
Targeting Interleukin 13 for the Treatment of Atopic Dermatitis

Abstract
:1. Introduction
2. Methods
3. The Role of Interleukin 13 in the Pathogenesis of Atopic Dermatitis
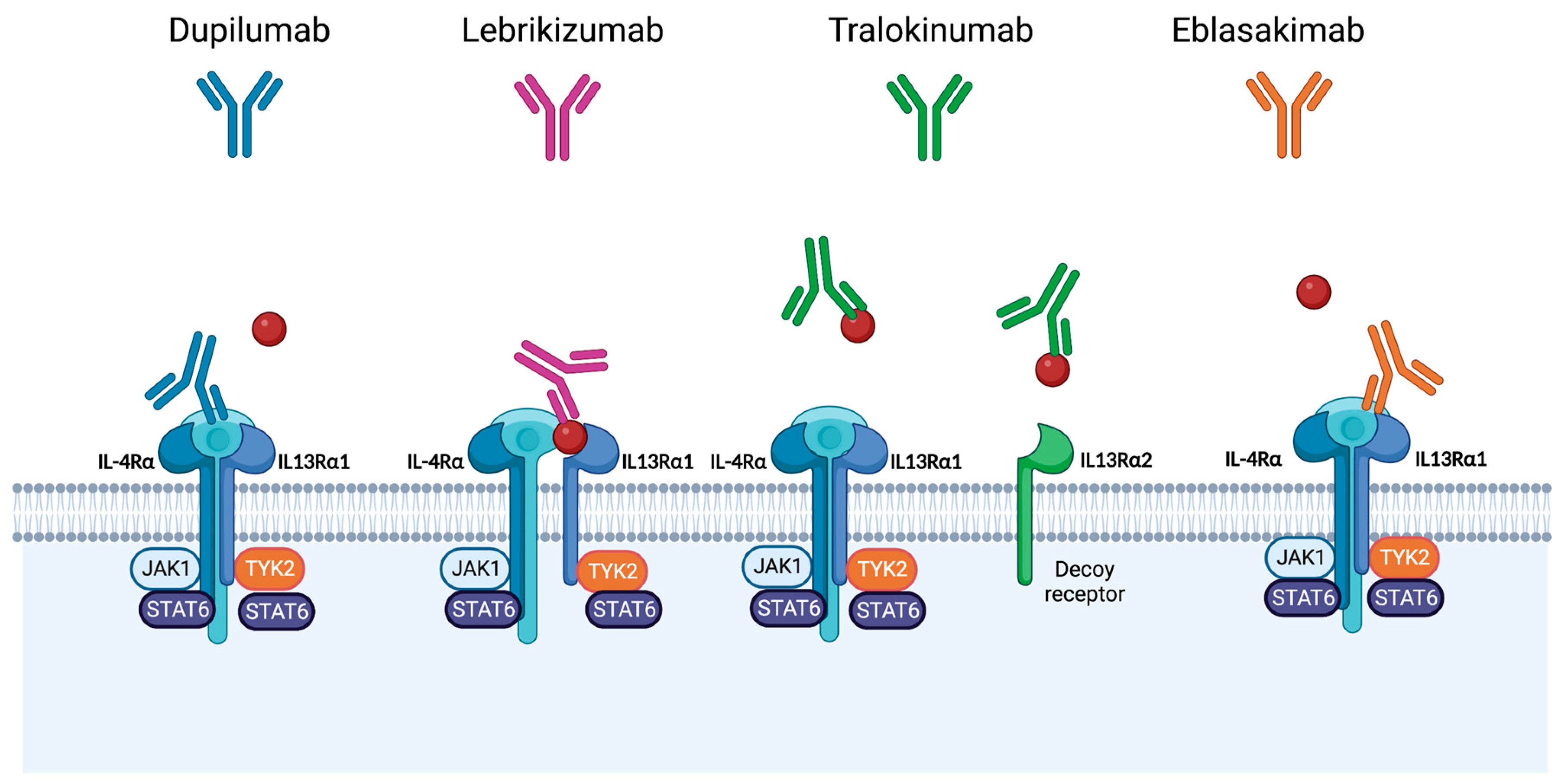
4. Therapies Targeting Interleukin-13 for Management of Atopic Dermatitis
5. Use of Tralokinumab to Manage Patients with Atopic Dermatitis
5.1. Overview of Tralokinumab
5.2. Clinical Efficacy of Tralokinumab in Patients with Atopic Dermatitis
5.3. Safety of Tralokinumab in Patients with Atopic Dermatitis
6. Use of Lebrikizumab to Manage Patients with Atopic Dermatitis
6.1. Overview of Lebrikizumab
6.2. Clinical Efficacy of Lebrikizumab in Patients with Atopic Dermatitis
6.3. Safety of Lebrikizumab in Patients with Atopic Dermatitis
6.4. Future IL-13 Inhibitors
7. Discussion
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deckers, I.A.; McLean, S.; Linssen, S.; Mommers, M.; van Schayck, C.P.; Sheikh, A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: A systematic review of epidemiological studies. PLoS ONE 2012, 7, e39803. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.J.; Johns, N.E.; Williams, H.C.; Bolliger, I.W.; Dellavalle, R.P.; Margolis, D.J.; Marks, R.; Naldi, L.; Weinstock, M.A.; Wulf, S.K.; et al. The global burden of skin disease in 2010: An analysis of the prevalence and impact of skin conditions. J. Investig. Dermatol. 2014, 134, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, U.; Vestergaard, C.; Deleuran, M. Emerging Treatment Options in Atopic Dermatitis: Systemic Therapies. Dermatology 2017, 233, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Fabbrocini, G.; Napolitano, M.; Megna, M.; Balato, N.; Patruno, C. Treatment of Atopic Dermatitis with Biologic Drugs. Dermatol. Ther. 2018, 8, 527–538. [Google Scholar] [CrossRef]
- Bieber, T. Atopic dermatitis. N. Engl. J. Med. 2008, 358, 1483–1494. [Google Scholar] [CrossRef]
- Rønnstad, A.T.M.; Halling-Overgaard, A.S.; Hamann, C.R.; Skov, L.; Egeberg, A.; Thyssen, J.P. Association of atopic dermatitis with depression, anxiety, and suicidal ideation in children and adults: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2018, 79, 448–456.e30. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Guttman-Yassky, E. What’s New in Atopic Dermatitis. Dermatol. Clin. 2019, 37, 205–213. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Kantor, R. The Role of Interleukins 4 and/or 13 in the Pathophysiology and Treatment of Atopic Dermatitis. Dermatol. Clin. 2017, 35, 327–334. [Google Scholar] [CrossRef]
- Yaghmaie, P.; Koudelka, C.W.; Simpson, E.L. Mental health comorbidity in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2013, 131, 428–433. [Google Scholar] [CrossRef]
- Drucker, A.M.; Wang, A.R.; Li, W.Q.; Sevetson, E.; Block, J.K.; Qureshi, A.A. The Burden of Atopic Dermatitis: Summary of a Report for the National Eczema Association. J. Investig. Dermatol. 2017, 137, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.; Torres, T. Dupilumab for the Treatment of Atopic Dermatitis. Actas Dermosifiliogr. (Engl. Ed.) 2018, 109, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Jones, S. Quality of life and childhood atopic dermatitis: The misery of living with childhood eczema. Int. J. Clin. Pract. 2006, 60, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Thaçi, D.; Pangan, A.L.; Hong, H.C.; Papp, K.A.; Reich, K.; Beck, L.A.; Mohamed, M.F.; Othman, A.A.; Anderson, J.K.; et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 2020, 145, 877–884. [Google Scholar] [CrossRef]
