
Response of the Endogenous Antioxidant Defense System Induced in RAW 264.7 Macrophages upon Exposure to Dextran-Coated Iron Oxide Nanoparticles

 , ,
, , 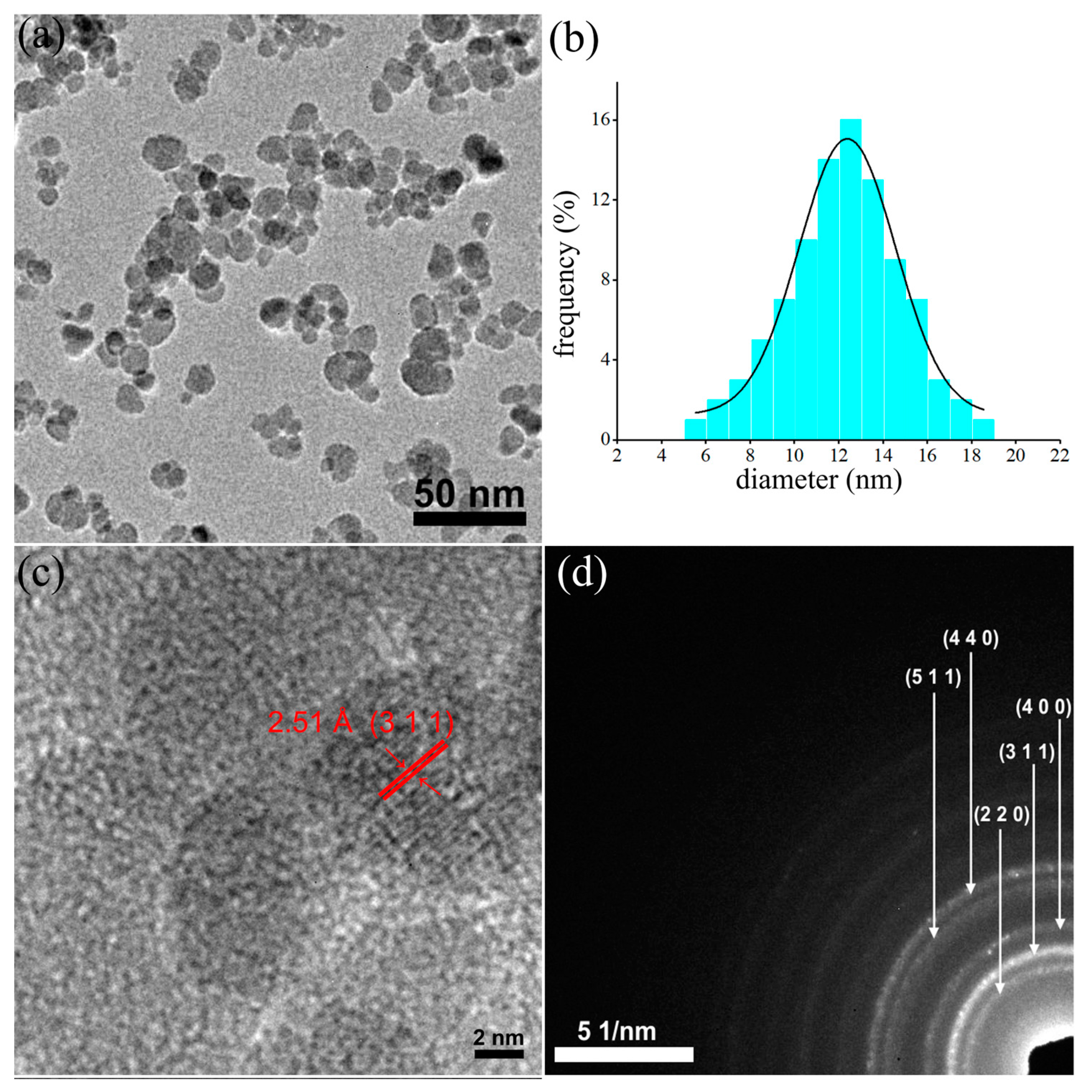{kind=link}
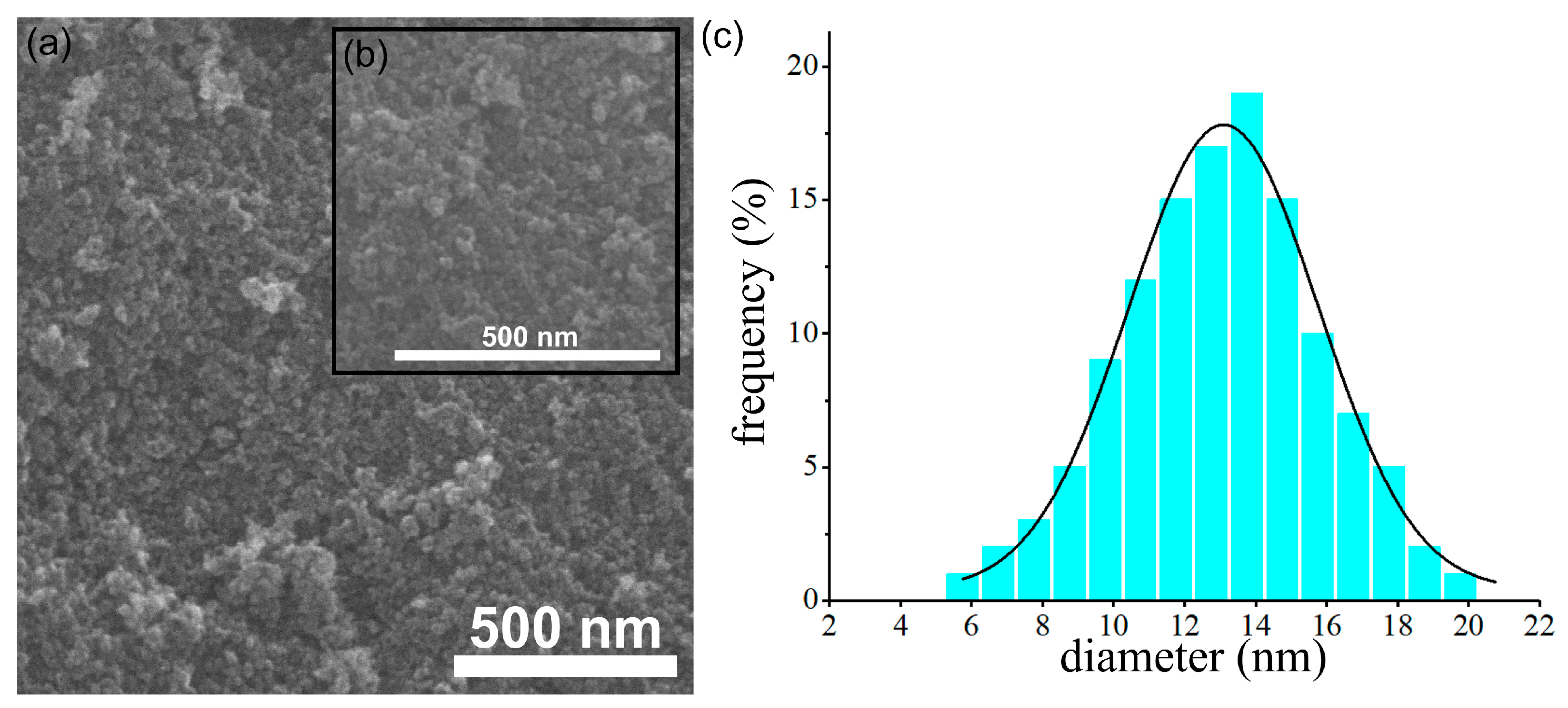{kind=link}
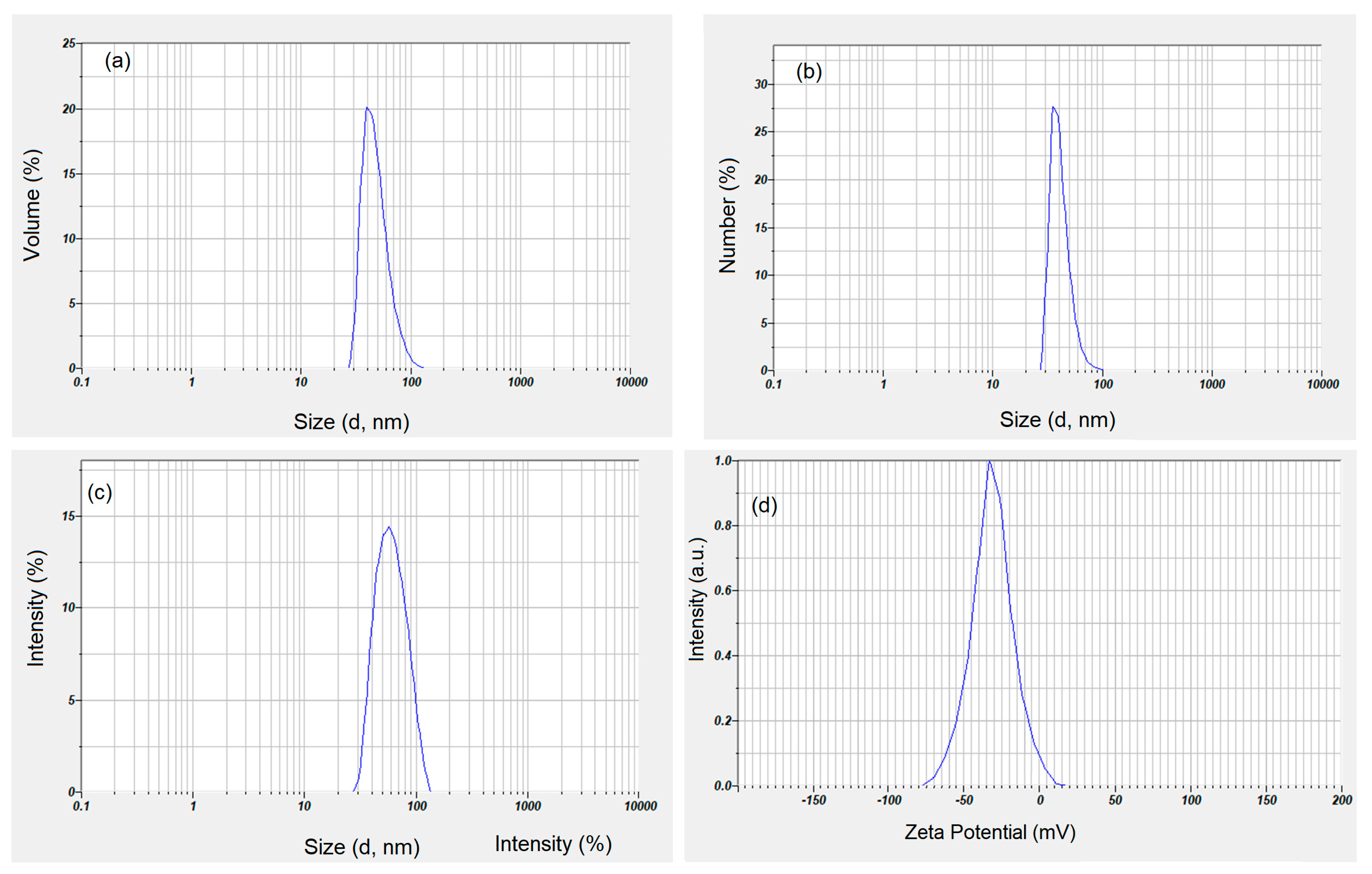{kind=link}
{kind=link}
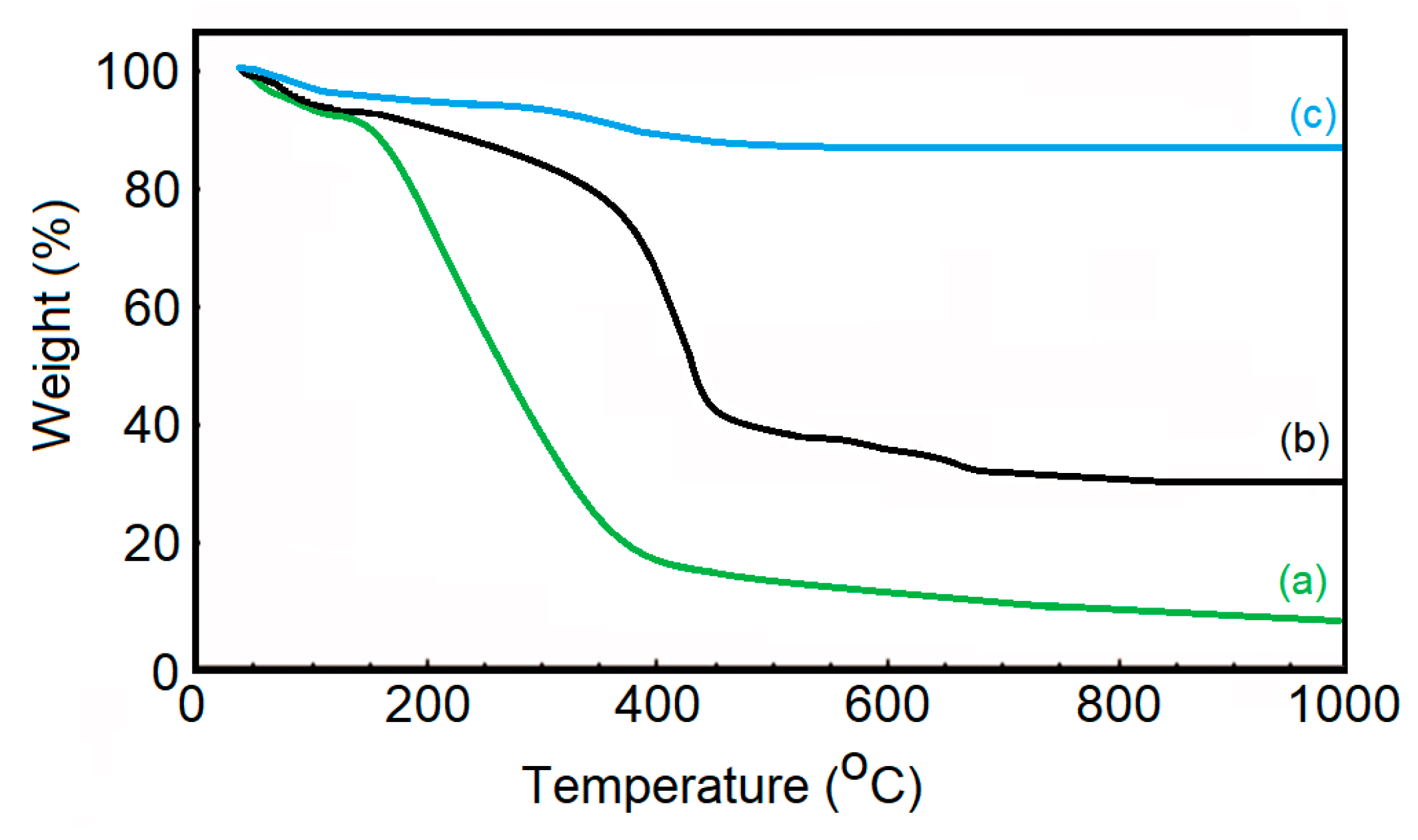{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Physicochemical Characterization
2.3. Biological Assays
2.3.1. Cell Culture and Treatment
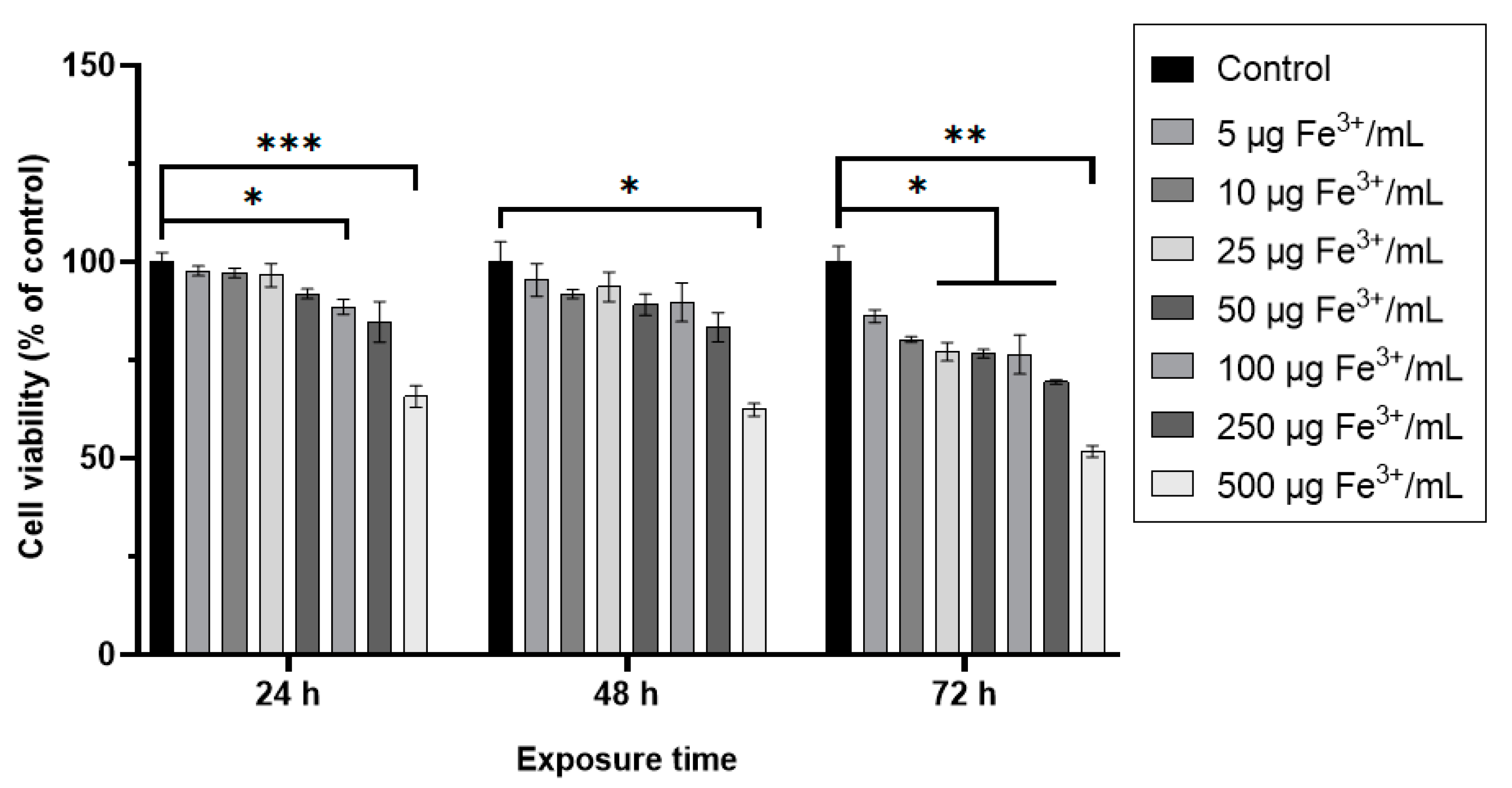
2.3.2. MTT Assay
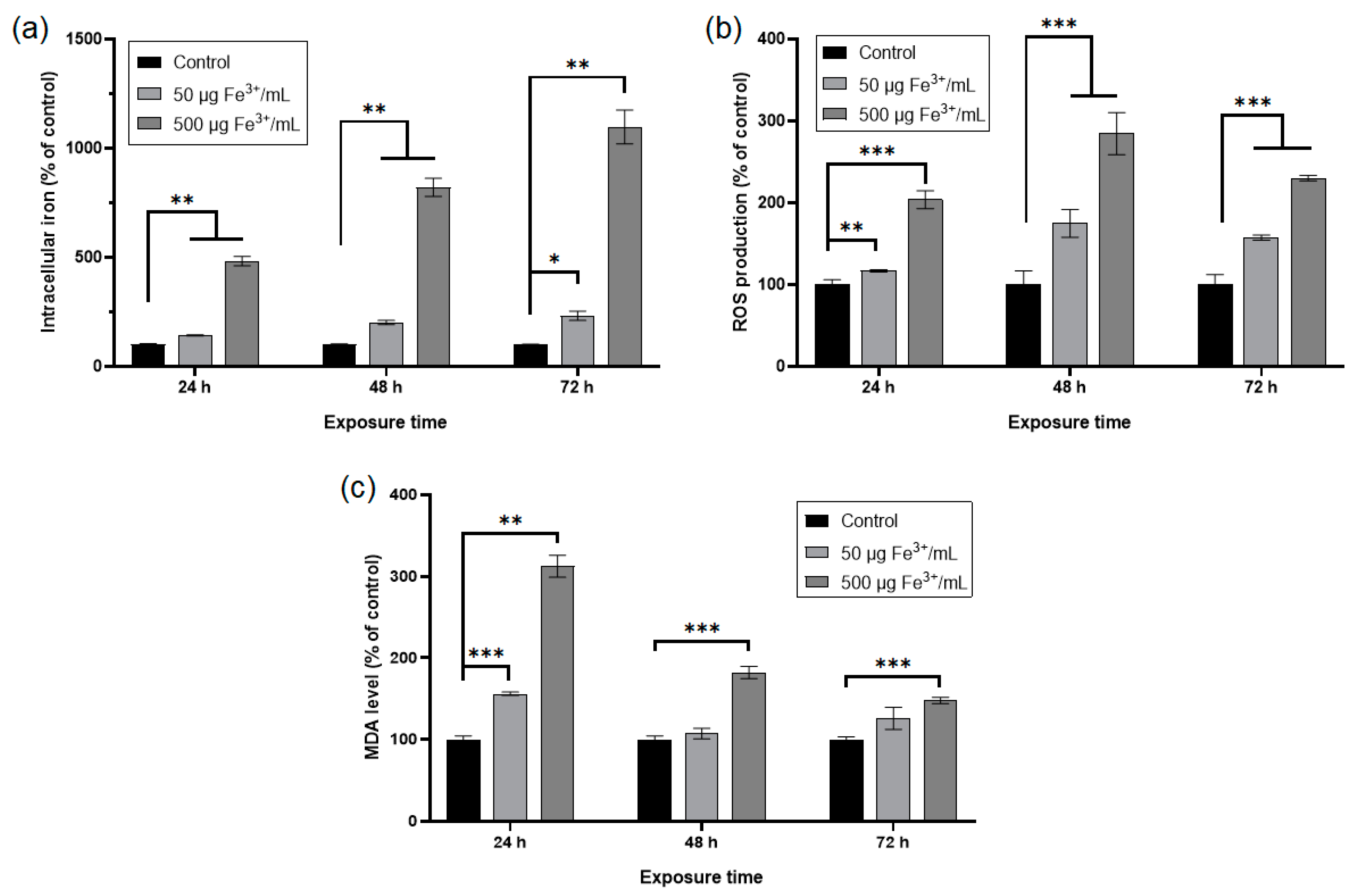
2.3.3. Quantification of Intracellular Iron Content
2.3.4. Measurement of ROS Production
2.3.5. Preparation of Cell Lysate
2.3.6. Detection of Lipid Peroxidation Products
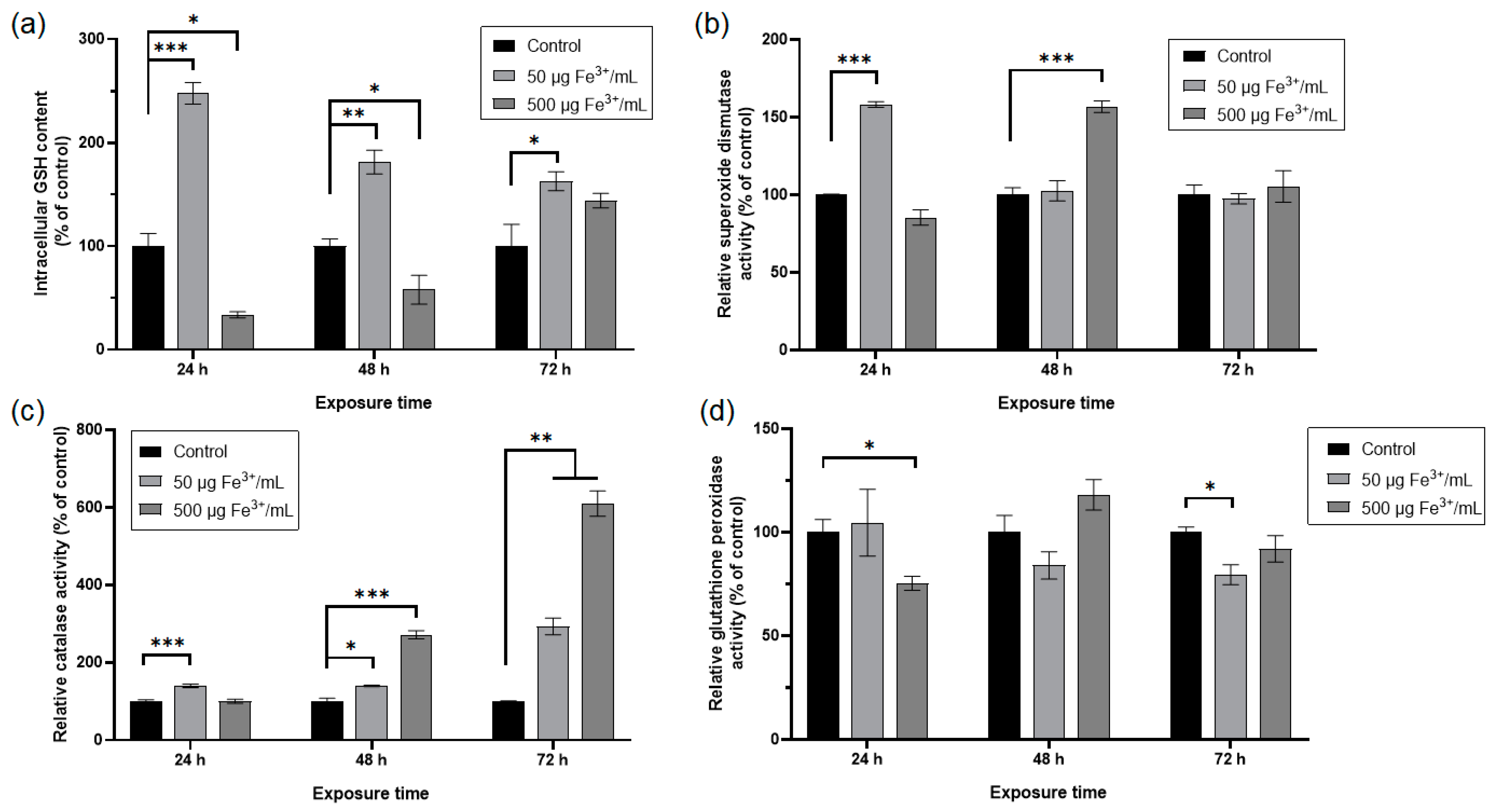
2.3.7. Quantification of Reduced Glutathione (GSH) Concentration
2.3.8. Measurement of Antioxidant Enzymatic Activity
