Mo, Q.; Huang, S.; Lesperance, J.; et al. Regorafenib is effective against neuroblastoma in vitro and in vivo and inhibits the RAS/MAPK, PI3K/Akt/mTOR and Fos/Jun pathways. Br. J. Cancer 2020, 123, 568–579. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.-W. Dacomitinib, an emerging HER-targeted therapy for non-small cell lung cancer. J. Thorac. Dis. 2012, 4, 639–642. [Google Scholar] [CrossRef]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef]
- Popat, S.; Yap, T. Toward precision medicine with next-generation EGFR inhibitors in non-small-cell lung cancer. Pharmacogenomics Pers. Med. 2014, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endersby, R.; Whitehouse, J.; Hii, H.; Greenall, S.A.; Johns, T.G.; Gottardo, N.G. A Pre-Clinical Assessment of the Pan-ERBB Inhibitor Dacomitinib in Pediatric and Adult Brain Tumors. Neoplasia 2018, 20, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.; Kaklamani, V. Lapatinib and breast cancer: Current indications and outlook for the future. Expert Rev. Anticancer. Ther. 2010, 10, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Bouchalova, K.; Cizkova, M.; Cwiertka, K.; Trojanec, R.; Friedecký, D.; Hajduch, M. Lapatinib in breast cancer—the predictive significance of her1 (egfr), her2, pten and pik3ca genes and lapatinib plasma level assessment. Biomed. Pap. 2010, 154, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Gorlick, R.; Kolb, E.A.; Houghton, P.J.; Morton, C.L.; Phelps, D.; Schaiquevich, P.; Stewart, C.; Keir, S.T.; Lock, R.; Carol, H.; et al. Initial testing (stage 1) of lapatinib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2009, 53, 594–598. [Google Scholar] [CrossRef] [Green Version]
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB family in cancer: Couples therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Herrmann, D.; Seitz, G.; Warmann, S.W.; Bonin, M.; Fuchs, J.; Armeanu-Ebinger, S. Cetuximab Promotes Immunotoxicity Against Rhabdomyosarcoma In Vitro. J. Immunother. 2010, 33, 279–286. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Fukuda, K.; Fuchimoto, Y.; Matsuzaki, Y.; Saikawa, Y.; Kitagawa, Y.; Morikawa, Y.; Kuroda, T. Cetuximab promotes anticancer drug toxicity in rhabdomyosarcomas with EGFR amplification in vitro. Oncol. Rep. 2013, 30, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, A.-M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W.H.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A.; et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003, 101, 3597–3605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Smith, K.M.; Chong, A.L.; Stempak, D.; Yeger, H.; Marrano, P.; Thorner, P.S.; Irwin, M.S.; Kaplan, D.R.; Baruchel, S. In Vivo Antitumor and Antimetastatic Activity of Sunitinib in Preclinical Neuroblastoma Mouse Model. Neoplasia 2009, 11, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M.; Bs, J.C.; Houghton, P.J.; Bs, C.L.M.; Kolb, E.A.; Lock, R.; Bs, M.T.; Reynolds, C.P.; Keir, S.T.; Wu, J.; et al. Initial testing (stage 1) of sunitinib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2008, 51, 42–48. [Google Scholar] [CrossRef]
- Hao, Z.; Wang, P. Lenvatinib in Management of Solid Tumors. Oncologist 2020, 25, e30. [Google Scholar] [CrossRef] [Green Version]
- Matsui, J.; Yamamoto, Y.; Funahashi, Y.; Tsuruoka, A.; Watanabe, T.; Wakabayashi, T.; Uenaka, T.; Asada, M. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int. J. Cancer 2007, 122, 664–671. [Google Scholar] [CrossRef]
- Capozzi, M.; De Divitiis, C.; Ottaiano, A.; von Arx, C.; Scala, S.; Tatangelo, F.; Delrio, P.; Tafuto, S. Lenvatinib, a molecule with versatile application: From preclinical evidence to future development in anti-cancer treatment. Cancer Manag. Res. 2019, 11, 3847–3860. [Google Scholar] [CrossRef] [Green Version]
- Bruheim, S.; Kristian, A.; Uenaka, T.; Suo, Z.; Tsuruoka, A.; Nesland, J.M.; Fodstad, A. Antitumour activity of oral E7080, a novel inhibitor of multiple tyrosine kinases, in human sarcoma xenografts. Int. J. Cancer 2011, 129, 742–750. [Google Scholar] [CrossRef]
- Glen, H.; Mason, S.; Patel, H.; MacLeod, K.; Brunton, V.G. E7080, a multi-targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Cancer 2011, 11, 309. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Campbell-Hewson, Q.; Melcon, S.G.; Locatelli, F.; Venkatramani, R.; Hecker-Nolting, S.; Gambart, M.; Bautista, F.; Thebaud, E.; Aerts, I.; et al. Phase I/II study of single-agent lenvatinib in children and adolescents with refractory or relapsed solid malignancies and young adults with osteosarcoma (ITCC-050). ESMO Open 2021, 6, 100250. [Google Scholar] [CrossRef]
- Wilky, B.A.; Meyer, C.F.; Trent, J.C. Pazopanib in sarcomas. Curr. Opin. Oncol. 2013, 25, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mokhtari, R.B.; Oliveira, I.D.; Islam, S.; Toledo, S.R.C.; Yeger, H.; Baruchel, S. Tumor Dynamics in Response to Antiangiogenic Therapy with Oral Metronomic Topotecan and Pazopanib in Neuroblastoma Xenografts. Transl. Oncol. 2013, 6, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mokhtari, R.B.; Sheikh, R.; Wu, B.; Zhang, L.; Xu, P.; Man, S.; Oliveira, I.D.; Yeger, H.; Kerbel, R.S.; et al. Metronomic Oral Topotecan with Pazopanib Is an Active Antiangiogenic Regimen in Mouse Models of Aggressive Pediatric Solid Tumor. Clin. Cancer Res. 2011, 17, 5656–5667. [Google Scholar] [CrossRef] [Green Version]
- Chiabotto, G.; Grignani, G.; Todorovic, M.; Martin, V.; Centomo, M.L.; Prola, E.; Giordano, G.; Merlini, A.; Miglio, U.; Berrino, E.; et al. Pazopanib and Trametinib as a Synergistic Strategy against Osteosarcoma: Preclinical Activity and Molecular Insights. Cancers 2020, 12, 1519. [Google Scholar] [CrossRef] [PubMed]
- Keir, S.T.; Bs, C.L.M.; Wu, J.; Kurmasheva, R.T.; Houghton, P.J.; Smith, M.A. Initial testing of the multitargeted kinase inhibitor pazopanib by the pediatric preclinical testing program. Pediatr. Blood Cancer 2011, 59, 586–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggerholm-Pedersen, N.; Rossen, P.; Rose, H.; Safwat, A. Pazopanib in the Treatment of Bone Sarcomas: Clinical Experience. Transl. Oncol. 2020, 13, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Kinoshita, S.; Kanamori, T.; Kataoka, H.; Joh, T.; Iida, S.; Takemoto, M.; Kondo, M.; Kuroda, J.; Komatsu, H. The Successful Treatment of Metastatic Extraosseous Ewing Sarcoma with Pazopanib. Intern. Med. 2018, 57, 2753–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donson, A.M.; Amani, V.; Warner, E.A.; Griesinger, A.M.; Witt, D.A.; Levy, J.M.M.; Hoffman, L.M.; Hankinson, T.C.; Handler, M.H.; Vibhakar, R.; et al. Identification of FDA-Approved Oncology Drugs with Selective Potency in High-Risk Childhood Ependymoma. Mol. Cancer Ther. 2018, 17, 1984–1994. [Google Scholar] [CrossRef] [Green Version]
- Schoen, L.F.; Craveiro, R.B.; Pietsch, T.; Moritz, T.; Troeger, A.; Jordans, S.; Dilloo, D. The PI3K inhibitor pictilisib and the multikinase inhibitors pazopanib and sorafenib have an impact on Rac1 level and migration of medulloblastoma in vitro. J. Cell. Mol. Med. 2022, 26, 5832–5845. [Google Scholar] [CrossRef]
- Craveiro, R.B.; Ehrhardt, M.; Holst, M.I.; Pietsch, T.; Dilloo, D. In Comparative Analysis of Multi-Kinase Inhibitors for Targeted Medulloblastoma Therapy Pazopanib Exhibits Promising In Vitro and In Vivo Efficacy, 2014. Available online: www.impactjournals.com/oncotarget (accessed on 22 October 2022).
- Scott, B.J.; Quant, E.C.; McNamara, M.B.; Ryg, P.A.; Batchelor, T.T.; Wen, P.Y. Bevacizumab salvage therapy following progression in high-grade glioma patients treated with VEGF receptor tyrosine kinase inhibitors. Neuro-Oncology 2010, 12, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Duke, E.S.; Barone, A.K.; Chatterjee, S.; Mishra-Kalyani, P.S.; Shen, Y.-L.; Isikwei, E.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, N.A.; et al. FDA Approval Summary: Cabozantinib for Differentiated Thyroid Cancer. Clin. Cancer Res. 2022, 28, 4173–4177. [Google Scholar] [CrossRef]
- Leavitt, J.; Copur, M.S. FDA Approved Uses of Cabozantinib. Oncology 2019, 33, 685004. [Google Scholar]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Bentzien, F.; Zuzow, M.; Heald, N.; Gibson, A.; Shi, Y.; Goon, L.; Yu, P.; Engst, S.; Zhang, W.; Huang, D.; et al. In Vitro and In Vivo Activity of Cabozantinib (XL184), an Inhibitor of RET, MET, and VEGFR2, in a Model of Medullary Thyroid Cancer. Thyroid 2013, 23, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Santoni, M.; Iacovelli, R.; Colonna, V.; Klinz, S.; Mauri, G.; Nuti, M. Antitumor effects of the multi-target tyrosine kinase inhibitor cabozantinib: A comprehensive review of the preclinical evidence. Expert Rev. Anticancer. Ther. 2021, 21, 1029–1054. [Google Scholar] [CrossRef]
- Fioramonti, M.; Fausti, V.; Pantano, F.; Iuliani, M.; Ribelli, G.; Lotti, F.; Pignochino, Y.; Grignani, G.; Santini, D.; Tonini, G.; et al. Cabozantinib Affects Osteosarcoma Growth Through A Direct Effect On Tumor Cells and Modifications In Bone Microenvironment. Sci. Rep. 2018, 8, 4177. [Google Scholar] [CrossRef] [Green Version]
- Pagnuzzi-Boncompagni, M.; Picco, V.; Vial, V.; Planas-Bielsa, V.; Vandenberghe, A.; Daubon, T.; Derieppe, M.-A.; Montemagno, C.; Durivault, J.; Grépin, R.; et al. Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma. Cancers 2021, 14, 70. [Google Scholar] [CrossRef]
- Daudigeos-Dubus, E.; Le Dret, L.; Bawa, O.; Opolon, P.; Vievard, A.; Villa, I.; Bosq, J.; Vassal, G.; Geoerger, B. Dual inhibition using cabozantinib overcomes HGF/MET signaling mediated resistance to pan-VEGFR inhibition in orthotopic and metastatic neuroblastoma tumors. Int. J. Oncol. 2016, 50, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Wind, S.; Schmid, U.; Freiwald, M.; Marzin, K.; Lotz, R.; Ebner, T.; Stopfer, P.; Dallinger, C. Clinical Pharmacokinetics and Pharmacodynamics of Nintedanib. Clin. Pharmacokinet. 2019, 58, 1131–1147. [Google Scholar] [CrossRef] [Green Version]
- A Kharlamova, L. Study of the radiobiological reactions of Amoeba proteus. 2. The effect of the binuclear state on the radiosensitivity of amebae. Radiobiologiia 1972, 12, 934–937. [Google Scholar]
- Lin, T.; Gong, L. Inhibition of lymphangiogenesis in vitro and in vivo by the multikinase inhibitor nintedanib. Drug Des. Dev. Ther. 2017, 11, 1147–1158. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, P.P.; Musi, E.; Schwartz, G.K. Preclinical Evaluation of Nintedanib, a Triple Angiokinase Inhibitor, in Soft-tissue Sarcoma: Potential Therapeutic Implication for Synovial Sarcoma. Mol. Cancer Ther. 2018, 17, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, J.-M.; Lin, J.; Hu, C.-Z.; Zhang, W.-B.; Yang, W.-L.; Zhang, J.; Zhu, J. Adaptive Fibrogenic Reprogramming of Osteosarcoma Stem Cells Promotes Metastatic Growth. Cell Rep. 2018, 24, 1266–1277.e5. [Google Scholar] [CrossRef] [Green Version]