- Ring, J.; Alomar, A.; Bieber, T.; Deleuran, M.; Fink-Wagner, A.; Gelmetti, C.; Gieler, U.; Lipozencic, J.; Luger, T.; Oranje, A.P.; et al. Guidelines for treatment of atopic eczema (atopic dermatitis) part I. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Roekevisch, E.; Spuls, P.I.; Kuester, D.; Limpens, J.; Schmitt, J. Efficacy and safety of systemic treatments for moderate-to-severe atopic dermatitis: A systematic review. J. Allergy Clin. Immunol. 2014, 133, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Baghoomian, W.; Na, C.; Simpson, E.L. New and Emerging Biologics for Atopic Dermatitis. Am. J. Clin. Dermatol. 2020, 21, 457–465. [Google Scholar] [CrossRef]
- Silverberg, J.I. Atopic dermatitis treatment: Current state of the art and emerging therapies. Allergy Asthma Proc. 2017, 38, 243–249. [Google Scholar] [CrossRef]
- Moyle, M.; Cevikbas, F.; Harden, J.L.; Guttman-Yassky, E. Understanding the immune landscape in atopic dermatitis: The era of biologics and emerging therapeutic approaches. Exp. Dermatol. 2019, 28, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Hanifin, J.M. Adult eczema prevalence and associations with asthma and other health and demographic factors: A US population-based study. J. Allergy Clin. Immunol. 2013, 132, 1132–1138. [Google Scholar] [CrossRef]
- Bieber, T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy 2020, 75, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Nadeau, K.C. IL-4Rα Inhibitor for Atopic Disease. Cell 2017, 170, 222. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.P.; et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; de Bruin-Weller, M.; Gooderham, M.; Cather, J.C.; Weisman, J.; Pariser, D.; Simpson, E.L.; Papp, K.A.; Hong, H.C.; Rubel, D.; et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): A 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet 2017, 389, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Thaçi, D.; Simpson, E.L.; Deleuran, M.; Kataoka, Y.; Chen, Z.; Gadkari, A.; Eckert, L.; Akinlade, B.; Graham, N.M.H.; Pirozzi, G.; et al. Efficacy and safety of dupilumab monotherapy in adults with moderate-to-severe atopic dermatitis: A pooled analysis of two phase 3 randomized trials (LIBERTY AD SOLO 1 and LIBERTY AD SOLO 2). J. Dermatol. Sci. 2019, 94, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Kamata, M.; Mizukawa, I.; Watanabe, A.; Agematsu, A.; Nagata, M.; Fukaya, S.; Hayashi, K.; Fukuyasu, A.; Tanaka, T.; et al. Real-world effectiveness and safety of dupilumab for the treatment of atopic dermatitis in Japanese patients: A single-centre retrospective study. Br. J. Dermatol. 2019, 181, 1083–1085. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Blauvelt, A.; Eichenfield, L.F.; Paller, A.S.; Armstrong, A.W.; Drew, J.; Gopalan, R.; Simpson, E.L. Efficacy and Safety of Lebrikizumab, a High-Affinity Interleukin 13 Inhibitor, in Adults With Moderate to Severe Atopic Dermatitis: A Phase 2b Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 411–420. [Google Scholar] [CrossRef]
- Soria, A.; Du-Thanh, A.; Seneschal, J.; Jachiet, M.; Staumont-Sallé, D.; Barbarot, S. Development or Exacerbation of Head and Neck Dermatitis in Patients Treated for Atopic Dermatitis With Dupilumab. JAMA Dermatol. 2019, 155, 1312–1315. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef]
- Bao, K.; Reinhardt, R.L. The differential expression of IL-4 and IL-13 and its impact on type-2 immunity. Cytokine 2015, 75, 25–37. [Google Scholar] [CrossRef]
- Oetjen, L.K.; Mack, M.R.; Feng, J.; Whelan, T.M.; Niu, H.; Guo, C.J.; Chen, S.; Trier, A.M.; Xu, A.Z.; Tripathi, S.V.; et al. Sensory Neurons Co-opt Classical Immune Signaling Pathways to Mediate Chronic Itch. Cell 2017, 171, 217–228.e13. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, M.; Marasca, C.; Fabbrocini, G.; Patruno, C. Adult atopic dermatitis: New and emerging therapies. Expert Rev. Clin. Pharm. 2018, 11, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Misery, L.; Huet, F.; Gouin, O.; Ständer, S.; Deleuran, M. Current pharmaceutical developments in atopic dermatitis. Curr. Opin. Pharm. 2019, 46, 7–13. [Google Scholar] [CrossRef]