2.4. Statistical Analysis
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Edge, D.; Shortt, C.M.; Gobbo, O.L.; Teughels, S.; Prina-Mello, A.; Volkov, Y.; MacEneaney, P.; Radomski, M.W.; Markos, F. Pharmacokinetics and bio-distribution of novel super paramagnetic iron oxide nanoparticles (SPIONs) in the anaesthetized pig. Clin. Exp. Pharmacol. Physiol. 2016, 43, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kievit, F.M.; Veiseh, O.; Bhattarai, N.; Fang, C.; Gunn, J.W.; Lee, D.; Ellenbogen, R.G.; Olson, J.M.; Zhang, M. PEI-PEG-Chitosan Copolymer Coated Iron Oxide Nanoparticles for Safe Gene Delivery: Synthesis, complexation, and transfection. Adv. Funct. Mater. 2009, 19, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Arami, H.; Stephen, Z.; Veiseh, O.; Zhang, M. Chitosan-Coated Iron Oxide Nanoparticles for Molecular Imaging and Drug Delivery. Adv. Polym. Sci. 2011, 243, 163–184. [Google Scholar]
- Berman, S.M.C.; Walczak, P.; Bulte, J.W.M. Tracking stem cells using magnetic nanoparticles. Nanomed. Nanobiotechnol. 2011, 3, 343–355. [Google Scholar] [CrossRef]
- Lu, M.; Cohen, M.H.; Rieves, D.; Pazdur, R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am. J. Hematol. 2010, 85, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kicheeva, A.G.; Sushko, E.S.; Bondarenko, L.S.; Kydralieva, K.A.; Pankratov, D.A.; Tropskaya, N.S.; Dzeranov, A.A.; Dzhardimalieva, G.I.; Zarrelli, M.; Kudryasheva, N.S. Functionalized Magnetite Nanoparticles: Characterization, Bioeffects, and Role of Reactive Oxygen Species in Unicellular and Enzymatic Systems. Int. J. Mol. Sci. 2023, 24, 1133. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Elst, L.V.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef]