- Milton, C.I.; Selfe, J.; Aladowicz, E.; Man, S.Y.K.; Bernauer, C.; Missiaglia, E.; Walters, Z.S.; Gatz, S.A.; Kelsey, A.; Generali, M.; et al. FGF7–FGFR2 autocrine signaling increases growth and chemoresistance of fusion-positive rhabdomyosarcomas. Mol. Oncol. 2021, 16, 1272–1289. [Google Scholar] [CrossRef]
- Loetsch, D.; Gojo, J.; Kirchhofer, D.; Van Schoonhoven, S.; Pajtler, K.; Kool, M.; Haberler, C.; Czech, T.; Slavc, I.; Berger, W. OS5.2 FGFR a novel target in malignant pediatric ependymoma. Neuro-Oncology 2018, 20, iii224. [Google Scholar] [CrossRef]
- Lötsch, D.; Kirchhofer, D.; Englinger, B.; Jiang, L.; Okonechnikov, K.; Senfter, D.; Laemmerer, A.; Gabler, L.; Pirker, C.; Donson, A.M.; et al. Targeting fibroblast growth factor receptors to combat aggressive ependymoma. Acta Neuropathol. 2021, 142, 339–360. [Google Scholar] [CrossRef] [PubMed]
- Kasamon, Y.L.; Ko, C.-W.; Subramaniam, S.; Ma, L.; Yang, Y.; Nie, L.; Shord, S.; Przepiorka, D.; Farrell, A.T.; McKee, A.E.; et al. FDA Approval Summary: Midostaurin for the Treatment of Advanced Systemic Mastocytosis. Oncologist 2018, 23, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midostaurin Gets FDA Nod for AML. Cancer Discov. 2017, 7, OF5. [CrossRef] [Green Version]
- Kawamoto, T.; Akisue, T.; Kishimoto, K.; Hara, H.; Imabori, M.; Fujimoto, T.; Kurosaka, M.; Hitora, T.; Kawaguchi, Y.; Yamamoto, T. Inhibition of PKCalpha activation in human bone and soft tissue sarcoma cells by the selective PKC inhibitor PKC412. Anticancer. Res. 2008, 28, 825–832. [Google Scholar]
- Boro, A.; Prêtre, K.; Rechfeld, F.; Thalhammer, V.; Oesch, S.; Wachtel, M.; Schäfer, B.W.; Niggli, F.K. Small-molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing’s sarcoma. Int. J. Cancer 2012, 131, 2153–2164. [Google Scholar] [CrossRef]
- Brounais, B.; Chipoy, C.; Mori, K.; Charrier, C.; Battaglia, S.; Pilet, P.; Richards, C.D.; Heymann, D.; Rédini, F.; Blanchard, F. Oncostatin M Induces Bone Loss and Sensitizes Rat Osteosarcoma to the Antitumor Effect of Midostaurin In vivo. Clin. Cancer Res. 2008, 14, 5400–5409. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.J.; Rixe, O. Axitinib (AG-013736). Small Mol. Oncol. 2009, 184, 33–44. [Google Scholar] [CrossRef]
- Lu, L.; Saha, D.; Martuza, R.L.; Rabkin, S.; Wakimoto, H. Single agent efficacy of the VEGFR kinase inhibitor axitinib in preclinical models of glioblastoma. J. Neuro-Oncology 2014, 121, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, M.; Craveiro, R.B.; Velz, J.; Olschewski, M.; Casati, A.; Schönberger, S.; Pietsch, T.; Dilloo, D. The FDA approved PI 3K inhibitor GDC -0941 enhances in vitro the anti-neoplastic efficacy of Axitinib against c-myc-amplified high-risk medulloblastoma. J. Cell. Mol. Med. 2018, 22, 2153–2161. [Google Scholar] [CrossRef] [Green Version]
- Suri, A.; Bailey, A.W.; Tavares, M.T.; Gunosewoyo, H.; Dyer, C.P.; Grupenmacher, A.T.; Piper, D.R.; Horton, R.A.; Tomita, T.; Kozikowski, A.P.; et al. Evaluation of Protein Kinase Inhibitors with PLK4 Cross-Over Potential in a Pre-Clinical Model of Cancer. Int. J. Mol. Sci. 2019, 20, 2112. [Google Scholar] [CrossRef] [Green Version]
- Schwinn, S.; Mokhtari, Z.; Thusek, S.; Schneider, T.; Sirén, A.-L.; Tiemeyer, N.; Caruana, I.; Miele, E.; Schlegel, P.G.; Beilhack, A.; et al. Cytotoxic effects and tolerability of gemcitabine and axitinib in a xenograft model for c-myc amplified medulloblastoma. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin. Cancer Res. 2018, 24, 3409–3422. [Google Scholar] [CrossRef] [Green Version]
- Rössler, J.; Monnet, Y.; Farace, F.; Opolon, P.; Daudigeos-Dubus, E.; Bourredjem, A.; Vassal, G.; Geoerger, B. The selective VEGFR1-3 inhibitor axitinib (AG-013736) shows antitumor activity in human neuroblastoma xenografts. Int. J. Cancer 2010, 128, 2748–2758. [Google Scholar] [CrossRef]
- Lu, D.; Jimenez, X.; Zhang, H.; Bohlen, P.; Witte, L.; Zhu, Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int. J. Cancer 2001, 97, 393–399. [Google Scholar] [CrossRef]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Renschler, M.; Perez, L.V.; Stephens, J.; Pytowski, B.; Wasserstrom, H.; Stancato, L.F.; Falcon, B. Anti-VEGFR2 therapy delays growth of preclinical pediatric tumor models and enhances anti-tumor activity of chemotherapy. Oncotarget 2019, 10, 5523–5533. [Google Scholar] [CrossRef] [Green Version]
- Casak, S.J.; Fashoyin-Aje, I.; Lemery, S.J.; Zhang, L.; Jin, R.; Li, H.; Zhao, L.; Zhao, H.; Zhang, H.; Chen, H.; et al. FDA Approval Summary: Ramucirumab for Gastric Cancer. Clin. Cancer Res. 2015, 21, 3372–3376. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y. Ramucirumab: A Review in Hepatocellular Carcinoma. Drugs 2020, 80, 315–322. [Google Scholar] [CrossRef]
- Tiwari, P. Ramucirumab: Boon or bane. J. Egypt. Natl. Cancer Inst. 2016, 28, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Debeuckelaere, C.; Murgioni, S.; Lonardi, S.; Girardi, N.; Alberti, G.; Fano, C.; Gallimberti, S.; Magro, C.; Ahcene-Djaballah, S.; Daniel, F.; et al. Ramucirumab: The long and winding road toward being an option for mCRC treatment. Expert Opin. Biol. Ther. 2019, 19, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Larkins, E.; Blumenthal, G.M.; Chen, H.; He, K.; Agarwal, R.; Gieser, G.; Stephens, O.; Zahalka, E.; Ringgold, K.; Helms, W.; et al. FDA Approval: Alectinib for the Treatment of Metastatic, ALK-Positive Non–Small Cell Lung Cancer Following Crizotinib. Clin. Cancer Res. 2016, 22, 5171–5176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Hayashi, M.; Aikawa, H.; Okamoto, I.; Fujiwara, Y.; Hamada, A. Heterogeneous distribution of alectinib in neuroblastoma xenografts revealed by matrix-assisted laser desorption ionization mass spectrometry imaging: A pilot study. Br. J. Pharmacol. 2017, 175, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Humphreys, A.; Turnbull, L.; Bellini, A.; Schleiermacher, G.; Salwen, H.; Cohn, S.L.; Bown, N.; Tweddle, D.A. Identification of differentALKmutations in a pair of neuroblastoma cell lines established at diagnosis and relapse. Oncotarget 2016, 7, 87301–87311. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Guan, S.; Zhao, Y.; Yu, Y.; Woodfield, S.E.; Zhang, H.; Yang, K.L.; Bieerkehazhi, S.; Qi, L.; Li, X.; et al. The second-generation ALK inhibitor alectinib effectively induces apoptosis in human neuroblastoma cells and inhibits tumor growth in a TH-MYCN transgenic neuroblastoma mouse model. Cancer Lett. 2017, 400, 61–68. [Google Scholar] [CrossRef]
- Yang, K.; Chen, Y.; Wang, F. Correction: Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. Exp. Mol. Med. 2020, 52, 989–990. [Google Scholar] [CrossRef]
- Brunac, A.; Laprie, A.; Castex, M.; Laurent, C.; Le Loarer, F.; Karanian, M.; Le Guellec, S.; Guillemot, D.; Pierron, G.; Gomez-Brouchet, A. The combination of radiotherapy and ALK inhibitors is effective in the treatment of intraosseous rhabdomyosarcoma with FUS-TFCP2 fusion transcript. Pediatr. Blood Cancer 2020, 67, e28185. [Google Scholar] [CrossRef]
- Hagiwara, K.; Tokunaga, T.; Iida, H.; Nagai, H. Combined Inhibition of ALK and HDAC Induces Synergistic Cytotoxicity in Neuroblastoma Cell Lines. Anticancer. Res. 2019, 39, 3579–3584. [Google Scholar] [CrossRef]
- Berezowska, S.; Diermeier-Daucher, S.; Brockhoff, G.; Busch, R.; Duyster, J.; Grosu, A.-L.; Schlegel, J. Effect of additional inhibition of human epidermal growth factor receptor 2 with the bispecific tyrosine kinase inhibitor AEE788 on the resistance to specific EGFR inhibition in glioma cells. Int. J. Mol. Med. 2010, 26, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Servidei, T.; Meco, D.; Trivieri, N.; Patriarca, V.; Vellone, V.G.; Zannoni, G.F.; Lamorte, G.; Pallini, R.; Riccardi, R. Effects of epidermal growth factor receptor blockade on ependymoma stem cells in vitro and in orthotopic mouse models. Int. J. Cancer 2012, 131, E791–E803. [Google Scholar] [CrossRef]
- Park, Y.W.; Younes, M.N.; Jasser, S.A.; Yigitbasi, O.G.; Zhou, G.; Bucana, C.D.; Bekele, B.N.; Myers, J.N. AEE788, a Dual Tyrosine Kinase Receptor Inhibitor, Induces Endothelial Cell Apoptosis in Human Cutaneous Squamous Cell Carcinoma Xenografts in Nude Mice. Clin. Cancer Res. 2005, 11, 1963–1973. [Google Scholar] [CrossRef] [Green Version]
- Meco, D.; Servidei, T.; Zannonit, G.F.; Martinelli, E.; Prisco, M.G.; de Waure, C.; Riccardi, R. Dual Inhibitor AEE78 Reduces Tumor Growth in Preclinical Models of Medulloblastoma. Transl. Oncol. 2010, 3, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Heigener, D.F.; Reck, M. Crizotinib. In Recent Results in Cancer Research; Springer LLC: New York, NY, USA, 2018; Volume 211, pp. 57–65. [Google Scholar] [CrossRef]
- Zomerman, W.W.; Plasschaert, S.L.A.; Diks, S.H.; Lourens, H.-J.; Boer, T.M.-D.; Hoving, E.W.; Dunnen, W.F.A.D.; De Bont, E.S.J.M. Exogenous HGF Bypasses the Effects of ErbB Inhibition on Tumor Cell Viability in Medulloblastoma Cell Lines. PLoS ONE 2015, 10, e0141381. [Google Scholar] [CrossRef]
- Sie, M.; Dunnen, W.F.A.D.; Lourens, H.J.; Boer, T.G.J.M.-D.; Scherpen, F.J.G.; Zomerman, W.W.; Kampen, K.; Hoving, E.W.; De Bont, E.S.J.M. Growth-Factor-Driven Rescue to Receptor Tyrosine Kinase (RTK) Inhibitors through Akt and Erk Phosphorylation in Pediatric Low Grade Astrocytoma and Ependymoma. PLoS ONE 2015, 10, e0122555. [Google Scholar] [CrossRef]
- Megiorni, F.; McDowell, H.P.; Camero, S.; Mannarino, O.; Ceccarelli, S.; Paiano, M.; Losty, P.D.; Pizer, B.; Shukla, R.; Pizzuti, A.; et al. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J. Exp. Clin. Cancer Res. 2015, 34, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef]
- Heuckmann, J.M.; Hölzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grütter, C.; et al. ALK Mutations Conferring Differential Resistance to Structurally Diverse ALK Inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [Green Version]
- George, R.E.; Sanda, T.; Hanna, M.; Fröhling, S.; Luther, W., II; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [Green Version]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Crizotinib resistance: Implications for therapeutic strategies. Ann. Oncol. 2016, 27 (Suppl. S3), iii42–iii50. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, M.; He, C.; Andrews, J.; Sloan-Henry, R.; Bianski, B.; Xie, J.; Wang, Y.; Twarog, N.; Onar-Thomas, A.; Ernst, K.; et al. MODL-06. Targeting c-MET in combination with radiation is effective in MET-fusion driven high-grade glioma. Neuro-Oncology 2022, 24, i169. [Google Scholar] [CrossRef]