- Fujii, M. Current Understanding of Pathophysiological Mechanisms of Atopic Dermatitis: Interactions among Skin Barrier Dysfunction, Immune Abnormalities and Pruritus. Biol. Pharm. Bull. 2020, 43, 12–19. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Ulzii, D.; Vu, Y.H.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology 2019, 158, 281–286. [Google Scholar] [CrossRef]
- Zaniboni, M.C.; Samorano, L.P.; Orfali, R.L.; Aoki, V. Skin barrier in atopic dermatitis: Beyond filaggrin. Bras. Dermatol. 2016, 91, 472–478. [Google Scholar] [CrossRef]
- Bonness, S.; Bieber, T. Molecular basis of atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2007, 7, 382–386. [Google Scholar] [CrossRef]
- Loh, T.Y.; Hsiao, J.L.; Shi, V.Y. Therapeutic Potential of Lebrikizumab in the Treatment of Atopic Dermatitis. J. Asthma Allergy 2020, 13, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; Maurelli, M.; Gisondi, P.; Peris, K.; Yosipovitch, G.; Girolomoni, G. Pruritus as a Distinctive Feature of Type 2 Inflammation. Vaccines 2021, 9, 303. [Google Scholar] [CrossRef]
- Dubin, C.; Del Duca, E.; Guttman-Yassky, E. The IL-4, IL-13 and IL-31 pathways in atopic dermatitis. Expert Rev. Clin. Immunol. 2021, 17, 835–852. [Google Scholar] [CrossRef]
- Gooderham, M.J.; Hong, H.C.; Eshtiaghi, P.; Papp, K.A. Dupilumab: A review of its use in the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2018, 78, S28–S36. [Google Scholar] [CrossRef]
- Omori-Miyake, M.; Yamashita, M.; Tsunemi, Y.; Kawashima, M.; Yagi, J. In vitro assessment of IL-4- or IL-13-mediated changes in the structural components of keratinocytes in mice and humans. J. Investig. Dermatol. 2014, 134, 1342–1350. [Google Scholar] [CrossRef]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; DeBenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2009, 124, R7–R12. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fairchild, H.R.; Kim, B.E.; Bin, L.; Boguniewicz, M.; Redzic, J.S.; Hansen, K.C.; Leung, D.Y. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J. Investig. Dermatol. 2008, 128, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.; Goleva, E.; Bronova, I.; Dyjack, N.; Rios, C.; Jung, J.; Taylor, P.; Jeong, M.; Hall, C.F.; Richers, B.N.; et al. Lipid abnormalities in atopic skin are driven by type 2 cytokines. JCI Insight 2018, 3, e98006. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Woodfolk, J.A. Skin barrier defects in atopic dermatitis. Curr. Allergy Asthma Rep. 2014, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Nomura, I.; Goleva, E.; Howell, M.D.; Hamid, Q.A.; Ong, P.Y.; Hall, C.F.; Darst, M.A.; Gao, B.; Boguniewicz, M.; Travers, J.B.; et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol. 2003, 171, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Investig. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Suárez-Fariñas, M.; Shemer, A.; Fuentes-Duculan, J.; Chiricozzi, A.; Cardinale, I.; Zaba, L.C.; Kikuchi, T.; Ramon, M.; Bergman, R.; et al. Atopic dermatitis keratinocytes exhibit normal T(H)17 cytokine responses. J. Allergy Clin. Immunol. 2010, 125, 744–746. [Google Scholar] [CrossRef]
- Kim, J.; Kim, B.E.; Leung, D.Y.M. Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma Proc. 2019, 40, 84–92. [Google Scholar] [CrossRef]
- Woo, T.E.; Sibley, C.D. The emerging utility of the cutaneous microbiome in the treatment of acne and atopic dermatitis. J. Am. Acad. Dermatol. 2020, 82, 222–228. [Google Scholar] [CrossRef]
- Tubau, C.; Puig, L. Therapeutic targeting of the IL-13 pathway in skin inflammation. Expert Rev. Clin. Immunol. 2021, 17, 15–25. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.M.; Heller, N.M. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 2015, 75, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Hussein, Y.M.; Ahmad, A.S.; Ibrahem, M.M.; Elsherbeny, H.M.; Shalaby, S.M.; El-Shal, A.S.; Sabbah, N.A. Interleukin 13 receptors as biochemical markers in atopic patients. J. Investig. Allergol. Clin. Immunol. 2011, 21, 101–107. [Google Scholar]
