
Quality by Design as a Tool in the Optimisation of Nanoparticle Preparation—A Case Study of PLGA Nanoparticles

Abstract
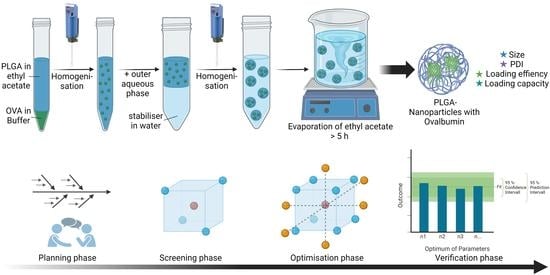
:
1. Introduction
- Definition of quality attributes and selection of outcome parameters;
- Identification and evaluation of process variables that might have an impact on PLGA-NP properties;
- Identification of significant process parameters by screening of the most promising variables;
- First approach of optimisation using the results of the screening;
- Further optimisation and controlled adjustments of the products quality by varying the most influencing process parameters within a response surface design;
- Prediction and verification of the optimal process parameters.
2. Materials and Methods
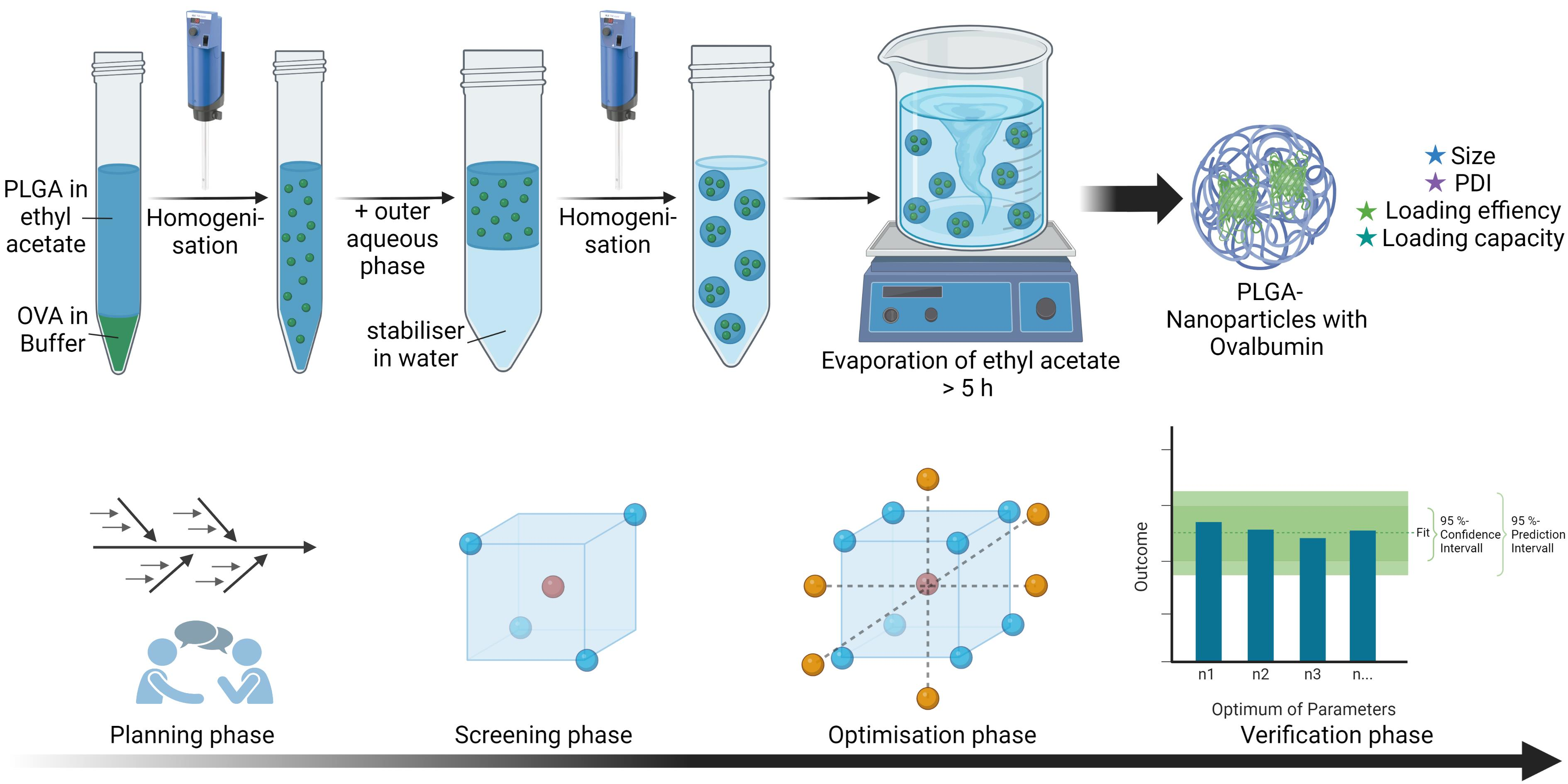
2.1. Nanoparticle Preparation
2.2. Purification
2.3. Measurement of Size, Size Distribution and Zeta-Potential
2.4. Protein Quantification
2.5. Scanning Electron Microscopy (SEM)
2.6. Design of Experiements
3. Results and Discussion
3.1. Definition of Quality Attributes and Selection of Outcome Parameters
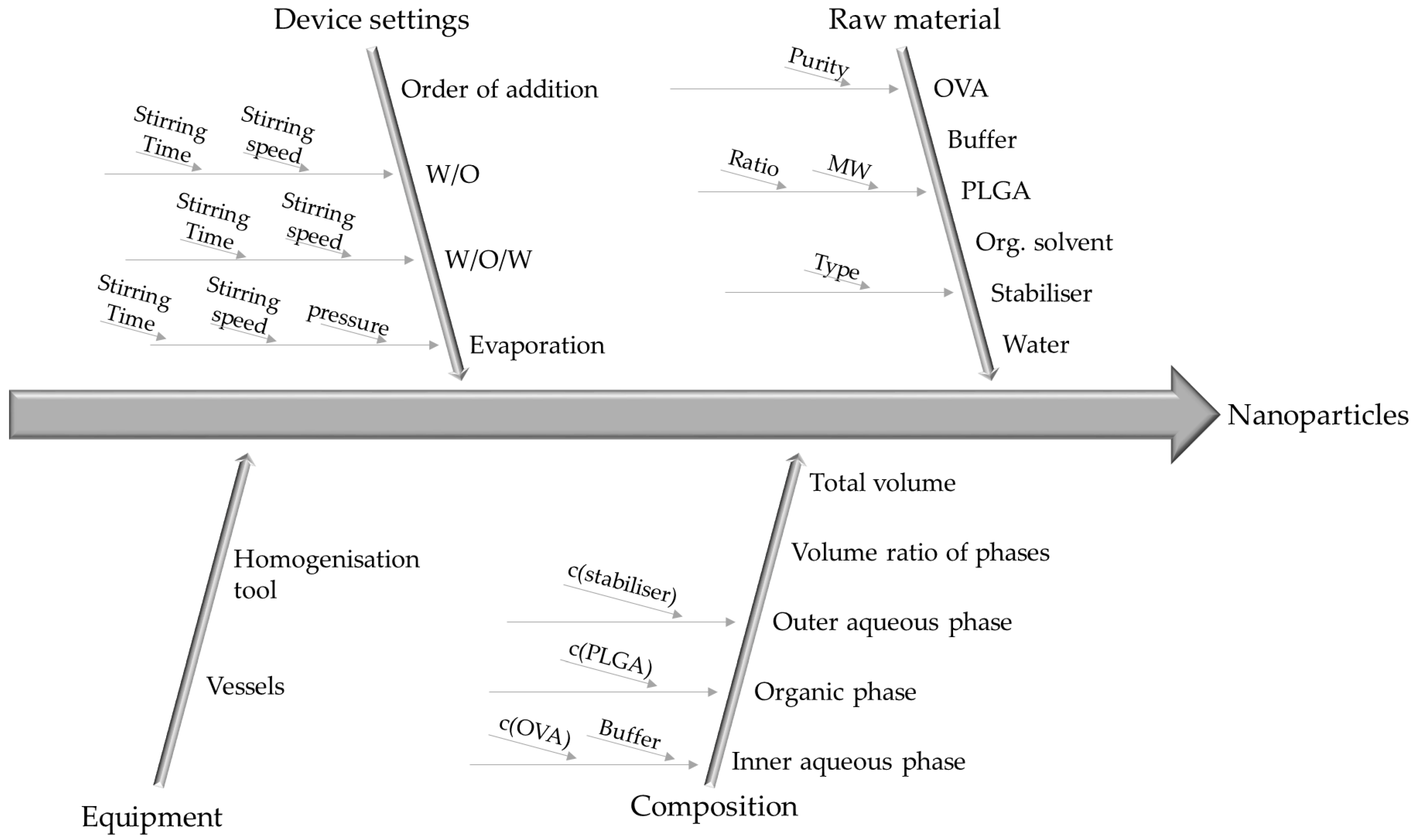
3.2. Identification and Evaluation of Process Parameters
3.3. Identification of Significant Process Parameters by Screening Design
3.3.1. Definitive Screening Design
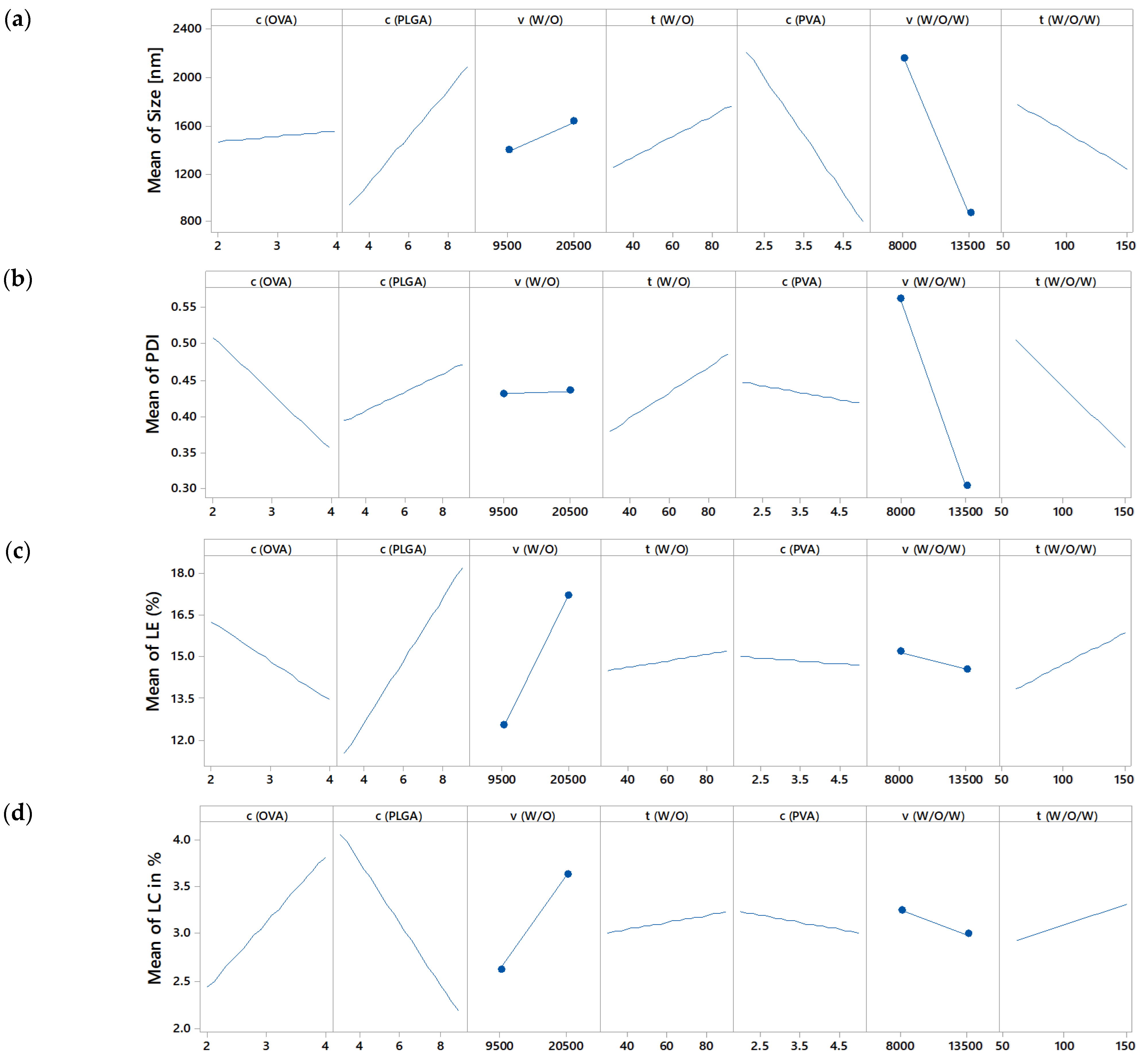
3.3.2. Effect on Size and Size Distribution