- Esaki, T.; Hirai, F.; Makiyama, A.; Seto, T.; Bando, H.; Naito, Y.; Yoh, K.; Ishihara, K.; Kakizume, T.; Natsume, K.; et al. Phase I dose-escalation study of capmatinib ( INC 280) in Japanese patients with advanced solid tumors. Cancer Sci. 2019, 110, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tepotinib: First Approval. Drugs 2020, 80, 829–833. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; Burgess, K.; Qiu, M.; Engstrom, L.D.; Yamazaki, S.; Parker, M.; Timofeevski, S.; et al. Sensitivity of Selected Human Tumor Models to PF-04217903, a Novel Selective c-Met Kinase Inhibitor. Mol. Cancer Ther. 2012, 11, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Niswander, L.M.; Guenther, L.M.; Mendoza, A.; Khanna, C.; Christensen, J.G.; Helman, L.J.; Kim, S. Effect of modulation of MET with the small molecule inhibitor PF-04217903 on osteosarcoma metastasis in vivo. J. Clin. Oncol. 2010, 28, 9567. [Google Scholar] [CrossRef]
- Lock, R.; Ingraham, R.; Maertens, O.; Miller, A.L.; Weledji, N.; Legius, E.; Konicek, B.M.; Yan, S.-C.B.; Graff, J.R.; Cichowski, K. Cotargeting MNK and MEK kinases induces the regression of NF1-mutant cancers. J. Clin. Investig. 2016, 126, 2181–2190. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-Hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic Activity of Tivantinib (ARQ 197) Is Not Due Solely to c-MET Inhibition. Cancer Res 2013, 73, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Geller, J.I.; Perentesis, J.P.; Liu, X.; Minard, C.G.; Kudgus, R.A.; Reid, J.M.; Fox, E.; Blaney, S.M.; Weigel, B.J. A phase 1 study of the c-Met inhibitor, tivantinib (ARQ197) in children with relapsed or refractory solid tumors: A Children’s Oncology Group study phase 1 and pilot consortium trial (ADVL1111). Pediatr. Blood Cancer 2017, 64, e26565. [Google Scholar] [CrossRef]
- Goldberg, J.M.; Gavcovich, T.; Saigal, G.; Goldman, J.W.; Rosen, L.S. Extended Progression-Free Survival in Two Patients With Alveolar Soft Part Sarcoma Exposed to Tivantinib. J. Clin. Oncol. 2014, 32, e114–e116. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.-L.; Dinh, D.; et al. Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][]2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [CrossRef]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Tucker, E.R.; Wan, H.; Chand, D.; Danielson, L.S.; Ruuth, K.; El Wakil, A.; Witek, B.; Jamin, Y.; Umapathy, G.; et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis. Model. Mech. 2016, 9, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Collier, T.L.; Maresca, K.P.; Normandin, M.D.; Richardson, P.; McCarthy, T.J.; Liang, S.H.; Waterhouse, R.N.; Vasdev, N. Brain Penetration of the ROS1/ALK Inhibitor Lorlatinib Confirmed by PET. Mol. Imaging 2017, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, A.; Orr, B.A.; Campagne, O.; Dhanda, S.; Nair, S.; Tran, Q.; Christensen, A.M.; Gajjar, A.; Furtado, L.V.; Vasilyeva, A.; et al. Lorlatinib in a Child with ALK-Fusion–Positive High-Grade Glioma. New Engl. J. Med. 2021, 385, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Merguerian, M.D.; Rowe, S.P.; Pratilas, C.A.; Chen, A.R.; Ladle, B.H. Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Mol. Case Stud. 2021, 7, a006064. [Google Scholar] [CrossRef] [PubMed]
- Khozin, S.; Blumenthal, G.M.; Zhang, L.; Tang, S.; Brower, M.; Fox, E.; Helms, W.; Leong, R.; Song, P.; Pan, Y.; et al. FDA Approval: Ceritinib for the Treatment of Metastatic Anaplastic Lymphoma Kinase–Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 2436–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Fransson, S.; Siaw, J.T. Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib Running title: Clinical response to ceritinib in ALK-positive neuroblastoma. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002550. [Google Scholar] [CrossRef]
- Mittal, A.; Gupta, A.; Rastogi, S.; Barwad, A.; Sharma, S. Near-complete response to low-dose ceritinib in recurrent infantile inflammatory myofibroblastic tumour. Ecancermedicalscience 2021, 15. [Google Scholar] [CrossRef]
- Russo, A.; Paret, C.; Lehmann, N.; Bender, H.; Wingerter, A.; Neu, M.A.; Alt, F.; Backes, N.; Roth, L.; Seidmann, L.; et al. Epen-29. individualized therapy of an anaplastic ependymoma pediatric patient with a notch1 germline mutation. Neuro-Oncology 2018, 20, i79. [Google Scholar] [CrossRef]
- Russo, A.; Paret, C.; Alt, F.; Burhenne, J.; Fresnais, M.; Wagner, W.; Glaser, M.; Bender, H.; Huprich, S.; Harter, P.N.; et al. Ceritinib-Induced Regression of an Insulin-Like Growth Factor-Driven Neuroepithelial Brain Tumor. Int. J. Mol. Sci. 2019, 20, 4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.Y.; Erkek, S.; Tong, Y.; Yin, L.; Federation, A.J.; Zapatka, M.; Haldipur, P.; Kawauchi, D.; Risch, T.; Warnatz, H.-J.; et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016, 530, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoli, M.; Wadham, C.; Pinese, M.; Failes, T.; Joshi, S.; Mould, E.; Yin, J.X.; Gayevskiy, V.; Kumar, A.; Kaplan, W.; et al. Integration of genomics, high throughput drug screening, and personalized xenograft models as a novel precision medicine paradigm for high risk pediatric cancer. Cancer Biol. Ther. 2018, 19, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Beck, O.; Paret, C.; Russo, A.; Burhenne, J.; Fresnais, M.; Steimel, K.; Seidmann, L.; Wagner, D.-C.; Vewinger, N.; Lehmann, N.; et al. Safety and Activity of the Combination of Ceritinib and Dasatinib in Osteosarcoma. Cancers 2020, 12, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting Anaplastic Lymphoma Kinase (ALK) in Rhabdomyosarcoma (RMS) with the Second-Generation ALK Inhibitor Ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgikh, N.; Fulda, S. Rhabdomyosarcoma cells are susceptible to cell death by LDK378 alone or in combination with sorafenib independently of anaplastic lymphoma kinase status. Anti-Cancer Drugs 2017, 28, 1118–1125. [Google Scholar] [CrossRef]
- Huang, W.-S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood–Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef]
- Spencer, S.A.; Riley, A.C.; Matthew, A.; Di Pasqua, A.J. Brigatinib: Novel ALK Inhibitor for Non–Small-Cell Lung Cancer. Ann. Pharmacother. 2019, 53, 621–626. [Google Scholar] [CrossRef]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells,Drosophilaand mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef]
- Drilon, A.; De Braud, F.G.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.-J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Liu, S.V.; et al. Abstract CT007: Entrectinib, an oral pan-Trk, ROS1, and ALK inhibitor in TKI-naïve patients with advanced solid tumors harboring gene rearrangements: Updated phase I results. Cancer Res 2016, 76, CT007. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, M.; Li, H.; Chen, L. Entrectinib, a new multi-target inhibitor for cancer therapy. Biomed. Pharmacother. 2022, 150, 112974. [Google Scholar] [CrossRef]
- Iyer, R.; Wehrmann, L.; Golden, R.L.; Naraparaju, K.; Croucher, J.L.; MacFarland, S.P.; Guan, P.; Kolla, V.; Wei, G.; Cam, N.; et al. Entrectinib is a potent inhibitor of Trk-driven neuroblastomas in a xenograft mouse model. Cancer Lett. 2016, 372, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.V.; Robinson, G.W.; Gauvain, K.; Basu, E.M.; E Macy, M.; Maese, L.; Whipple, N.S.; Sabnis, A.J.; Foster, J.H.; Shusterman, S.; et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro-Oncology 2022, 24, 1776–1789. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Naraparaju, K.; Iyer, R.; Guan, P.; Kolla, V.; Hu, Y.; Tan, K.; Brodeur, G.M. Mechanisms of Entrectinib Resistance in a Neuroblastoma Xenograft Model. Mol. Cancer Ther. 2020, 19, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Spitaleri, G.; Passaro, A.; de Marinis, F. Ensartinib (X-396) a novel drug for anaplastic lymphoma kinase-positive non-small cell lung cancer patients: We need smart trials to avoid wasting good bullets. Chin. Clin. Oncol. 2019, 8, S1. [Google Scholar] [CrossRef]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-Driven Cancers Revealed through Development of Novel ALK Tyrosine Kinase Inhibitors. Cancer Res 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, D.; Yang, D.; Pastorino, F.; Emionite, L.; Cilli, M.; Daga, A.; Destafanis, E.; Di Fiore, A.; Piaggio, F.; Brignole, C.; et al. New therapeutic strategies in neuroblastoma: Combined targeting of a novel tyrosine kinase inhibitor and liposomal siRNAs against ALK. Oncotarget 2015, 6, 28774–28789. [Google Scholar] [CrossRef] [Green Version]
- Perera, T.P.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Luzzi, S.; Brambilla, I.; Mantelli, S.S.; Mosconi, M.; Foiadelli, T.; Savasta, S. Targeting the medulloblastoma: A molecular-based approach. Acta Biomed. 2020, 91, 79–100. [Google Scholar] [CrossRef]
- Musumeci, F.; Cianciusi, A.; D’Agostino, I.; Grossi, G.; Carbone, A.; Schenone, S. Synthetic Heterocyclic Derivatives as Kinase Inhibitors Tested for the Treatment of Neuroblastoma. Molecules 2021, 26, 7069. [Google Scholar] [CrossRef] [PubMed]
- Holzhauser, S.; Lukoseviciute, M.; Papachristofi, C.; Vasilopoulou, C.; Herold, N.; Wickström, M.; Kostopoulou, O.N.; Dalianis, T. Effects of PI3K and FGFR inhibitors alone and in combination, and with/without cytostatics in childhood neuroblastoma cell lines. Int. J. Oncol. 2021, 58, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Lukoseviciute, M.; Maier, H.; Poulou-Sidiropoulou, E.; Rosendahl, E.; Holzhauser, S.; Dalianis, T.; Kostopoulou, O.N. Targeting PI3K, FGFR, CDK4/6 Signaling Pathways Together With Cytostatics and Radiotherapy in Two Medulloblastoma Cell Lines. Front. Oncol. 2021, 11, 748657. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Lopez-Martin, J.A.; Lin, C.-C.; Gschwend, J.E.; Harzstark, A.; Castellano, D.; Soria, J.-C.; Sen, P.; Chang, J.; Shi, M.; et al. Phase I Study of Dovitinib (TKI258), an Oral FGFR, VEGFR, and PDGFR Inhibitor, in Advanced or Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Arnz, E.; Hertwig, F.; Krämer, A.; Ikram, F.; Roels, F.; Kocak, H.; Engesser, A.; Thomas, R.; Peifer, M.; Ackermann, S.; et al. Fibroblast growth factor receptors as therapeutic targets in neuroblastoma. Klin. Pädiatrie 2014, 226, A22. [Google Scholar] [CrossRef]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting Wild-Type and Mutationally Activated FGFR4 in Rhabdomyosarcoma with the Inhibitor Ponatinib (AP24534). PLoS ONE 2013, 8, e76551. [Google Scholar] [CrossRef] [Green Version]
- Schramm, K.; Iskar, M.; Statz, B.; Jäger, N.; Haag, D.; Słabicki, M.; Pfister, S.M.; Zapatka, M.; Gronych, J.; Jones, D.T.W.; et al. DECIPHER pooled shRNA library screen identifies PP2A and FGFR signaling as potential therapeutic targets for diffuse intrinsic pontine gliomas. Neuro-Oncology 2019, 21, 867–877. [Google Scholar] [CrossRef]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; A Joyce, J. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef] [Green Version]