- Hao, R.; Xing, R.; Xu, Z.; Hou, Y.; Gao, S.; Sun, S. Synthesis, functionalization, and biomedical applications of multifunctional magnetic nanoparticles. Adv. Mater. 2010, 22, 2729–2742. [Google Scholar] [CrossRef]
- Xie, J.; Liu, G.; Eden, H.S.; Ai, H.; Chen, X. Surface-engineered magnetic nanoparticle platforms for cancer imaging and therapy. Acc. Chem. Res. 2011, 44, 883–892. [Google Scholar] [CrossRef]
- Bondarenko, L.S.; Kovel, E.S.; Kydralieva, K.A.; Dzhardimalieva, G.I.; Illés, E.; Tombácz, E.; Kicheeva, A.G.; Kudryasheva, N.S. Effects of Modified Magnetite Nanoparticles on Bacterial Cells and Enzyme Reactions. Nanomaterials 2020, 10, 1499. [Google Scholar] [CrossRef]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of Engineered Nanoparticles for Drug Delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef]
- Gamucci, O.; Bertero, A.; Gagliardi, M.; Bardi, G. Biomedical Nanoparticles: Overview of Their Surface Immune-Compatibility. Coatings 2014, 4, 139–159. [Google Scholar] [CrossRef]
- Ghosh, S.; Ghosh, I.; Chakrabarti, M.; Mukherjee, A. Genotoxicity and biocompatibility of superparamagnetic iron oxide nanoparticles: Influence of surface modification on biodistribution, retention, DNA damage and oxidative stress. Food Chem. Toxicol. 2020, 136, 110989. [Google Scholar] [CrossRef] [PubMed]
- Kievit, F.M.; Zhang, M.Q. Surface Engineering of Iron Oxide Nanoparticles for Targeted Cancer Therapy. Acc. Chem. Res. 2011, 44, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Tassa, C.; Shaw, S.Y.; Weissleder, R. Dextran-coated Iron Oxide Nanoparticles: A Versatile Platform for Targeted Molecular Imaging, Molecular Diagnostics, and Therapy. Acc. Chem. Res. 2011, 44, 842–852. [Google Scholar] [CrossRef]
- Cole, A.J.; David, A.E.; Wang, J.X.; Galban, C.J.; Hill, H.L.; Yang, V.C. Polyethylene Glycol Modified, Cross-Linked Starch-Coated Iron Oxide Nanoparticles for Enhanced Magnetic Tumor Targeting. Biomaterials 2011, 32, 2183–2193. [Google Scholar] [CrossRef]