3.3.3. Effect on Drug Loading and Loading Efficiency
3.4. First Approach of Optimisation with Screening Results
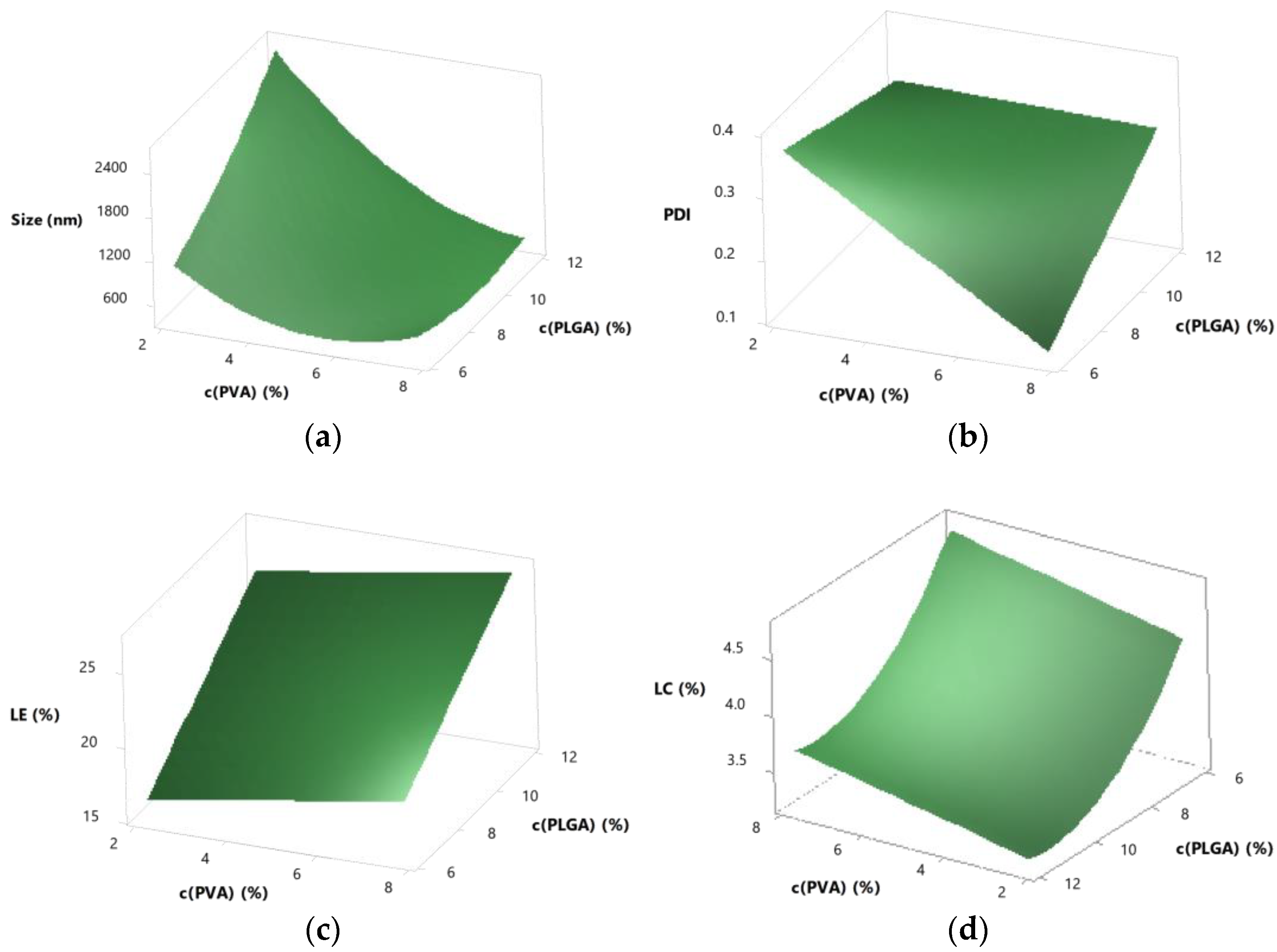
3.5. Further Optimisation by Response Surface Design
3.5.1. Central Composite Response Surface Design
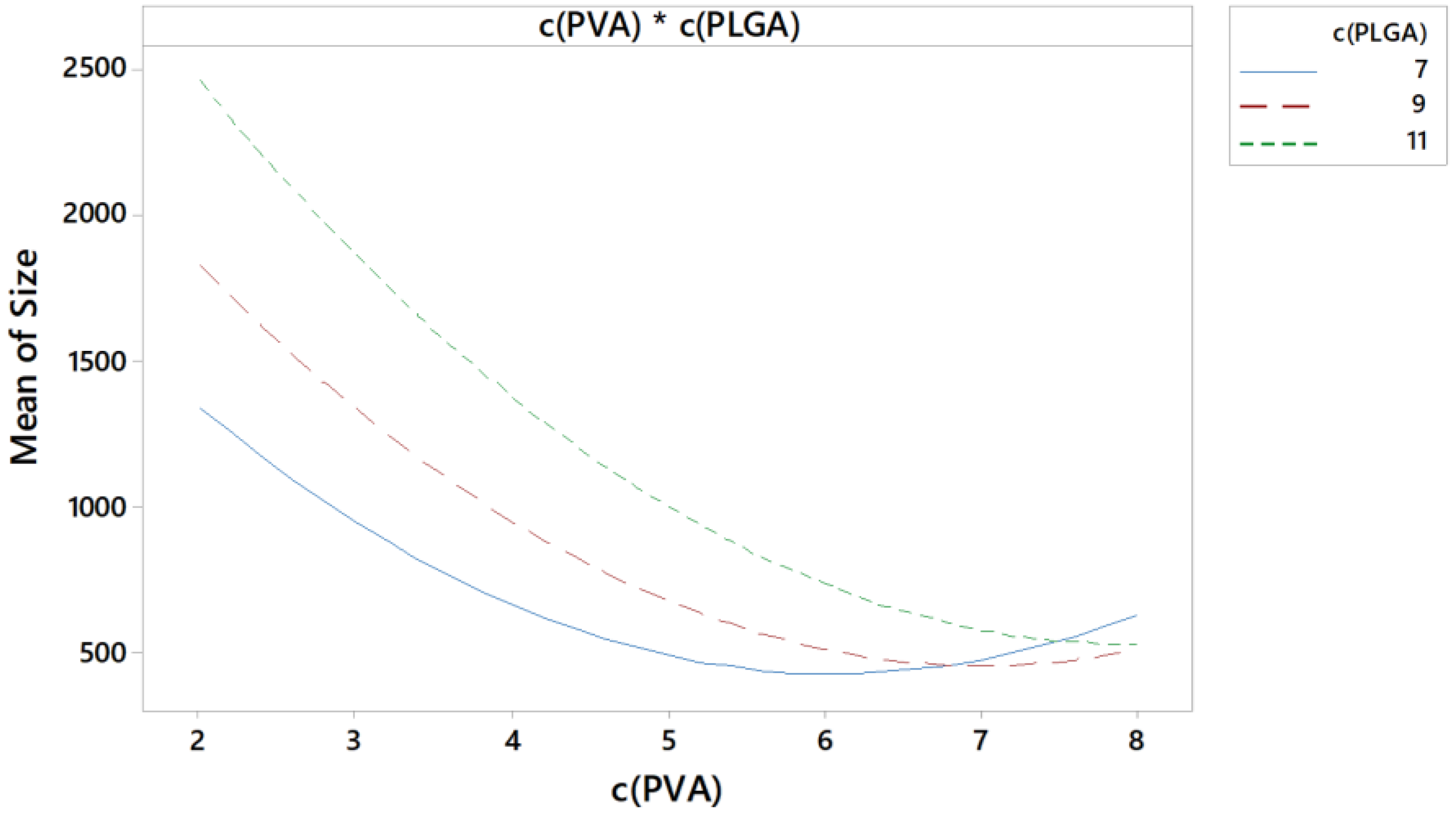
3.5.2. Interactions
3.5.3. Predictability
3.5.4. Optimisation
3.6. Verification of the Optimal Process Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scherließ, R.; Janke, J. Preparation of Poly-Lactic-Co-Glycolic Acid Nanoparticles in a Dry Powder Formulation for Pulmonary Antigen Delivery. Pharmaceutics 2021, 13, 1196. [Google Scholar] [CrossRef] [PubMed]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: The promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, E.C.; Ward, R.W. Mucosal vaccines—Fortifying the frontiers. Nat. Rev. Immunol. 2022, 22, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Hellfritzsch, M.; Scherließ, R. Mucosal Vaccination via the Respiratory Tract. Pharmaceutics 2019, 11, 375. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, F.; Ghahremani, M.H.; Esmaeili, B.; Khoshayand, M.R.; Atyabi, F.; Dinarvand, R. PLGA nanoparticles of different surface properties: Preparation and evaluation of their body distribution. Int. J. Pharm. 2008, 349, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Desai, N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ICH Guideline Q8 (R2) on Pharmaceutical Development; European Medicines Agency: London, UK, 2017. [Google Scholar]
- Yu, L.X. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2008, 25, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Lionberger, R.A.; Lee, S.L.; Lee, L.; Raw, A.; Yu, L.X. Quality by design: Concepts for ANDAs. AAPS J. 2008, 10, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Draheim, C.; De Crécy, F.; Hansen, S.; Collnot, E.-M.; Lehr, C.-M. A Design of Experiment Study of Nanoprecipitation and Nano Spray Drying as Processes to Prepare PLGA Nano- and Microparticles with Defined Sizes and Size Distributions. Pharm. Res. 2015, 32, 2609–2624. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Wu, J.; Li, H.; Ma, G. Recent research and development of PLGA/PLA microspheres/nanoparticles: A review in scientific and industrial aspects. Front. Chem. Sci. Eng. 2019, 13, 14–27. [Google Scholar] [CrossRef]
- Sadat Tabatabaei Mirakabad, F.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y.; et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac. J. Cancer Prev. 2014, 15, 517–535. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm?event=BasicSearch.page (accessed on 4 November 2022).
- Janke, J. PLGA-Nanopartikel für die Inhalative Vakzinierung, Dissertation, Kiel University. 2014. Available online: https://macau.uni-kiel.de/receive/diss_mods_00015686 (accessed on 4 November 2022).