- Georgin-Lavialle, S.; Lhermitte, L.; Suarez, F.; Yang, Y.; Létard, S.; Hanssens, K.; Feger, F.; Renand, A.; Brouze, C.; Canioni, D.; et al. Mast cell leukemia: Identification of a new c-Kit mutation, dup(501-502), and response to masitinib, a c-Kit tyrosine kinase inhibitor. Eur. J. Haematol. 2012, 89, 47–52. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Marech, I.; Patruno, R.; Zizzo, N.; Gadaleta, C.; Introna, M.; Zito, A.F.; Gadaleta, C.D.; Ranieri, G. Masitinib (AB1010), from canine tumor model to human clinical development: Where we are? Crit. Rev. Oncol. 2014, 91, 98–111. [Google Scholar] [CrossRef]
- Fleming, T.; Cunningham, C.; Keir, S. The Effect of Masitinib on Pediatric Glioblastoma; Duke University: Durham, NC, USA, 2014. [Google Scholar]
- Buti, S.; Leonetti, A.; Dallatomasina, A.; Bersanelli, M. Everolimus in the management of metastatic renal cell carcinoma: An evidence-based review of its place in therapy. Core Évid. 2016, 11, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Nashan, B. Review of the proliferation inhibitor everolimus. Expert Opin. Investig. Drugs 2002, 11, 1845–1857. [Google Scholar] [CrossRef]
- Pignochino, Y.; Dell’Aglio, C.; Basiricò, M.; Capozzi, F.; Soster, M.; Marchiò, S.; Bruno, S.; Gammaitoni, L.; Sangiolo, D.; Torchiaro, E.; et al. The Combination of Sorafenib and Everolimus Abrogates mTORC1 and mTORC2 Upregulation in Osteosarcoma Preclinical Models. Clin. Cancer Res. 2013, 19, 2117–2131. [Google Scholar] [CrossRef] [Green Version]
- Miklja, Z.; Yadav, V.N.; Cartaxo, R.T.; Siada, R.; Thomas, C.C.; Cummings, J.R.; Mullan, B.; Stallard, S.; Paul, A.; Bruzek, A.K.; et al. Everolimus improves the efficacy of dasatinib in PDGFRα-driven glioma. J. Clin. Investig. 2020, 130, 5313–5325. [Google Scholar] [CrossRef]
- Poore, B.; Yuan, M.; Arnold, A.; Price, A.; Alt, J.; A Rubens, J.; Slusher, B.S.; Eberhart, C.G.; Raabe, E.H. Inhibition of mTORC1 in pediatric low-grade glioma depletes glutathione and therapeutically synergizes with carboplatin. Neuro-Oncology 2018, 21, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Salussolia, C.L.; Klonowska, K.; Kwiatkowski, D.J.; Sahin, M. Genetic Etiologies, Diagnosis, and Treatment of Tuberous Sclerosis Complex. Annu. Rev. Genom. Hum. Genet. 2019, 20, 217–240. [Google Scholar] [CrossRef]
- Rosset, C.; Netto, C.B.O.; Ashton-Prolla, P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: A review. Genet. Mol. Biol. 2017, 40, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for Subependymal Giant-Cell Astrocytomas in Tuberous Sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef]
- Franz, D.N.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Voi, M.; et al. Everolimus for treatment-refractory seizures in TSC. Neurol. Clin. Pract. 2018, 8, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Hopkins, B.; Perruzzi, C.; Udayakumar, D.; Sherris, D.; Benjamin, L.E. Palomid 529, a Novel Small-Molecule Drug, Is a TORC1/TORC2 Inhibitor That Reduces Tumor Growth, Tumor Angiogenesis, and Vascular Permeability. Cancer Res 2008, 68, 9551–9557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Buil, L.; Sherris, D.; Beijnen, J.H.; van Tellingen, O. Dual mTORC1 and mTORC2 inhibitor Palomid 529 penetrates the Blood-Brain Barrier without restriction by ABCB1 and ABCG2. Int. J. Cancer 2013, 133, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Monache, S.D.; Sferra, R.; Pompili, S.; Vitale, F.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Brain Penetrating and Dual TORC1/TORC2 Inhibitor, RES529, Elicits Anti-Glioma Activity and Enhances the Therapeutic Effects of Anti-Angiogenetic Compounds in Preclinical Murine Models. Cancers 2019, 11, 1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerna, D.; Carter, D.; Flaherty, S.; Cao, L.; Sherris, D.; Yoo, S.S. Abstract 2506: Palomid 529, a PI3K/Akt/mTOR dual TORC1/2 inhibitor, is a radiosensitizer with effect in both subcutaneous and orthotopic U251 glioblastoma tumor xenograft models. Cancer Res. 2010, 70, 2506. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Z.; Chen, M.; Chen, X.; Liang, W. The anti-osteosarcoma cell activity by a mTORC1/2 dual inhibitor RES-529. Biochem. Biophys. Res. Commun. 2018, 497, 499–505. [Google Scholar] [CrossRef]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical Characterization of OSI-027, a Potent and Selective Inhibitor of mTORC1 and mTORC2: Distinct from Rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef] [Green Version]
- Eckerdt, F.; Clymer, J.; Bell, J.B.; Beauchamp, E.M.; Blyth, G.T.; Goldman, S.; Platanias, L.C. Pharmacological mTOR targeting enhances the antineoplastic effects of selective PI3Kα inhibition in medulloblastoma. Sci. Rep. 2019, 9, 12822. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.K.; Guroji, P.; Jin, L.; Mukhtar, M.S.; Athar, M. Combined inhibition of BET bromodomain and mTORC1/2 provides therapeutic advantage for rhabdomyosarcoma by switching cell death mechanism. Mol. Carcinog. 2022, 61, 737–751. [Google Scholar] [CrossRef]
- Eckerdt, F.; Beauchamp, E.; Bell, J.; Iqbal, A.; Su, B.; Fukunaga, R.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Regulatory effects of a Mnk2-eIF4E feedback loop during mTORC1 targeting of human medulloblastoma cells. Oncotarget 2014, 5, 8442–8451. [Google Scholar] [CrossRef] [Green Version]
- Clymer, J.; Eckerd, F.; Bell, J.; Lulla, R.; Goldman, S.; Platanias, L. MEDU-44. TARGETING SHH SIGNALING VIA PI3K/MTOR INHIBITION IN MEDULLOBLASTOMA AND EWING SARCOMA. Neuro-Oncology 2017, 19, iv47. [Google Scholar] [CrossRef]
- Calimeri, T.; Ferreri, A.J.M. m-TOR inhibitors and their potential role in haematological malignancies. Br. J. Haematol. 2017, 177, 684–702. [Google Scholar] [CrossRef]
- Kolev, V.N.; Wright, Q.G.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR Dual Inhibitor VS-5584 Preferentially Targets Cancer Stem Cells. Cancer Res 2015, 75, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-Y.; Hou, Y.-J.; Cui, H.-J.; Zhang, C.; Yang, M.-F.; Wang, F.-Z.; Sun, Z.; Fan, C.-D.; Sun, B.-L.; Oh, J.R. VS-5584 Inhibits Human Osteosarcoma Cells Growth by Induction of G1- phase Arrest through Regulating PI3K/mTOR and MAPK Pathways. Curr. Cancer Drug Targets 2020, 20, 616–623. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Hou, Y.-J.; Yin, Y.-B.; Wang, F.-Z.; Yang, M.-F.; Zhang, Y.-Y.; Fan, C.-D.; Sun, B.-L. CCT128930 induces G1-phase arrest and apoptosis and synergistically enhances the anticancer efficiency of VS5584 in human osteosarcoma cells. Biomed. Pharmacother. 2020, 130, 110544. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Manara, M.C.; Nicoletti, G.; Zambelli, D.; Ventura, S.; Guerzoni, C.; Landuzzi, L.; Lollini, P.-L.; Maira, S.-M.; García-Echeverría, C.; Mercuri, M.; et al. NVP-BEZ235 as a New Therapeutic Option for Sarcomas. Clin. Cancer Res. 2010, 16, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Boro, A.; Rechfeld, F.; Lopez-Garcia, L.A.; Gierisch, M.E.; Schäfer, B.W.; Niggli, F.K. PI3K/AKT signaling modulates transcriptional expression of EWS/FLI1 through specificity protein 1. Oncotarget 2015, 6, 28895–28910. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.-R.; Min, H.; Fang, J.-F.; Zhou, F.; Deng, X.-W.; Zhang, Y.-Q. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against osteosarcoma. Cancer Biol. Ther. 2015, 16, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.; Wang, B.; Mao, W.; Wang, J.; Zhao, Y.; Li, Q.; Zhang, C.; Ma, J. Enhanced efficacy of histone deacetylase inhibitor panobinostat combined with dual PI3K/mTOR inhibitor BEZ235 against glioblastoma. Nagoya J. Med. Sci. 2019, 81, 93–102. [Google Scholar] [CrossRef]
- Vazquez, N.; Lopez, A.; Cuello, V.; Persans, M.; Schuenzel, E.; Innis-Whitehouse, W.; Keniry, M. NVP-BEZ235 or JAKi Treatment leads to decreased survival of examined GBM and BBC cells. Cancer Treat. Res. Commun. 2021, 27, 100340. [Google Scholar] [CrossRef]
- Xie, C.; Freeman, M.J.; Lu, H.; Wang, X.; Forster, C.L.; Sarver, A.L.; Hallstrom, T.C. Retinoblastoma cells activate the AKT pathway and are vulnerable to the PI3K/mTOR inhibitor NVP-BEZ235. Oncotarget 2017, 8, 38084–38098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzhauser, S.; Lukoseviciute, M.; Andonova, T.; Ursu, R.G.; Dalianis, T.; Wickström, M.; Kostopoulou, O.N. Targeting Fibroblast Growth Factor Receptor (FGFR) and Phosphoinositide 3-kinase (PI3K) Signaling Pathways in Medulloblastoma Cell Lines. Anticancer. Res. 2019, 40, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Gobin, B.; Battaglia, S.; Lanel, R.; Chesneau, J.; Amiaud, J.; Rédini, F.; Ory, B.; Heymann, D. NVP-BEZ235, a dual PI3K/mTOR inhibitor, inhibits osteosarcoma cell proliferation and tumor development in vivo with an improved survival rate. Cancer Lett. 2014, 344, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.K.; Kling, M.J.; Griggs, C.N.; Kesherwani, V.; Shukla, M.; McIntyre, E.M.; Ray, S.; Liu, Y.; McGuire, T.R.; Sharp, J.G.; et al. A Novel Combination Approach Targeting an Enhanced Protein Synthesis Pathway in MYC-driven (Group 3) Medulloblastoma. Mol. Cancer Ther. 2020, 19, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with Resistance to Smoothened Antagonists by Inhibition of the PI3K Pathway in Medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [Green Version]
- Garlich, J.R.; De, P.; Dey, N.; Su, J.D.; Peng, X.; Miller, A.; Murali, R.; Lu, Y.; Mills, G.B.; Kundra, V.; et al. A Vascular Targeted Pan Phosphoinositide 3-Kinase Inhibitor Prodrug, SF1126, with Antitumor and Antiangiogenic Activity. Cancer Res 2008, 68, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Chiorean, E.; Harris, W.; Von Hoff, D.; Stejskal-Barnett, A.; Qi, W.; Anthony, S.; Younger, A.; Rensvold, D.; Cordova, F.; et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer 2012, 48, 3319–3327. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.R.; Joshi, S.; Zulcic, M.; Alcaraz, M.; Garlich, J.R.; Morales, G.A.; Cho, Y.J.; Bao, L.; Levy, M.L.; Newbury, R.; et al. PI-3K Inhibitors Preferentially Target CD15+ Cancer Stem Cell Population in SHH Driven Medulloblastoma. PLoS ONE 2016, 11, e0150836. [Google Scholar] [CrossRef]
- Goldin, A.N.; Singh, A.; Joshi, S.; Jamieson, C.; Durden, D.L.M. Augmented Antitumor Activity for Novel Dual PI3K/BDR4 Inhibitors, SF2523 and SF1126 in Ewing Sarcoma. J. Pediatr. Hematol. 2021, 43, e304–e311. [Google Scholar] [CrossRef]
- Peirce, S.; Durden, D.; Garlich, J.; Findley, H. SF1126, a novel pan-PI3K inhibitor, inhibits activation of Mdm2 and in-creases sensitivity to doxorubicin in wild type p53 neuroblastoma cell lines. Cancer Res. 2007, 67, LB-294. [Google Scholar]