- Auerbach, M.; Pappadakis, J.A.; Bahrain, H.; Auerbach, S.A.; Ballard, H.; Dahl, N.V. Safety and Efficacy of Rapidly Administered (One Hour) One Gram of Low Molecular Weight Iron Dextran (INFeD) for the Treatment of Iron Deficient Anemia. Am. J. Hematol. 2011, 86, 860–862. [Google Scholar] [CrossRef] [PubMed]
- Ros, P.R.; Freeny, P.C.; Harms, S.E.; Seltzer, S.E.; Davis, P.L.; Chan, T.W.; Stillman, A.E.; Muroff, L.R.; Runge, V.M.; Nissenbaum, M.A. Hepatic MR Imaging With Ferumoxides: A Multicenter Clinical Trial of the Safety and Efficacy in the Detection of Focal Hepatic Lesions. Radiology 1995, 196, 481–488. [Google Scholar] [CrossRef]
- Peng, M.; Li, H.; Luo, Z.; Kong, J.; Wan, J.; Zheng, Z.; Zhang, Q.; Niu, H.; Vermorken, A.; Van de Ven, W.; et al. Dextran-coated superparamagnetic nanoparticles as potential cancer drug carriers in vivo. Nanoscale 2015, 7, 11155–11162. [Google Scholar] [CrossRef]