- Jones, B.; Nachtsheim, C.J. A Class of Three-Level Designs for Definitive Screening in the Presence of Second-Order Effects. J. Qual. Technol. 2011, 43, 1–5. [Google Scholar] [CrossRef]
- Jones, B.; Nachtsheim, C.J. Effective Design-Based Model Selection for Definitive Screening Designs. Technometrics 2017, 59, 319–329. [Google Scholar] [CrossRef]
- Anderson, M.J.; Whitcomb, P.J. RSM Simplified: Optimizing Processes Using Response Surface Methods for Design of Experiments; CRC Press: Boca Raton, FL, USA, 2005; ISBN 1563272970. [Google Scholar]
- Chigbu, P.E.; Ukaegbu, E.C.; Nwanya, J.C. On comparing the prediction variances of some central composite designs in spherical regions: A review. Statistica 2009, 69, 285–298. [Google Scholar]
- Khuri, A.I.; Mukhopadhyay, S. Response surface methodology. WIREs Comput. Stat. 2010, 2, 128–149. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.S.; Abdullah, N.; Fakru’l-razi, A. Optimizing the Process Parameters for Encapsulation of Linamarin into PLGA Nanoparticles Using Double Emulsion Solvent Evaporation Technique. Adv. Polym. Technol. 2013, 32, E486–E504. [Google Scholar] [CrossRef]
- Mainardes, R.M.; Evangelista, R.C. PLGA nanoparticles containing praziquantel: Effect of formulation variables on size distribution. Int. J. Pharm. 2005, 290, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Minitab 18. Analysis of variance table for Analyze Definitive Screening Design. Available online: https://support.minitab.com/en-us/minitab/18/help-and-how-to/modeling-statistics/doe/how-to/screening/analyze-screening-design/interpret-the-results/all-statistics-and-graphs/analysis-of-variance-table/ (accessed on 17 June 2021).
- El Hami, A.; Pougnet, P. Embedded Mechatronic Systems, Volume 2: Analysis of Failures, Modeling, Simulation and Optimization; Elsevier Science: Burlington, NJ, USA, 2015; ISBN 9781785480140. [Google Scholar]
- Minitab 18. What Is an Interaction? Available online: https://support.minitab.com/en-us/minitab/18/help-and-how-to/modeling-statistics/anova/supporting-topics/anova-models/what-is-an-interaction/ (accessed on 4 November 2022).
- Minitab 18. Interpret the Key Results for Analyze Definitive Screening Design. Available online: https://support.minitab.com/en-us/minitab/18/help-and-how-to/modeling-statistics/doe/how-to/screening/analyze-screening-design/interpret-the-results/key-results/ (accessed on 4 November 2022).
- Minitab 18. Model Summary Table for Analyze Response Surface Design. Available online: https://support.minitab.com/en-us/minitab/18/help-and-how-to/modeling-statistics/doe/how-to/response-surface/analyze-response-surface-design/interpret-the-results/all-statistics-and-graphs/model-summary-table/ (accessed on 4 November 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Composition | Quantity |
|---|---|---|
| Internal aqueous phase (W1) | Ovalbumin in phosphate buffer, pH 7.4 | 1600 µL |
| Organic phase (O) | PLGA in ethyl acetate | 4 mL |
| External aqueous phase (W2) | PVA in water | 12 g |
| Stabiliser solution | PVA (1% (w/w)) in water | 40 g |
| Parameter | Low | Medium | High |
|---|---|---|---|
| c (OVA) [%] | 2 | 3 | 4 |
| c (PLGA) [%] | 3 | 6 | 9 |