- Erdreich-Epstein, A.; Singh, A.R.; Joshi, S.; Vega, F.M.; Guo, P.; Xu, J.; Groshen, S.; Ye, W.; Millard, M.; Campan, M.; et al. Association of high microvessel αvβ3 and low PTEN with poor outcome in stage 3 neuroblastoma: Rationale for using first in class dual PI3K/BRD4 inhibitor, SF1126. Oncotarget 2016, 8, 52193–52210. [Google Scholar] [CrossRef] [Green Version]
- Beljanski, V. Triciribine. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1–5. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Mouhieddine, T.H.; Chalhoub, R.M.; Assi, S.; Araji, T.; Chamaa, F.; Itani, M.M.; Nokkari, A.; Kobeissy, F.; Daoud, G.; et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget 2018, 9, 33549–33561. [Google Scholar] [CrossRef] [Green Version]
- Cubitt, C.L.; Menth, J.; Dawson, J.; Martinez, G.V.; Foroutan, P.; Morse, D.L.; Bui, M.M.; Letson, G.D.; Sullivan, D.M.; Reed, D.R. Rapid Screening of Novel Agents for Combination Therapy in Sarcomas. Sarcoma 2013, 2013, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Smeester, B.A.; Draper, G.M.; Slipek, N.J.; Larsson, A.T.; Stratton, N.; Pomeroy, E.J.; Becklin, K.L.; Yamamoto, K.; Williams, K.B.; Laoharawee, K.; et al. Implication of ZNF217 in Accelerating Tumor Development and Therapeutically Targeting ZNF217-Induced PI3K–AKT Signaling for the Treatment of Metastatic Osteosarcoma. Mol. Cancer Ther. 2020, 19, 2528–2541. [Google Scholar] [CrossRef]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef]
- Slotkin, E.K.; Patwardhan, P.P.; Vasudeva, S.D.; de Stanchina, E.; Tap, W.D.; Schwartz, G.K. MLN0128, an ATP-Competitive mTOR Kinase Inhibitor with Potent In Vitro and In Vivo Antitumor Activity, as Potential Therapy for Bone and Soft-Tissue Sarcoma. Mol. Cancer Ther. 2015, 14, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zeng, Z. Dual mTORC1/2 inhibition by INK-128 results in antitumor activity in preclinical models of osteosarcoma. Biochem. Biophys. Res. Commun. 2015, 468, 255–261. [Google Scholar] [CrossRef]
- Maynard, R.E.; Poore, B.; Hanaford, A.R.; Pham, K.; James, M.; Alt, J.; Park, Y.; Slusher, B.S.; Tamayo, P.; Mesirov, J.; et al. TORC1/2 kinase inhibition depletes glutathione and synergizes with carboplatin to suppress the growth of MYC-driven medulloblastoma. Cancer Lett. 2021, 504, 137–145. [Google Scholar] [CrossRef]
- Zhang, H.; Dou, J.; Yu, Y.; Zhao, Y.; Fan, Y.; Cheng, J.; Xu, X.; Liu, W.; Guan, S.; Chen, Z.; et al. mTOR ATP-competitive inhibitor INK128 inhibits neuroblastoma growth via blocking mTORC signaling. Apoptosis 2014, 20, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Arnold, A.; Yuan, M.; Price, A.; Harris, L.; Eberhart, C.G.; Raabe, E.H. Synergistic activity of mTORC1/2 kinase and MEK inhibitors suppresses pediatric low-grade glioma tumorigenicity and vascularity. Neuro-Oncology 2019, 22, 563–574. [Google Scholar] [CrossRef]
- Tang, L.; Fu, Y.; Song, J.; Hu, T.; Li, K.; Li, Z. mTOR inhibition by TAK-228 is effective against growth, survival and angiogenesis in preclinical retinoblastoma models. Pharmacol. Res. Perspect. 2022, 10, e00930. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Infante, J.R.; Brail, L.H.; Simon, G.R.; Cooksey, J.F.; Jones, S.F.; Farrington, D.L.; Yeo, A.; Jackson, K.A.; Chow, K.H.; et al. A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Investig. New Drugs 2015, 33, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Kunnimalaiyaan, S.; Schwartz, V.K.; Jackson, I.A.; Gamblin, T.C.; Kunnimalaiyaan, M. Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 2018, 18, 560. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Zhu, X.; Li, S.; Liu, G.; Wang, Y.; Wang, W.; Zhang, Q.; Jiang, S. Tideglusib suppresses stem-cell-like features and progression of osteosarcoma by inhibiting GSK-3β/NOTCH1 signaling. Biochem. Biophys. Res. Commun. 2021, 554, 206–213. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Chalhoub, R.M.; Harati, H.; Bou-Gharios, J.; Assi, S.; Ballout, F.; Monzer, A.; Msheik, H.; Araji, T.; Elajami, M.K.; et al. Tideglusib attenuates growth of neuroblastoma cancer stem/progenitor cells in vitro and in vivo by specifically targeting GSK-3β. Pharmacol. Rep. 2020, 73, 211–226. [Google Scholar] [CrossRef]
- Mathuram, T.L.; Ravikumar, V.; Reece, L.M.; Karthik, S.; Sasikumar, C.S.; Cherian, K.M. Tideglusib induces apoptosis in human neuroblastoma IMR32 cells, provoking sub-G 0 /G 1 accumulation and ROS generation. Environ. Toxicol. Pharmacol. 2016, 46, 194–205. [Google Scholar] [CrossRef]
- Bharathy, N.; Svalina, M.N.; Settelmeyer, T.P.; Cleary, M.M.; Berlow, N.E.; Airhart, S.D.; Xiang, S.; Keck, J.; Hayden, J.B.; Shern, J.F.; et al. Preclinical testing of the glycogen synthase kinase-3β inhibitor tideglusib for rhabdomyosarcoma. Oncotarget 2017, 8, 62976–62983. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S.; et al. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs In vitro and In vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Gorlick, R.; Maris, J.M.; Houghton, P.J.; Lock, R.; Carol, H.; Kurmasheva, R.T.; Kolb, E.A.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; et al. Testing of the Akt/PKB inhibitor MK-2206 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2011, 59, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; E Perez, R.; Hansen, M.; Gitelis, S.; Maki, C.G. Increasing cisplatin sensitivity by schedule-dependent inhibition of AKT and Chk1. Cancer Biol. Ther. 2014, 15, 1600–1612. [Google Scholar] [CrossRef]
- Santo, E.E.; Stroeken, P.; Sluis, P.V.; Koster, J.; Versteeg, R.; Westerhout, E.M. FOXO3a Is a Major Target of Inactivation by PI3K/AKT Signaling in Aggressive Neuroblastoma. Cancer Res 2013, 73, 2189–2198. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Toyoda, H.; Xu, D.-Q.; Zhou, Y.; Sakurai, N.; Amano, K.; Kihira, K.; Hori, H.; Azuma, E.; Komada, Y. PDK1-mTOR signaling pathway inhibitors reduce cell proliferation in MK2206 resistant neuroblastoma cells. Cancer Cell Int. 2015, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yan, S.; Attayan, N.; Ramalingam, S.; Thiele, C.J. Combination of an Allosteric Akt Inhibitor MK-2206 with Etoposide or Rapamycin Enhances the Antitumor Growth Effect in Neuroblastoma. Clin. Cancer Res. 2012, 18, 3603–3615. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.W.; Chau, I. Molecular target: Pan-AKT in gastric cancer. ESMO Open 2020, 5, e000728. [Google Scholar] [CrossRef]
- Choo, F.; Odintsov, I.; Nusser, K.; Nicholson, K.S.; Davis, L.; Corless, C.L.; Stork, L.; Somwar, R.; Ladanyi, M.; Davis, J.L.; et al. Functional impact and targetability of PI3KCA, GNAS, and PTEN mutations in a spindle cell rhabdomyosarcoma with MYOD1 L122R mutation. Mol. Case Stud. 2022, 8, a006140. [Google Scholar] [CrossRef]
- Abdelgalil, A.A.; Alkahtani, H.M.; Al-Jenoobi, F.I. Sorafenib. Profiles Drug Subst. Excip. Relat. Methodol. 2019, 44, 239–266. [Google Scholar] [CrossRef]
- Yang, F.; Van Meter, T.E.; Buettner, R.; Hedvat, M.; Liang, W.; Kowolik, C.M.; Mepani, N.; Mirosevich, J.; Nam, S.; Chen, M.Y.; et al. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol. Cancer Ther. 2008, 7, 3519–3526. [Google Scholar] [CrossRef] [Green Version]
- Chai, H.; Luo, A.Z.; Weerasinghe, P.; E Brown, R. Sorafenib downregulates ERK/Akt and STAT3 survival pathways and induces apoptosis in a human neuroblastoma cell line. Int. J. Clin. Exp. Pathol. 2010, 3, 408–415. [Google Scholar]
- Kakodkar, N.C.; Peddinti, R.R.; Tian, Y.; Guerrero, L.J.; Chlenski, A.; Yang, Q.; Salwen, H.R.; Maitland, M.L.; Cohn, S.L. Sorafenib inhibits neuroblastoma cell proliferation and signaling, blocks angiogenesis, and impairs tumor growth. Pediatr. Blood Cancer 2011, 59, 642–647. [Google Scholar] [CrossRef]
- Karajannis, M.A.; Legault, G.; Fisher, M.J.; Milla, S.S.; Cohen, K.J.; Wisoff, J.H.; Harter, D.H.; Goldberg, J.D.; Hochman, T.; Merkelson, A.; et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro-Oncology 2014, 16, 1408–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarrán, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; Román, M.S.; Calvo, J.C.; de Aguado, P.P.; Moreno, J.; Guerrero, P.; et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Niu, X.; Yao, W. Receptor Tyrosine Kinases in Osteosarcoma Treatment: Which Is the Key Target? Front. Oncol. 2020, 10, 1642. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Lin, K.-H.; Fu, B.-S.; Hsu, F.-T.; Tsai, J.-J.; Weng, M.-C.; Pan, P.-J. Sorafenib Induces Apoptosis and Inhibits NF-κB-mediated Anti-apoptotic and Metastatic Potential in Osteosarcoma Cells. Anticancer. Res. 2021, 41, 1251–1259. [Google Scholar] [CrossRef]
- Dumont, S.; Yang, D.; Dumont, A.; Reynoso, D.; Blay, J.-Y.; Trent, J. Targeted polytherapy in small cell sarcoma and its association with doxorubicin. Mol. Oncol. 2014, 8, 1458–1468. [Google Scholar] [CrossRef]
- Higuchi, T.; Igarashi, K.; Yamamoto, N.; Hayashi, K.; Kimura, H.; Miwa, S.; Bouvet, M.; Tsuchiya, H.; Hoffman, R.M. Osteosarcoma Patient-derived Orthotopic Xenograft (PDOX) Models Used to Identify Novel and Effective Therapeutics: A Review. Anticancer. Res. 2021, 41, 5865–5871. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, Y.; Yu, Y.; Pang, J.C.; Woodfield, S.E.; Tao, L.; Guan, S.; Zhang, H.; Bieerkehazhi, S.; Shi, Y.; et al. Small molecule inhibitor regorafenib inhibits RET signaling in neuroblastoma cells and effectively suppresses tumor growth in vivo. Oncotarget 2017, 8, 104090–104103. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.J.; Gill, J.D.; Roth, M.E.; Zhang, W.; Teicher, B.; Erickson, S.; Gatto, G.; Kurmasheva, R.T.; Houghton, P.J.; Smith, M.A.; et al. Initial in vivo testing of a multitarget kinase inhibitor, regorafenib, by the Pediatric Preclinical Testing Consortium. Pediatr. Blood Cancer 2020, 67, e28222. [Google Scholar] [CrossRef]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Shailubhai, K.; Bahleda, R.; Gazzah, A.; Varga, A.; Hollebecque, A.; Massard, C.; Spreafico, A.; Reni, M.; Soria, J.-C. Phase I dose-escalation study of milciclib in combination with gemcitabine in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 1257–1265. [Google Scholar] [CrossRef]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Albanese, C.; Alzani, R.; Amboldi, N.; Degrassi, A.; Festuccia, C.; Fiorentini, F.; Gravina, G.L.; Mercurio, C.; Pastori, W.; Brasca, M.; et al. Anti-tumour efficacy on glioma models of PHA-848125, a multi-kinase inhibitor able to cross the blood-brain barrier. Br. J. Pharmacol. 2013, 169, 156–166. [Google Scholar] [CrossRef]
- Martínez-Chávez, A.; Broeders, J.; Lebre, M.C.; Tibben, M.T.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. The role of drug efflux and uptake transporters ABCB1 (P-gp), ABCG2 (BCRP) and OATP1A/1B and of CYP3A4 in the pharmacokinetics of the CDK inhibitor milciclib. Eur. J. Pharm. Sci. 2021, 159, 105740. [Google Scholar] [CrossRef]