- Kwak, E.J.; Hong, J.Y.; Kim, M.N.; Kim, S.Y.; Kim, S.H.; Park, C.O.; Kim, K.W.; Lee, C.G.; Elias, J.A.; Jee, H.M.; et al. Chitinase 3-like 1 drives allergic skin inflammation via Th2 immunity and M2 macrophage activation. Clin. Exp. Allergy 2019, 49, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, T.; Sugiura, H.; Sugiura, Y.; Uehara, M. Relative importance of IL-4 and IL-13 in lesional skin of atopic dermatitis. Arch. Dermatol. Res. 2004, 295, 459–464. [Google Scholar] [CrossRef] [PubMed]
- La Grutta, S.; Richiusa, P.; Pizzolanti, G.; Mattina, A.; Pajno, G.B.; Citarrella, R.; Passalacqua, G.; Giordano, C. CD4(+)IL-13(+) cells in peripheral blood well correlates with the severity of atopic dermatitis in children. Allergy 2005, 60, 391–395. [Google Scholar] [CrossRef]
- Hijnen, D.; Knol, E.F.; Gent, Y.Y.; Giovannone, B.; Beijn, S.J.; Kupper, T.S.; Bruijnzeel-Koomen, C.A.; Clark, R.A. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J. Investig. Dermatol. 2013, 133, 973–979. [Google Scholar] [CrossRef]
- Bangert, C.; Rindler, K.; Krausgruber, T.; Alkon, N.; Thaler, F.M.; Kurz, H.; Ayub, T.; Demirtas, D.; Fortelny, N.; Vorstandlechner, V.; et al. Persistence of mature dendritic cells, T(H)2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Rα blockade. Sci. Immunol. 2021, 6, eabe2749. [Google Scholar] [CrossRef]
- Mashiko, S.; Mehta, H.; Bissonnette, R.; Sarfati, M. Increased frequencies of basophils, type 2 innate lymphoid cells and Th2 cells in skin of patients with atopic dermatitis but not psoriasis. J. Dermatol. Sci. 2017, 88, 167–174. [Google Scholar] [CrossRef]
- Szegedi, K.; Lutter, R.; Res, P.C.; Bos, J.D.; Luiten, R.M.; Kezic, S.; Middelkamp-Hup, M.A. Cytokine profiles in interstitial fluid from chronic atopic dermatitis skin. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2136–2144. [Google Scholar] [CrossRef]
- Choy, D.F.; Hsu, D.K.; Seshasayee, D.; Fung, M.A.; Modrusan, Z.; Martin, F.; Liu, F.T.; Arron, J.R. Comparative transcriptomic analyses of atopic dermatitis and psoriasis reveal shared neutrophilic inflammation. J. Allergy Clin. Immunol. 2012, 130, 1335–1343.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungar, B.; Garcet, S.; Gonzalez, J.; Dhingra, N.; Correa da Rosa, J.; Shemer, A.; Krueger, J.G.; Suarez-Farinas, M.; Guttman-Yassky, E. An Integrated Model of Atopic Dermatitis Biomarkers Highlights the Systemic Nature of the Disease. J. Investig. Dermatol. 2017, 137, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Popovic, B.; Breed, J.; Rees, D.G.; Gardener, M.J.; Vinall, L.M.; Kemp, B.; Spooner, J.; Keen, J.; Minter, R.; Uddin, F.; et al. Structural Characterisation Reveals Mechanism of IL-13-Neutralising Monoclonal Antibody Tralokinumab as Inhibition of Binding to IL-13Rα1 and IL-13Rα2. J. Mol. Biol. 2017, 429, 208–219. [Google Scholar] [CrossRef]
- May, R.D.; Fung, M. Strategies targeting the IL-4/IL-13 axes in disease. Cytokine 2015, 75, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Tollenaere, M.A.X.; Litman, T.; Moebus, L.; Rodriguez, E.; Stölzl, D.; Drerup, K.; Werfel, T.; Schmitt, J.; Norsgaard, H.; Weidinger, S. Skin Barrier and Inflammation Genes Associated with Atopic Dermatitis are Regulated by Interleukin-13 and Modulated by Tralokinumab In vitro. Acta Dermatol. Venereol. 2021, 101, adv00447. [Google Scholar] [CrossRef]
- Beck, L.A.; Bieber, T.; Weidinger, S.; Tauber, M.; Saeki, H.; Irvine, A.D.; Eichenfield, L.F.; Werfel, T.; Arlert, P.; Jiang, L.; et al. Tralokinumab treatment improves the skin microbiota by increasing the microbial diversity in adults with moderate-to-severe atopic dermatitis: Analysis of microbial diversity in ECZTRA 1, a randomized controlled trial. J. Am. Acad. Dermatol. 2022. [Google Scholar] [CrossRef]