| Stirring speed W/O | 9500 rpm | - | 20,500 rpm |
| Stirring time W/O [s] | 30 | 60 | 90 |
| c (PVA) [%] | 2 | 3.5 | 5 |
| Stirring speed W/O/W | 8000 rpm | - | 13,500 rpm |
| Stirring time W/O/W [s] | 60 | 105 | 150 |
| Outcome | z-Average | PDI | LE | LC | |
|---|---|---|---|---|---|
| Parameter | |||||
| OVA conc. (%) | not sign. | not sign. | significant (p = 0.007) | significant (p = 0.000) | |
| PLGA conc. (%) | significant (p = 0.0257) | not sign. | significant (p = 0.000) | significant (p = 0.000) | |
| w/o stirring speed (rpm) | not sign. | not sign. | significant (p = 0.000) | significant (p = 0.002) | |
| w/o stirring time (s) | not sign. | not sign. | not sign. | not sign. | |
| PVA conc. (%) | significant (p = 0.00936) | not sign. | not sign. | not sign. | |
| w/o/w stirring speed (rpm) | significant (p = 0.00893) | significant (p = 0.004) | not sign. | not sign. | |
| w/o/w stirring time (s) | not sign. | not sign. | significant (p = 0.034) | not sign. | |
| p-Value of model | 0.010 | 0.041 | 0.000 | 0.000 | |
| R² (%) | 70.56 | 70.22 | 93.56 | 89.48 | |
| Response | Fit | Result (n = 3) ± SD |
|---|---|---|
| z-Average (d.nm) | 702 | 682.21 ± 11.94 |
| PDI | 0.1989 | 0.30 ± 0.03 |
| LE (%) | 19.88 | 24.25 ± 2.55 |
| LC (%) | 3.3299 | 4.07 ± 0.41 |
| Parameter | Low Axial | Low | Medium | High | High Axial |
|---|---|---|---|---|---|
| c (PLGA) [%] | 6.17 | 7 | 9 | 11 | 11.83 |
| c (PVA) [%] | 2.17 | 3 | 5 | 7 | 7.83 |
| Run | PLGA Conc. (%) | PVA Conc. (%) | z-Average (d.nm) | PDI | LE (%) | LC (%) |
|---|---|---|---|---|---|---|
| 1 | 5 | 9 | 713.97 | 0.232 | 20.32 | 3.49 |
| 2 | 5 | 11.83 | 1204.67 | 0.353 | 27.17 | 3.54 |
| 3 | 3 | 11 | 1807.00 | 0.293 | 23.08 | 3.25 |
| 4 | 5 | 9 | 650.20 | 0.286 | 22.48 | 3.84 |
| 5 | 5 | 9 | 707.30 | 0.295 | 18.58 | 3.20 |
| 6 | 7 | 7 | 446.63 | 0.192 | 19.64 | 4.30 |
| 7 | 7.83 | 9 | 503.43 | 0.180 | 22.71 | 3.88 |
| 8 | 2.17 | 9 | 1810.33 | 0.311 | 19.53 | 3.36 |
| 9 | 5 | 9 | 606.73 | 0.255 | 23.21 | 3.96 |
| 10 | 7 | 11 | 565.50 | 0.257 | 25.20 | 3.54 |
| 11 | 5 | 6.17 | 506.23 | 0.242 | 18.20 | 4.51 |
| 12 | 3 | 7 | 873.43 | 0.351 | 18.90 | 4.14 |
| 13 | 5 | 9 | 695.43 | 0.301 | 22.61 | 3.86 |
| Response | Fit | 95% CI | 95% PI |
|---|---|---|---|
| z-Average (d.nm) | 687.94 | 563.1; 812.7 | 496.0; 879.9 |
| PDI | 0.291 | 0.236; 0.347 | 0.201; 0.382 |
| LE (%) | 25.90 | 24.03; 27.77 | 22.24; 29.56 |
| LC | 3.57 | 3.22; 3.93 | 2.96; 4.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Struzek, A.-M.; Scherließ, R. Quality by Design as a Tool in the Optimisation of Nanoparticle Preparation—A Case Study of PLGA Nanoparticles. Pharmaceutics 2023, 15, 617. https://doi.org/10.3390/pharmaceutics15020617
Struzek A-M, Scherließ R. Quality by Design as a Tool in the Optimisation of Nanoparticle Preparation—A Case Study of PLGA Nanoparticles. Pharmaceutics. 2023; 15(2):617. https://doi.org/10.3390/pharmaceutics15020617
Chicago/Turabian StyleStruzek, Anna-Maria, and Regina Scherließ. 2023. "Quality by Design as a Tool in the Optimisation of Nanoparticle Preparation—A Case Study of PLGA Nanoparticles" Pharmaceutics 15, no. 2: 617. https://doi.org/10.3390/pharmaceutics15020617