- Smolewski, P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. IDrugs. 2008, 11, 204–214. [Google Scholar]
- Castro-Gamero, A.M.; Borges, K.S.; Moreno, D.A.; Suazo, V.K.; Fujinami, M.M.; Queiroz, R.D.P.G.; de Oliveira, H.F.; Carlotti, C.G.; Scrideli, C.; Tone, L.G. Tetra-O-methyl nordihydroguaiaretic acid, an inhibitor of Sp1-mediated survivin transcription, induces apoptosis and acts synergistically with chemo-radiotherapy in glioblastoma cells. Investig. New Drugs 2013, 31, 858–870. [Google Scholar] [CrossRef] [Green Version]
- Akinaga, S.; Sugiyama, K.; Akiyama, T. UCN-01 (7-hydroxystaurosporine) and other indolocarbazole compounds: A new generation of anti-cancer agents for the new century? Anti-Cancer Drug Des. 2000, 15, 43–52. [Google Scholar]
- Shao, R.-G.; Shimizu, T.; Pommier, Y. 7-Hydroxystaurosporine (UCN-01) Induces Apoptosis in Human Colon Carcinoma and Leukemia Cells Independently of p53. Exp. Cell Res. 1997, 234, 388–397. [Google Scholar] [CrossRef]
- Lien, W.C.; Chen, T.Y.; Sheu, S.Y.; Lin, T.C.; Kang, F.C.; Yu, C.H.; Kuan, T.S.; Huang, B.M.; Wang, C.Y. 7-hydroxy-staurosporine, UCN-01, induces DNA damage response, and autophagy in human osteosarcoma U2-OS cells. J. Cell. Biochem. 2018, 119, 4729–4741. [Google Scholar] [CrossRef]
- Shankar, S.L.; Krupski, M.; Parashar, B.; Okwuaka, C.; O’Guin, K.; Mani, S.; Shafit-Zagardo, B. UCN-01 alters phosphorylation of Akt and GSK3beta and induces apoptosis in six independent human neuroblastoma cell lines. J. Neurochem. 2004, 90, 702–711. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, S.; Ye, Q.; Pan, J. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, C.; Sun, X.; Shi, X.; Jin, B.; Ding, K.; Yeung, S.-C.J.; Pan, J. Cyclin-Dependent Kinase 7/9 Inhibitor SNS-032 Abrogates FIP1-like-1 Platelet-Derived Growth Factor Receptor α and Bcr-Abl Oncogene Addiction in Malignant Hematologic Cells. Clin. Cancer Res. 2012, 18, 1966–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Tang, H.; Wu, S.; Chen, J.; Liu, J.; Liao, C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int. J. Oncol. 2014, 45, 804–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrace, S.F.; Kierstan, P.; Borgognoni, J.; Denny, S.; Wayne, J.; Bentley, C.; Cansfield, A.D.; Jackson, P.S.; Lockie, A.M.; Curtin, N.J.; et al. Transient treatment with CDK inhibitors eliminates proliferative potential even when their abilities to evoke apoptosis and DNA damage are blocked. Cell Cycle 2008, 7, 3898–3907. [Google Scholar] [CrossRef] [PubMed]
- Löschmann, N.; Michaelis, M.; Rothweiler, F.; Voges, Y.; Balónová, B.; Blight, B.A.; Jr, J.C. ABCB1 as predominant resistance mechanism in cells with acquired SNS-032 resistance. Oncotarget 2016, 7, 58051–58064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löschmann, N.; Michaelis, M.; Rothweiler, F.; Zehner, R.; Cinatl, J.; Voges, Y.; Sharifi, M.; Riecken, K.; Meyer, J.; von Deimling, A.; et al. Testing of SNS-032 in a Panel of Human Neuroblastoma Cell Lines with Acquired Resistance to a Broad Range of Drugs. Transl. Oncol. 2013, 6, 685–IN18. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Ali, M.A.; Reis, A.; Ding, L.-H.; Story, M.D.; Habib, A.A.; Chattopadhyay, A. SNS-032 prevents hypoxia-mediated glioblastoma cell invasion by inhibiting hypoxia inducible factor-1α expression. Int. J. Oncol. 2009, 34, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Choy, H.; Habib, A.A.; Saha, D. SNS-032 Prevents Tumor Cell-Induced Angiogenesis By Inhibiting Vascular Endothelial Growth Factor. Neoplasia 2007, 9, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Gaevyĭ, M.D.; Seĭf, M.N. Cerebral blood circulation during angiotensin-converting enzyme inhibition. Fiziol Zh Im I M Sechenova 1993, 79, 74–78. [Google Scholar]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Aldoss, I.T.; Tashi, T.; Ganti, A.K. Seliciclib in malignancies. Expert Opin. Investig. Drugs 2009, 18, 1957–1965. [Google Scholar] [CrossRef]
- Wu, T.; Qin, Z.; Tian, Y.; Wang, J.; Xu, C.; Li, Z.; Bian, J. Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. J. Med. Chem. 2020, 63, 13228–13257. [Google Scholar] [CrossRef]
- Khalil, H.S.; Mitev, V.; Vlaykova, T.; Cavicchi, L.; Zhelev, N. Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. J. Biotechnol. 2015, 202, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Tirado, O.M.; Mateo-Lozano, S.; Notario, V. Roscovitine Is an Effective Inducer of Apoptosis of Ewing’s Sarcoma Family Tumor Cells In vitro and In vivo. Cancer Res 2005, 65, 9320–9327. [Google Scholar] [CrossRef] [Green Version]
- Iurisci, I.; Filipski, E.; Reinhardt, J.; Bach, S.; Gianella-Borradori, A.; Iacobelli, S.; Meijer, L.; Lévi, F. Improved Tumor Control through Circadian Clock Induction by Seliciclib, a Cyclin-Dependent Kinase Inhibitor. Cancer Res 2006, 66, 10720–10728. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, J.G.; Folch, J.; Junyent, F.; Verdaguer, E.; Auladell, C.; Beas-Zarate, C.; Pallàs, M.; Camins, A. Antiapoptotic effects of roscovitine on camptothecin-induced DNA damage in neuroblastoma cells. Apoptosis 2011, 16, 536–550. [Google Scholar] [CrossRef]
- Garrofé-Ochoa, X.; Cosialls, A.M.; Ribas, J.; Gil, J.; Boix, J. Transcriptional modulation of apoptosis regulators by roscovitine and related compounds. Apoptosis 2011, 16, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tsai, Y.-H.; Tseng, S.-H. Inhibition of cyclin-dependent kinase 1–induced cell death in neuroblastoma cells through the microRNA-34a–MYCN–survivin pathway. Surgery 2012, 153, 4–16. [Google Scholar] [CrossRef]
- Ribas, J.; Boix, J.; Meijer, L. (R)-Roscovitine (CYC202, Seliciclib) sensitizes SH-SY5Y neuroblastoma cells to nutlin-3-induced apoptosis. Exp. Cell Res. 2006, 312, 2394–2400. [Google Scholar] [CrossRef] [Green Version]
- Krüger, K.; Geist, K.; Stuhldreier, F.; Schumacher, L.; Blümel, L.; Remke, M.; Wesselborg, S.; Stork, B.; Klöcker, N.; Bormann, S.; et al. Multiple DNA damage-dependent and DNA damage-independent stress responses define the outcome of ATR/Chk1 targeting in medulloblastoma cells. Cancer Lett. 2018, 430, 34–46. [Google Scholar] [CrossRef]
- Bhatia, B.; Hsieh, M.; Kenney, A.M.; Nahlé, Z. Mitogenic Sonic hedgehog signaling drives E2F1-dependent lipogenesis in progenitor cells and medulloblastoma. Oncogene 2010, 30, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2020, 81, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L., II; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 1343–1357. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Monleon, A.; Öberg, H.K.; Gaarder, J.; Berbegall, A.P.; Javanmardi, N.; Djos, A.; Ussowicz, M.; Taschner-Mandl, S.; Ambros, I.M.; Øra, I.; et al. Amplification of CDK4 and MDM2: A detailed study of a high-risk neuroblastoma subgroup. Sci. Rep. 2022, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Guntner, A.S.; Peyrl, A.; Mayr, L.; Englinger, B.; Berger, W.; Slavc, I.; Buchberger, W.; Gojo, J. Cerebrospinal fluid penetration of targeted therapeutics in pediatric brain tumor patients. Acta Neuropathol. Commun. 2020, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.-L.; Hsieh, T.-H.; Liu, Y.-R.; Chen, Y.-W.; Lee, Y.-Y.; Chang, F.-C.; Lin, S.-C.; Huang, M.-C.; Ho, D.M.-T.; Wong, T.-T.; et al. Significance of cyclin D1 overexpression in progression and radio-resistance of pediatric ependymomas. Oncotarget 2017, 9, 2527–2542. [Google Scholar] [CrossRef] [Green Version]
- D’Oto, A.; Fang, J.; Jin, H.; Xu, B.; Singh, S.; Mullasseril, A.; Jones, V.; Abu-Zaid, A.; von Buttlar, X.; Cooke, B.; et al. KDM6B promotes activation of the oncogenic CDK4/6-pRB-E2F pathway by maintaining enhancer activity in MYCN-amplified neuroblastoma. Nat. Commun. 2021, 12, 7204. [Google Scholar] [CrossRef]
- Swadi, R.R.; Sampat, K.; Herrmann, A.; Losty, P.D.; See, V.; Moss, D.J. CDK inhibitors reduce cell proliferation and reverse hypoxia-induced metastasis of neuroblastoma tumours in a chick embryo model. Sci. Rep. 2019, 9, 9136. [Google Scholar] [CrossRef] [Green Version]
- Rihani, A.; Vandesompele, J.; Speleman, F.; Van Maerken, T. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma. Cancer Cell Int. 2015, 15, 76. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.; Muñoz-Galván, S.; Jiménez-García, M.P.; Marín, J.J.; Carnero, A. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget 2015, 6, 40557–40574. [Google Scholar] [CrossRef] [Green Version]
- Böhm, M.J.; Marienfeld, R.; Jäger, D.; Mellert, K.; von Witzleben, A.; Brüderlein, S.; Wittau, M.; von Baer, A.; Schultheiss, M.; Mayer-Steinacker, R.; et al. Analysis of the CDK4/6 Cell Cycle Pathway in Leiomyosarcomas as a Potential Target for Inhibition by Palbociclib. Sarcoma 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Singh, A.S.; Kiyuna, T.; Dry, S.M.; Li, Y.; James, A.W.; Igarashi, K.; Kawaguchi, K.; DeLong, J.C.; Zhang, Y.; et al. Effective molecular targeting of CDK4/6 and IGF-1R in a rare FUS-ERG fusion CDKN2A-deletion doxorubicin-resistant Ewing’s sarcoma patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 2016, 7, 47556–47564. [Google Scholar] [CrossRef]
- Tramontana, T.F.; Marshall, M.S.; Helvie, A.E.; Schmitt, M.R.; Ivanovich, J.; Carter, J.L.; Renbarger, J.L.; Ferguson, M.J. Sustained Complete Response to Palbociclib in a Refractory Pediatric Sarcoma With BCOR-CCNB3 Fusion and Germline CDKN2B Variant. JCO Precis. Oncol. 2020, 466–471. [Google Scholar] [CrossRef]
- Riess, C.; Koczan, D.; Schneider, B.; Linke, C.; del Moral, K.; Classen, C.F.; Maletzki, C. Cyclin-dependent kinase inhibitors exert distinct effects on patient-derived 2D and 3D glioblastoma cell culture models. Cell Death Discov. 2021, 7, 1–15. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, Y.; Yan, K.; Li, Z.; Xu, C.; Geng, Y.; Pan, C.; Chen, X.; Zhang, L.; Xi, Q. Potent anti-tumor efficacy of palbociclib in treatment-naïve H3.3K27M-mutant diffuse intrinsic pontine glioma. Ebiomedicine 2019, 43, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Morris-Hanon, O.; Marazita, M.C.; Romorini, L.; Isaja, L.; Fernandez-Espinosa, D.D.; Sevlever, G.E.; Scassa, M.E.; Videla-Richardson, G.A. Palbociclib Effectively Halts Proliferation but Fails to Induce Senescence in Patient-Derived Glioma Stem Cells. Mol. Neurobiol. 2019, 56, 7810–7821. [Google Scholar] [CrossRef]
- Huillard, E.; Hashizume, R.; Phillips, J.J.; Griveau, A.; Ihrie, R.A.; Aoki, Y.; Nicolaides, T.; Perry, A.; Waldman, T.; McMahon, M.; et al. Cooperative interactions of BRAF V600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc. Natl. Acad. Sci. USA 2012, 109, 8710–8715. [Google Scholar] [CrossRef] [Green Version]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 Inhibitor, Significantly Prolongs Survival in a Genetically Engineered Mouse Model of Brainstem Glioma. PLoS ONE 2013, 8, e77639. [Google Scholar] [CrossRef] [Green Version]