- Oh, C.K.; Faggioni, R.; Jin, F.; Roskos, L.K.; Wang, B.; Birrell, C.; Wilson, R.; Molfino, N.A. An open-label, single-dose bioavailability study of the pharmacokinetics of CAT-354 after subcutaneous and intravenous administration in healthy males. Br. J. Clin. Pharm. 2010, 69, 645–655. [Google Scholar] [CrossRef]
- Wollenberg, A.; Blauvelt, A.; Guttman-Yassky, E.; Worm, M.; Lynde, C.; Lacour, J.P.; Spelman, L.; Katoh, N.; Saeki, H.; Poulin, Y.; et al. Tralokinumab for moderate-to-severe atopic dermatitis: Results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br. J. Dermatol. 2021, 184, 437–449. [Google Scholar] [CrossRef]
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141. [Google Scholar] [CrossRef]
- Simpson, E.L.; Flohr, C.; Eichenfield, L.F.; Bieber, T.; Sofen, H.; Taïeb, A.; Owen, R.; Putnam, W.; Castro, M.; DeBusk, K.; et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: A randomized, placebo-controlled phase II trial (TREBLE). J. Am. Acad. Dermatol. 2018, 78, 863–871.e11. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Toth, D.; Bieber, T.; Alexis, A.F.; Elewski, B.E.; Pink, A.E.; Hijnen, D.; Jensen, T.N.; Bang, B.; Olsen, C.K.; et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: Results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br. J. Dermatol. 2021, 184, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Adam, D.N.; Zirwas, M.; Kalia, S.; Gutermuth, J.; Pinter, A.; Pink, A.E.; Chiricozzi, A.; Barbarot, S.; Mark, T.; et al. Tralokinumab Plus Topical Corticosteroids as Needed Provides Progressive and Sustained Efficacy in Adults with Moderate-to-Severe Atopic Dermatitis Over a 32-Week Period: An ECZTRA 3 Post Hoc Analysis. Am. J. Clin. Dermatol. 2022, 23, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A. Atopic dermatitis: Therapy. In Proceedings of the Annual Meeting of the American Academy of Dermatology (AAD 2022), Boston, MA, USA, 28 March 2022. [Google Scholar]
- Wollenberg, A.; Beck, L.A.; de Bruin Weller, M.; Simpson, E.L.; Imafuku, S.; Boguniewicz, M.; Zachariae, R.; Olsen, C.K.; Thyssen, J.P. Conjunctivitis in adult patients with moderate-to-severe atopic dermatitis: Results from five tralokinumab clinical trials. Br. J. Dermatol. 2022, 186, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 2016, 15, 35–50. [Google Scholar] [CrossRef]
- Zhu, R.; Zheng, Y.; Dirks, N.L.; Vadhavkar, S.; Jin, J.Y.; Peng, K.; Holweg, C.T.J.; Olsson, J.; Matthews, J.G.; Putnam, W.S. Model-based clinical pharmacology profiling and exposure-response relationships of the efficacy and biomarker of lebrikizumab in patients with moderate-to-severe asthma. Pulm Pharm. Ther. 2017, 46, 88–98. [Google Scholar] [CrossRef]
- Simpson, E.L. Efficacy And Safety of Lebrikizumab in Moderate-to-Severe Atopic Dermatitis: Results from Two Phase 3, Randomized, Double-Blinded, Placebo-Controlled Trials. In Proceedings of the American Academy of Dermatology Annual Meeting, Boston, MA, USA, 26 March 2022. [Google Scholar]
- Blauvelt, B.; Thyssen, J.P.; Guttman-Yassky, E.; Bieber, T.; Serra-Baldrich, E.; Simpson, E.; Rosmarin, D.; Elmaraghy, H.; Meskimen, E.; Natalie, C.R.; et al. Efficacy and Safety of Lebrikizumab in Moderate-to-Severe Atopic Dermatitis: 52-Week Results of Two Randomized, Double-Blinded, Placebo-Controlled Phase 3 Trials (ADvocate1 and ADvocate2). In Proceedings of the EADV Congress, Milan, Italy, 7–10 September 2022. [Google Scholar]
- Safety and Efficacy of Lebrikizumab (LY3650150) in Combination With Topical Corticosteroid in Moderate-to-Severe Atopic Dermatitis. (ADhere). Available online: https://clinicaltrials.gov/ct2/show/study/NCT04250337 (accessed on 9 November 2022).