- Lallena, M.J.; Boehnke, K.; Torres, R.; Hermoso, A.; Amat, J.; Calsina, B.; De Dios, A.; Buchanan, S.; Du, J.; Beckmann, R.P.; et al. Abstract 3101: In-vitro characterization of Abemaciclib pharmacology in ER+ breast cancer cell lines. Cancer Res 2015, 75, 3101. [Google Scholar] [CrossRef]
- Torres-Guzmán, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef] [Green Version]
- Chong, Q.-Y.; Kok, Z.-H.; Bui, N.-L.; Xiang, X.; Wong, A.L.-A.; Yong, W.-P.; Sethi, G.; Lobie, P.E.; Wang, L.; Goh, B.-C. A unique CDK4/6 inhibitor: Current and future therapeutic strategies of abemaciclib. Pharmacol. Res. 2020, 156, 104686. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Dowless, M.S.; Lowery, C.D.; Shackleford, T.J.; Renschler, M.; Stephens, J.R.; Flack, R.; Blosser, W.; Gupta, S.; Stewart, J.; Webster, Y.; et al. Abemaciclib Is Active in Preclinical Models of Ewing Sarcoma via Multipronged Regulation of Cell Cycle, DNA Methylation, and Interferon Pathway Signaling. Clin. Cancer Res. 2018, 24, 6028–6039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Bao, H. Abemaciclib is synergistic with doxorubicin in osteosarcoma pre-clinical models via inhibition of CDK4/6–Cyclin D–Rb pathway. Cancer Chemother. Pharmacol. 2021, 89, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Schubert, N.A.; Schild, L.; van Oirschot, S.; Keller, K.M.; Alles, L.K.; Vernooij, L.; Nulle, M.E.; Dolman, M.E.M.; van den Boogaard, M.L.; Molenaar, J.J. Combined targeting of the p53 and pRb pathway in neuroblastoma does not lead to synergistic responses. Eur. J. Cancer 2020, 142, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, X.; Kong, S.; Shang, S.; Qi, Y. CDK4/6 inhibition suppresses tumour growth and enhances the effect of temozolomide in glioma cells. J. Cell. Mol. Med. 2020, 24, 5135–5145. [Google Scholar] [CrossRef] [Green Version]
- Mayr, L.; Guntner, A.S.; Madlener, S.; Schmook, M.T.; Peyrl, A.; Azizi, A.A.; Dieckmann, K.; Reisinger, D.; Stepien, N.M.; Schramm, K.; et al. Cerebrospinal Fluid Penetration and Combination Therapy of Entrectinib for Disseminated ROS1/NTRK-Fusion Positive Pediatric High-Grade Glioma. J. Pers. Med. 2020, 10, 290. [Google Scholar] [CrossRef]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 2018, 34, 411–426.e19. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Kour, S.; Sonawane, Y.A.; Robb, C.M.; Contreras, J.I.; Kizhake, S.; Zahid, M.; Karpf, A.R.; Natarajan, A. Symbiotic prodrugs (SymProDs) dual targeting of NFkappaB and CDK. Chem. Biol. Drug Des. 2020, 96, 773–784. [Google Scholar] [CrossRef]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.-M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Oghabi, M.; Safaroghli-Azar, A.; Pourbagheri-Sigaroodi, A.; Sayyadi, M.; Hamidpour, M.; Mohammadi, M.H.; Bashash, D. Anti-proliferative effects of a small molecule inhibitor of CDK AT7519 on chronic myeloid leukemia (CML) cells through halting the transition of cells from G2/M phase of the cell cycle. Biocell 2020, 44, 183–192. [Google Scholar] [CrossRef]
- Dolman, M.E.M.; Poon, E.; Ebus, M.E.; Hartog, I.J.D.; van Noesel, C.J.; Jamin, Y.; Hallsworth, A.; Robinson, S.P.; Petrie, K.; Sparidans, R.W.; et al. Cyclin-Dependent Kinase Inhibitor AT7519 as a Potential Drug for MYCN-Dependent Neuroblastoma. Clin. Cancer Res. 2015, 21, 5100–5109. [Google Scholar] [CrossRef] [Green Version]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krššák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Pezuk, J.A.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; de Oliveira, H.F.; Scrideli, C.A.; Tone, L.G. Inhibition of Polo-Like Kinase 1 Induces Cell Cycle Arrest and Sensitizes Glioblastoma Cells to Ionizing Radiation. Cancer Biotherapy Radiopharm. 2013, 28, 516–522. [Google Scholar] [CrossRef] [Green Version]
- A Pezuk, J.; Brassesco, M.S.; Morales, A.G.; de Oliveira, J.C.; Queiroz, R.G.D.P.; Machado, H.R.; Carlotti, C.G.; Neder, L.; A Scrideli, C.; Tone, L.G. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer Gene Ther. 2013, 20, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Pezuk, J.A.; Brassesco, M.S.; Ramos, P.M.M.; Scrideli, C.A.; Tone, L.G.; Pezuk, M.S.B.J.A. Polo-Like Kinase 1 Pharmacological Inhibition as Monotherapy or in Combination: Comparative Effects of Polo-Like Kinase 1 Inhibition in Medulloblastoma Cells. Anti-Cancer Agents Med. Chem. 2017, 17, 1278–1291. [Google Scholar] [CrossRef]
- Yunoki, T.; Tabuchi, Y.; Hayashi, A.; Kondo, T. Inhibition of Polo-Like Kinase 1 Promotes Hyperthermia Sensitivity via Inactivation of Heat Shock Transcription Factor 1 in Human Retinoblastoma Cells. Investig. Opthalmology Vis. Sci. 2013, 54, 8353–8363. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, C.; Li, X.; Du, X.; Tao, Y.; Ren, J.; Fang, F.; Xie, Y.; Li, M.; Qian, G.; et al. The dual role of BI 2536, a small-molecule inhibitor that targets PLK1, in induction of apoptosis and attenuation of autophagy in neuroblastoma cells. J. Cancer 2020, 11, 3274–3287. [Google Scholar] [CrossRef] [Green Version]
- Grinshtein, N.; Datti, A.; Fujitani, M.; Uehling, D.; Prakesch, M.; Isaac, M.; Irwin, M.S.; Wrana, J.L.; Al-Awar, R.; Kaplan, D.R. Small Molecule Kinase Inhibitor Screen Identifies Polo-Like Kinase 1 as a Target for Neuroblastoma Tumor-Initiating Cells. Cancer Res 2011, 71, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-H.; Yeh, H.-N.; Huang, C.-T.; Wang, W.-H.; Hsu, W.-M.; Huang, H.-C.; Juan, H.-F. BI-2536 Promotes Neuroblastoma Cell Death via Minichromosome Maintenance Complex Components 2 and 10. Pharmaceuticals 2021, 15, 37. [Google Scholar] [CrossRef]
- Morales, A.G.; Brassesco, M.S.; Pezuk, J.A.; Oliveira, J.C.; Montaldi, A.P.; Sakamoto-Hojo, E.T.; Scrideli, C.A.; Tone, L.G. BI 2536-mediated PLK1 inhibition suppresses HOS and MG-63 osteosarcoma cell line growth and clonogenicity. Anti-Cancer Drugs 2011, 22, 995–1001. [Google Scholar] [CrossRef]
- Liu, X.; Choy, E.; Harmon, D.; Yang, S.; Yang, C.; Mankin, H.; Hornicek, F.J.; Duan, Z. Inhibition of polo-like kinase 1 leads to the suppression of osteosarcoma cell growth in vitro and in vivo. Anti-Cancer Drugs 2011, 22, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Thalhammer, V.; Lopez-Garcia, L.A.; Herrero-Martin, D.; Hecker, R.; Laubscher, D.; Gierisch, M.E.; Wachtel, M.; Bode, P.; Nanni, P.; Blank, B.; et al. PLK1 Phosphorylates PAX3-FOXO1, the Inhibition of Which Triggers Regression of Alveolar Rhabdomyosarcoma. Cancer Res 2015, 75, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehle, A.; Hugle, M.; Fulda, S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer Lett. 2015, 365, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an Orally Available, Specific Small-Molecule Polo-like Kinase 1 Inhibitor with Antitumor Activity in Solid and Hematologic Malignancies. Mol. Cancer Ther. 2012, 11, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Sero, V.; Tavanti, E.; Vella, S.; Hattinger, C.M.; Fanelli, M.; Michelacci, F.; Versteeg, R.; Valsasina, B.; Gudeman, B.; Picci, P.; et al. Targeting polo-like kinase 1 by NMS-P937 in osteosarcoma cell lines inhibits tumor cell growth and partially overcomes drug resistance. Investig. New Drugs 2014, 32, 1167–1180. [Google Scholar] [CrossRef]
- Wang, D.; Veo, B.; Pierce, A.; Fosmire, S.; Madhavan, K.; Balakrishnan, I.; Donson, A.; Alimova, I.; Sullivan, K.D.; Joshi, M.; et al. A novel PLK1 inhibitor onvansertib effectively sensitizes MYC-driven medulloblastoma to radiotherapy. Neuro-Oncology 2021, 24, 414–426. [Google Scholar] [CrossRef]
- Wu, C.-P.; Hsiao, S.-H.; Luo, S.-Y.; Tuo, W.-C.; Su, C.-Y.; Li, Y.-Q.; Huang, Y.-H.; Hsieh, C.-H. Overexpression of Human ABCB1 in Cancer Cells Leads to Reduced Activity of GSK461364, a Specific Inhibitor of Polo-like Kinase 1. Mol. Pharm. 2014, 11, 3727–3736. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Sadowski, N.; Ackermann, S.; Althoff, K.; Schönbeck, K.; Batzke, K.; Schäfers, S.; Odersky, A.; Heukamp, L.; Astrahantseff, K.; et al. The GSK461364 PLK1 inhibitor exhibits strong antitumoral activity in preclinical neuroblastoma models. Oncotarget 2016, 8, 6730–6741. [Google Scholar] [CrossRef]
- Chou, Y.-S.; Yen, C.-C.; Chen, W.-M.; Lin, Y.-C.; Wen, Y.-S.; Ke, W.-T.; Wang, J.-Y.; Liu, C.-Y.; Yang, M.-H.; Chen, T.-H.; et al. Cytotoxic mechanism of PLK1 inhibitor GSK461364 against osteosarcoma: Mitotic arrest, apoptosis, cellular senescence, and synergistic effect with paclitaxel. Int. J. Oncol. 2016, 48, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Bogado, R.F.; Pezuk, J.A.; de Oliveira, H.F.; Tone, L.G.; Brassesco, M.S. BI 6727 and GSK461364 suppress growth and radiosensitize osteosarcoma cells, but show limited cytotoxic effects when combined with conventional treatments. Anti-Cancer Drugs 2015, 26, 56–63. [Google Scholar] [CrossRef]
- Gorlick, R.; Kolb, E.A.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Lock, R.; Billups, C.A.; Kurmasheva, R.T.; et al. Initial testing (stage 1) of the polo-like kinase inhibitor volasertib (BI 6727), by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2013, 61, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Carol, H.; Boehm, I.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Wu, J.; et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother. Pharmacol. 2011, 68, 1291–1304. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M.; Bs, C.L.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing of the aurora kinase a inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr. Blood Cancer 2010, 55, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Boi, D.; Souvalidou, F.; Capelli, D.; Polverino, F.; Marini, G.; Montanari, R.; Pochetti, G.; Tramonti, A.; Contestabile, R.; Trisciuoglio, D.; et al. PHA-680626 Is an Effective Inhibitor of the Interaction between Aurora-A and N-Myc. Int. J. Mol. Sci. 2021, 22, 13122. [Google Scholar] [CrossRef]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Geron, L.; Borges, K.S.; Andrade, A.F.; Suazo, V.K.; Scrideli, C.A.; Tone, L.G. Antitumour activity of AMG 900 alone or in combination with histone deacetylase inhibitor SaHa on medulloblastoma cell lines. Neurol. Res. 2015, 37, 703–711. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 2022, 15, 1–28. [Google Scholar] [CrossRef]
- Chambers, A.; Kundranda, M.; Rao, S.; Niu, J. Anti-angiogenesis Revisited: Combination with Immunotherapy in Solid Tumors. Lung Cancer 1912, 23, 100. [Google Scholar] [CrossRef]
- Inaba, H.; Rubnitz, J.E.; Coustan-Smith, E.; Li, L.; Furmanski, B.D.; Mascara, G.P.; Heym, K.M.; Christensen, R.; Onciu, M.; Shurtleff, S.A.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the Multikinase Inhibitor Sorafenib in Combination With Clofarabine and Cytarabine in Pediatric Relapsed/Refractory Leukemia. J. Clin. Oncol. 2011, 29, 3293–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.G.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. 2011, 23, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Keino, D.; Yokosuka, T.; Hirose, A.; Sakurai, Y.; Nakamura, W.; Fujita, S.; Hayashi, A.; Miyagawa, N.; Iwasaki, F.; Hamanoue, S.; et al. Pilot study of the combination of sorafenib and fractionated irinotecan in pediatric relapse/refractory hepatic cancer (FINEX pilot study). Pediatr. Blood Cancer 2020, 67, e28655. [Google Scholar] [CrossRef] [PubMed]
- Geller, J.I.; Fox, E.; Turpin, B.K.; Goldstein, S.L.; Liu, X.; Minard, C.G.; Kudgus, R.A.; Reid, J.M.; Berg, S.L.; Weigel, B.J. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: A Children’s Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer 2018, 124, 4548–4555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daw, N.C.; Furman, W.L.; Stewart, C.F.; Iacono, L.C.; Krailo, M.; Bernstein, M.L.; Dancey, J.E.; Speights, R.A.; Blaney, S.M.; Croop, J.M.; et al. Phase I and Pharmacokinetic Study of Gefitinib in Children With Refractory Solid Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2005, 23, 6172–6180. [Google Scholar] [CrossRef]
- Pollack, I.F.; Stewart, C.F.; Kocak, M.; Poussaint, T.Y.; Broniscer, A.; Banerjee, A.; Douglas, J.G.; Kun, L.E.; Boyett, J.M.; Geyer, J.R. A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: A report from the Pediatric Brain Tumor Consortium. Neuro-Oncology 2011, 13, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): A phase 1/2, open-label, basket trial. Lancet Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef]
- Del Rivero, J.; Edgerly, M.; Ward, J.; Madan, R.A.; Balasubramaniam, S.; Fojo, T.; Gramza, A.W. Phase I/II Trial of Vandetanib and Bortezomib in Adults with Locally Advanced or Metastatic Medullary Thyroid Cancer. Oncol. 2018, 24, 16-e14. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.I.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.A.; Ohashi, K.; et al. Tumor agnostic efficacy of selpercatinib in patients with RET fusion+ solid tumors: A global, multicenter, registrational trial update (LIBRETTO-001). J. Clin. Oncol. 2022, 40, 3094. [Google Scholar] [CrossRef]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion–positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Fukano, R.; Mori, T.; Sekimizu, M.; Choi, I.; Kada, A.; Saito, A.M.; Asada, R.; Takeuchi, K.; Terauchi, T.; Tateishi, U.; et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci. 2020, 111, 4540–4547. [Google Scholar] [CrossRef]
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Adashek, J.J.; Menta, A.K.; Reddy, N.K.; Desai, A.P.; Roszik, J.; Subbiah, V. Tissue-Agnostic Activity of BRAF plus MEK Inhibitor in BRAF V600–Mutant Tumors. Mol. Cancer Ther. 2022, 21, 871–878. [Google Scholar] [CrossRef]
- Bouffet, E.; Hansford, J.; Garré, M.L.; Hara, J.; Plant-Fox, A.; Aerts, I.; Locatelli, F.; Van der Lugt, J.; Papusha, L.; Sahm, F.; et al. Primary analysis of a phase II trial of dabrafenib plus trametinib (dab + tram) in BRAF V600–mutant pediatric low-grade glioma (pLGG). J. Clin. Oncol. 2022, 40, LBA2002. [Google Scholar] [CrossRef]
- Hargrave, D.R.; Bouffet, E.; Tabori, U.; Broniscer, A.; Cohen, K.J.; Hansford, J.R.; Geoerger, B.; Hingorani, P.; Dunkel, I.J.; Russo, M.W.; et al. Efficacy and Safety of Dabrafenib in Pediatric Patients with BRAF V600 Mutation–Positive Relapsed or Refractory Low-Grade Glioma: Results from a Phase I/IIa Study. Clin. Cancer Res. 2019, 25, 7303–7311. [Google Scholar] [CrossRef]
- Lau, N.; Feldkamp, M.M.; Roncari, L.; Loehr, A.H.; Shannon, P.; Gutmann, D.H.; Guha, A. Loss of Neurofibromin Is Associated with Activation of RAS/MAPK and PI3-K/AKT Signaling in a Neurofibromatosis 1 Astrocytoma. J. Neuropathol. Exp. Neurol. 2000, 59, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef]
- Bautista, F.; Paoletti, X.; Rubino, J.; Brard, C.; Rezai, K.; Nebchi, S.; Andre, N.; Aerts, I.; De Carli, E.; van Eijkelenburg, N.; et al. Phase I or II Study of Ribociclib in Combination With Topotecan-Temozolomide or Everolimus in Children With Advanced Malignancies: Arms A and B of the AcSé-ESMART Trial. J. Clin. Oncol. 2021, 39, 3546–3560. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 1–36. [Google Scholar] [CrossRef]
- Van Mater, D.; Gururangan, S.; Becher, O.; Campagne, O.; Leary, S.; Phillips, J.J.; Huang, J.; Lin, T.; Poussaint, T.Y.; Goldman, S.; et al. A phase I trial of the CDK 4/6 inhibitor palbociclib in pediatric patients with progressive brain tumors: A Pediatric Brain Tumor Consortium study (PBTC-042). Pediatr. Blood Cancer 2021, 68, e28879. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Lipsitz, E.; Fox, E.; Teachey, D.T.; Maris, J.M.; Weigel, B.; Adamson, P.C.; Ingle, M.A.; Ahern, C.H.; Blaney, S.M. Pediatric Phase I Trial and Pharmacokinetic Study of MLN8237, an Investigational Oral Selective Small-Molecule Inhibitor of Aurora Kinase A: A Children’s Oncology Group Phase I Consortium Study. Clin. Cancer Res. 2012, 18, 6058–6064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, L.; Marshall, L.V.; Pearson, A.D.; Morland, B.; Elliott, M.; Campbell-Hewson, Q.; Makin, G.; Halford, S.E.; Acton, G.; Ross, P.; et al. A Phase I Trial of AT9283 (a Selective Inhibitor of Aurora Kinases) in Children and Adolescents with Solid Tumors: A Cancer Research UK Study. Clin. Cancer Res. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossé, Y.P.; Fox, E.; Teachey, D.T.; Reid, J.M.; Safgren, S.L.; Carol, H.; Lock, R.B.; Houghton, P.J.; Smith, M.A.; Hall, D.; et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin. Cancer Res. 2019, 25, 3229–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliciano, S.V.M.; Santos, M.D.O.; Pombo-De-Oliveira, M.S. Incidência e Mortalidade por Câncer entre Crianças e Adolescentes: Uma Revisão Narrativa. Rev. Bras. Cancerol. 2019, 64, 389–396. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. Biodrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Lee, J.; Park, Y.H. Trastuzumab deruxtecan for HER2+ advanced breast cancer. Futur. Oncol. 2022, 18, 7–19. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Lopes, L.F.; Bacchi, C.E. Imatinib treatment for gastrointestinal stromal tumour (GIST). J. Cell. Mol. Med. 2009, 14, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Fischer, P.M. The Design of Drug Candidate Molecules as Selective Inhibitors of Therapeutically Relevant Protein Kinases. Curr. Med. Chem. 2004, 11, 1563–1583. [Google Scholar] [CrossRef]
- A Bogoyevitch, M.; Fairlie, D. A new paradigm for protein kinase inhibition: Blocking phosphorylation without directly targeting ATP binding. Drug Discov. Today 2007, 12, 622–633. [Google Scholar] [CrossRef]
- Gaumann, A.K.; Kiefer, F.; Alfer, J.; Lang, S.A.; Geissler, E.K.; Breier, G. Receptor tyrosine kinase inhibitors: Are they real tumor killers? Int. J. Cancer 2015, 138, 540–554. [Google Scholar] [CrossRef]
- Roberti, M.; Bottegoni, G. Non-ATP Competitive Protein Kinase Inhibitors. Curr. Med. Chem. 2010, 17, 2804–2821. [Google Scholar] [CrossRef]
- Suttorp, M.; Bornhäuser, M.; Metzler, M.; Millot, F.; Schleyer, E. Pharmacology and pharmacokinetics of imatinib in pediatric patients. Expert Rev. Clin. Pharmacol. 2017, 11, 219–231. [Google Scholar] [CrossRef]
- Angel, M.C.-G.; Julia, A.P.; María, S.B.; Luiz, G.T. G2/M inhibitors as pharmacotherapeutic opportunities for glioblastoma: The old, the new, and the future. Cancer Biol. Med. 2018, 15, 354–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of Neuroblastomas Reveals a Skewed ALK Mutation Spectrum in Tumors with MYCN Amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L.; et al. Differential Inhibitor Sensitivity of Anaplastic Lymphoma Kinase Variants Found in Neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef] [Green Version]
- de Sousa, G.R.; Vieira, G.M.; das Chagas, P.F.; Pezuk, J.A.; Brassesco, M.S. Should we keep rocking? Portraits from targeting Rho kinases in cancer. Pharmacol. Res. 2020, 160, 105093. [Google Scholar] [CrossRef]
- Martinsson, T.; Eriksson, T.; Abrahamsson, J.; Caren, H.; Hansson, M.; Kogner, P.; Kamaraj, S.; Schönherr, C.; Weinmar, J.; Ruuth, K.; et al. Appearance of the Novel Activating F1174S ALK Mutation in Neuroblastoma Correlates with Aggressive Tumor Progression and Unresponsiveness to Therapy. Cancer Res 2011, 71, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Katayama, R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Cancer Sci. 2018, 109, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Rolfo, C.; Passiglia, F.; Castiglia, M.; E Raez, L.; Germonpre, P.; Gil-Bazo, I.; Zwaenepoel, K.; De Wilde, A.; Bronte, G.; Russo, A.; et al. ALK and crizotinib: After the honeymoon…what else? Resistance mechanisms and new therapies to overcome it. Transl. Lung Cancer Res. 2014, 3, 250–261. [Google Scholar] [CrossRef]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef]
- Massicotte, M.P.; Sofronas, M.; Deveber, G. Difficulties in performing clinical trials of antithrombotic therapy in neonates and children. Thromb. Res. 2006, 118, 153–163. [Google Scholar] [CrossRef]
- Bond, M.C.; Pritchard, S. Understanding clinical trials in childhood cancer. Paediatr. Child Health 2006, 11, 148–150. [Google Scholar] [CrossRef] [Green Version]
- Renfro, L.A.; Ji, L.; Piao, J.; Onar-Thomas, A.; Kairalla, J.A.; Alonzo, T.A. Trial Design Challenges and Approaches for Precision Oncology in Rare Tumors: Experiences of the Children’s Oncology Group. JCO Precis. Oncol. 2019, 1, 1–13. [Google Scholar] [CrossRef]
- Bavcar, S.; Argyle, D.J. Receptor tyrosine kinase inhibitors: Molecularly targeted drugs for veterinary cancer therapy. Veter- Comp. Oncol. 2012, 10, 163–173. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candido, M.F.; Medeiros, M.; Veronez, L.C.; Bastos, D.; Oliveira, K.L.; Pezuk, J.A.; Valera, E.T.; Brassesco, M.S. Drugging Hijacked Kinase Pathways in Pediatric Oncology: Opportunities and Current Scenario. Pharmaceutics 2023, 15, 664. https://doi.org/10.3390/pharmaceutics15020664
Candido MF, Medeiros M, Veronez LC, Bastos D, Oliveira KL, Pezuk JA, Valera ET, Brassesco MS. Drugging Hijacked Kinase Pathways in Pediatric Oncology: Opportunities and Current Scenario. Pharmaceutics. 2023; 15(2):664. https://doi.org/10.3390/pharmaceutics15020664
Chicago/Turabian StyleCandido, Marina Ferreira, Mariana Medeiros, Luciana Chain Veronez, David Bastos, Karla Laissa Oliveira, Julia Alejandra Pezuk, Elvis Terci Valera, and María Sol Brassesco. 2023. "Drugging Hijacked Kinase Pathways in Pediatric Oncology: Opportunities and Current Scenario" Pharmaceutics 15, no. 2: 664. https://doi.org/10.3390/pharmaceutics15020664