- Noonan, M.; Korenblat, P.; Mosesova, S.; Scheerens, H.; Arron, J.R.; Zheng, Y.; Putnam, W.S.; Parsey, M.V.; Bohen, S.P.; Matthews, J.G. Dose-ranging study of lebrikizumab in asthmatic patients not receiving inhaled steroids. J. Allergy. Clin. Immunol. 2013, 132, 567–574.e12. [Google Scholar] [CrossRef]
- Hanania, N.A.; Noonan, M.; Corren, J.; Korenblat, P.; Zheng, Y.; Fischer, S.K.; Cheu, M.; Putnam, W.S.; Murray, E.; Scheerens, H.; et al. Lebrikizumab in moderate-to-severe asthma: Pooled data from two randomised placebo-controlled studies. Thorax 2015, 70, 748–756. [Google Scholar] [CrossRef]
- Korenblat, P.; Kerwin, E.; Leshchenko, I.; Yen, K.; Holweg, C.T.J.; Anzures-Cabrera, J.; Martin, C.; Putnam, W.S.; Governale, L.; Olsson, J.; et al. Efficacy and safety of lebrikizumab in adult patients with mild-to-moderate asthma not receiving inhaled corticosteroids. Respir. Med. 2018, 134, 143–149. [Google Scholar] [CrossRef]
- Scheerens, H.; Arron, J.R.; Zheng, Y.; Putnam, W.S.; Erickson, R.W.; Choy, D.F.; Harris, J.M.; Lee, J.; Jarjour, N.N.; Matthews, J.G. The effects of lebrikizumab in patients with mild asthma following whole lung allergen challenge. Clin. Exp. Allergy 2014, 44, 38–46. [Google Scholar] [CrossRef]
- Hanania, N.A.; Korenblat, P.; Chapman, K.R.; Bateman, E.D.; Kopecky, P.; Paggiaro, P.; Yokoyama, A.; Olsson, J.; Gray, S.; Holweg, C.T.; et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): Replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir. Med. 2016, 4, 781–796. [Google Scholar] [CrossRef]
- Blauvelt, A. Eblasakimab, a human anti-IL-13 receptor monoclonal antibody, in adult patients with moderate-to-severe atopic dermatitis: A randomized, double-blinded, placebo-controlled, proof-of-concept study. In Proceedings of the American Academy of Dermatology Annual Meeting, Boston, MA, USA, 26 March 2022. [Google Scholar]
- Blauvelt, A.; Teixeira, H.D.; Simpson, E.L.; Costanzo, A.; De Bruin-Weller, M.; Barbarot, S.; Prajapati, V.H.; Lio, P.; Hu, X.; Wu, T.; et al. Efficacy and Safety of Upadacitinib vs Dupilumab in Adults With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2021, 157, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Akinlade, B.; Guttman-Yassky, E.; de Bruin-Weller, M.; Simpson, E.L.; Blauvelt, A.; Cork, M.J.; Prens, E.; Asbell, P.; Akpek, E.; Corren, J.; et al. Conjunctivitis in dupilumab clinical trials. Br. J. Dermatol. 2019, 181, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Torres, T. Conjunctivitis in patients with atopic dermatitis treated with dupilumab. Drugs Context 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Bakker, D.S.; Ariens, L.F.M.; van Luijk, C.; van der Schaft, J.; Thijs, J.L.; Schuttelaar, M.L.A.; van Wijk, F.; Knol, E.F.; Balak, D.M.W.; van Dijk, M.R.; et al. Goblet cell scarcity and conjunctival inflammation during treatment with dupilumab in patients with atopic dermatitis. Br. J. Dermatol. 2019, 180, 1248–1249. [Google Scholar] [CrossRef]
- Agnihotri, G.; Shi, K.; Lio, P.A. A Clinician’s Guide to the Recognition and Management of Dupilumab-Associated Conjunctivitis. Drugs R D 2019, 19, 311–318. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Clinical Trial | Eligibility (Sample Size) | Treatment Groups (Duration) | EASI | EASI 50 | EASI 75 | IGA 0 or 1 | SCORAD 50 | NRS | DLQI |
|---|---|---|---|---|---|---|---|---|---|
| Tralokinumab | |||||||||
| NCT02347176 [69] | 18–75 years SCORAD ≥ 25 EASI ≥ 12 BSA ≥ 10% IGA ≥ 3 (N = 204) | 45 mg tralokinumab Q2W + TCS (12 weeks) | −10.78 (p = 0.143) | 54.3% | N/A | 11.6% (p = 0.974) | 26.9% | −1.80 | N/A |
| 150 mg tralokinumab Q2W + TCS (12 weeks) | −15.14 (p = 0.027) | 67.3% | N/A | 19.5% (p = 0.281) | 44.2% | −1.59 | N/A | ||
| 300 mg tralokinumab Q2W + TCS (12 weeks) | −15.72 (p = 0.011) | 73.4% (p = 0.03) | 42.5% (p = 0.003) | 26.7% (p = 0.061) | 44.1% | −2.17 | N/A | ||
| Placebo + TCS (12 weeks) | −10.78 | 51.9% | 15.5% | 11.8% | 19.5% | −1.03 | N/A | ||
| Lebrikizumab | |||||||||
| TREBLE (NCT02340234) [70] | 18–75 years EASI ≥ 14 BSA ≥ 10% IGA ≥ 3 Pruritis VAS ≥ 3 (N = 212) | Lebrikizumab 125 mg single dose | N/A | 69.2% | 38.5% | 21.2% | N/A | N/A | N/A |
| Lebrikizumab 250 mg single dose | N/A | 69.5% | 49.1% | 28.3% | N/A | N/A | N/A | ||
| Lebrikizumab 125 mg Q4W + TCS (12 weeks) | N/A | 82.4% (p = 0.026) | 54.9% (p = 0.036) | 33.3% | N/A | N/A | N/A | ||
| Placebo + TCS (12 weeks) | N/A | 62.3% | 34.0% | 18.9% | N/A | N/A | N/A | ||
| NCT03443024 [26] | 18–75 years EASI ≥ 16 BSA ≥ 10% IGA ≥ 3 (N = 280) | Lebrikizumab 250 mg loading dose + 125 mg Q4W (16 weeks) | −62.34% (p = 0.0165) | 66.4% (p = 0.0554) | 43.3% (p = 0.0610) | 26.6% (p = 0.1917) | N/A | −35.94% (p = 0.0047) | N/A |
| Lebrikizumab 500 mg loading dose + 250 mg lebrikizumab Q4W (16 weeks) | −69.21% (p = 0.0022) | 77.0% (p = 0.0037) | 56.1% (p = 0.0021) | 33.7% (p = 0.0392) | N/A | −49.6% (p = 0.0002) | N/A | ||
| Lebrikizumab 500 mg loading dose + 250 mg lebrikizumab Q2W (16 weeks) | −72.09% (p = 0.0005) | 81.0% (p = 0.0008) | 60.6% (p = 0.0005) | 44.6% (p = 0.0023) | N/A | −60.63% (p < 0.0001) | N/A | ||
| Placebo (16 weeks) | −41.12% | 45.8% | 24.3% | 15.3% | N/A | 4.26 | N/A | ||
| Clinical Trial | Eligibility (Sample Size) | Treatment Groups (Duration) | EASI | EASI 90 | EASI 75 | IGA 0 or 1 | SCORAD Change | NRS Change | DLQI Change |
|---|---|---|---|---|---|---|---|---|---|
| Tralokinumab | |||||||||
| ECZTRA 1 (NCT03131648) | ≥18 years BSA ≥ 10% (N = 802) | Tralokinumab 300 mg Q2W (16 weeks) | −15.5 (p < 0.001) | 14.5% (p < 0.001) | 25.0% (p < 0.001) | 15.8% (p = 0.002) | −25.2 (p < 0.001) | −2.6 (p < 0.001) | −7.1 (p = 0.002) |
| Placebo (16 weeks) | −9.0 | 4.1% | 12.7% | 7.1% | −14.7 | −1.7 | −5.0 | ||
| Tralokinumab 300 mg Q2W (52 weeks) | N/A | N/A | 59.6% (p = 0.056) | 51.3% (p = 0.68) | N/A | N/A | N/A | ||
| Tralokinumab 300 mg Q4W (52 weeks) | N/A | N/A | 49.1% (p = 0.27) | 38.95% (p = 0.50) | N/A | N/A | N/A | ||
| Placebo (52 weeks) | N/A | N/A | 33.3% | 47.4% | N/A | N/A | N/A | ||
| ECZTRA 2 (NCT03160885) | ≥18 years BSA ≥ 10% (N = 794) | Tralokinumab 300 mg Q2W (16 weeks) | −16.9 (p < 0.001) | 18.3% (p < 0.001) | 33.2% (p < 0.001) | 22.2% (p < 0.001) | −28.1 (p < 0.001) | −2.9 (p < 0.001) | −8.8 (p < 0.001) |
| Placebo (16 weeks) | −7.0 | 5.5% | 11.4% | 10.9% | −14.0 | −1.6 | −4.9 | ||
| Tralokinumab 300 mg Q2W (52 weeks) | N/A | N/A | 55.8% (p < 0.001) | 59.3% (p = 0.004) | N/A | N/A | N/A | ||
| Tralokinumab 300 mg Q4W (52 weeks) | N/A | N/A | 51.4% (p = 0.001) | 44.9% (p = 0.084) | N/A | N/A | N/A | ||
| Placebo (52 weeks) | N/A | N/A | 21.4% | 25.0% | N/A | N/A | N/A | ||
| ECZTRA 3 (NCT03363854) | ≥18 years BSA ≥ 10% (N = 380) | Tralokinumab 600 mg loading dose + 300 mg Q2W + TCS (16 weeks) | −21.0 (p < 0.001) | 32.9% (p = 0.022) | 56.0% (p < 0.001) | 38.9% (p = 0.015) | −37.7 (p < 0.001) | −4.1 (p < 0.001) | −11.7 (p < 0.001) |
| Placebo + TCS (16 weeks) | −15.6 | 21.4% | 35.7% | 26.2% | −26.8 | −2.9 | −8.8 | ||
| Tralokinumab 600 mg loading dose + 300 mg Q2W + TCS (32 weeks) | N/A | N/A | 92.5% | 89.6% | N/A | N/A | N/A | ||
| Tralokinumab 600 mg loading dose + 300 mg Q4W + TCS (32 weeks) | N/A | N/A | 90.8% | 77.6% | N/A | N/A | N/A | ||
| Lebrikizumab | |||||||||
| ADVOCATE 1 (NCT04146363) | ≥12 years EASI ≥ 16 BSA ≥ 10% IGA ≥ 3 (N = 424) | Lebrikizumab 500 mg loading dose + 250 mg Q2W (16 weeks) | −64.75% (p < 0.001) | 38.2% (p < 0.001) | 59.3% (p < 0.001) | 43.0% (p < 0.001) | −47.26 (p < 0.001) | −45.75% (p < 0.001) | −8.78 (p < 0.001) |
| Placebo (16 weeks) | −26.16% | 9.1% | 16.4% | 12.8% | −16.79 | −15.24% | −2.94 | ||
| Lebrikizumab Q2W (52 weeks) | N/A | N/A | 79.2% (p < 0.05) | 75.8% (p < 0.001) | N/A | N/A | N/A | ||
| Lebrikizumab Q4W (52 weeks) | N/A | N/A | 77.4% (p < 0.05) | 74.2% (p < 0.001) | N/A | N/A | N/A | ||
| Lebrikizumab Withdrawal (52 weeks) | N/A | N/A | 61.3% | 46.5% | N/A | N/A | N/A | ||
| ADVOCATE 2 (NCT04178967) | ≥12 years EASI ≥ 16 BSA ≥ 10% IGA ≥ 3 (N = 445) | Lebrikizumab 500 mg loading dose + 250 mg Q2W (16 weeks) | −60.61% (p < 0.001) | 30.2% (p < 0.001) | 50.8% (p < 0.001) | 33.1% (p < 0.001) | −43.85% (p < 0.001) | −35.7% (p < 0.001) | −6.99% (p < 0.001) |
| Placebo (16 weeks) | −28.22% | 9.4% | 18.2% | 10.9% | −13.87% | −8.91% | −2.47% | ||
| Lebrikizumab Q2W (52 weeks) | N/A | N/A | 79.2% (p < 0.05) | 64.6% (p < 0.001) | N/A | N/A | N/A | ||
| Lebrikizumab Q4W (52 weeks) | N/A | N/A | 84.7% (p < 0.05) | 80.6% (p < 0.001) | N/A | N/A | N/A | ||
| Lebrikizumab Withdrawal (52 weeks) | N/A | N/A | 72.0% | 49.8% (p < 0.001) | N/A | N/A | N/A | ||
| ADhere (NCT04250337) | ≥12 years EASI ≥ 16 BSA ≥ 10% IGA ≥ 3 (N = 228) | Lebrikizumab 500 mg loading dose + 250 mg Q2W + TCS (16 weeks) | −76.76% (p < 0.001) | 41.2% (p = 0.008) | 69.5% (p < 0.001) | 41.2% (p = 0.011) | −55.04% (p < 0.001) | −50.68% (p = 0.017263) | −9.79 (p = 0.001031) |
| Placebo + TCS (16 weeks) | −53.12% | 21.7% | 42.2% | 22.1% | −37.35% | −35.47% | −6.46 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lytvyn, Y.; Gooderham, M. Targeting Interleukin 13 for the Treatment of Atopic Dermatitis. Pharmaceutics 2023, 15, 568. https://doi.org/10.3390/pharmaceutics15020568
Lytvyn Y, Gooderham M. Targeting Interleukin 13 for the Treatment of Atopic Dermatitis. Pharmaceutics. 2023; 15(2):568. https://doi.org/10.3390/pharmaceutics15020568
Chicago/Turabian StyleLytvyn, Yuliya, and Melinda Gooderham. 2023. "Targeting Interleukin 13 for the Treatment of Atopic Dermatitis" Pharmaceutics 15, no. 2: 568. https://doi.org/10.3390/pharmaceutics15020568