- Lewis, S.L. Medical Surgical Nursing, 8th ed.; Mosby: St. Louis, MI, USA, 2010; ISBN 978-0323079150. [Google Scholar]
- Turrina, C.; Milani, D.; Klassen, A.; Rojas-González, D.M.; Cookman, J.; Opel, M.; Sartori, B.; Mela, P.; Berensmeier, S.; Schwaminger, S.P. Carboxymethyl-Dextran-Coated Superparamagnetic Iron Oxide Nanoparticles for Drug Delivery: Influence of the Coating Thickness on the Particle Properties. Int. J. Mol. Sci. 2022, 23, 14743. [Google Scholar] [CrossRef]
- Weissleder, R.; Bogdanov, A.; Neuwelt, E.A.; Papisov, M. Long-circulating iron oxides for MR imaging. Adv. Drug Deliv. Rev. 1995, 16, 321–334. [Google Scholar] [CrossRef]
- Huang, Y.; Hsu, J.C.; Koo, H.; Cormode, D.P. Repurposing ferumoxytol: Diagnostic and therapeutic applications of an FDA-approved nanoparticle. Theranostics 2022, 12, 796–816. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.H.; van de Ven, A.L.; Godin, B.; Blanco, E.; Serda, R.E.; Grattoni, A.; Ziemys, A.; Bouamrani, A.; Hu, T.; Ranganathan, S.I.; et al. Enabling individualized therapy through nanotechnology. Pharmacol Res. 2010, 62, 57–89. [Google Scholar] [CrossRef] [PubMed]
- Iv, M.; Telischak, N.; Feng, D.; Holdsworth, S.J.; Yeom, K.W.; Daldrup-Link, H.E. Clinical applications of iron oxide nanoparticles for magnetic resonance imaging of brain tumors. Nanomedicine 2015, 10, 993–1018. [Google Scholar] [CrossRef] [PubMed]
- Gerb, J.; Strauss, W.; Derman, R.; Short, V.; Mendelson, B.; Bahrain, H. Ferumoxytol for the treatment of iron deficiency and iron-deficiency anemia of pregnancy. Ther. Adv. Hematol. 2021, 12, 20406207211018042. [Google Scholar] [CrossRef]
- Blumenstein, I.; Shanbhag, S.; Langguth, P.; Kalra, P.A.; Zoller, H.; Lim, W. Newer formulations of intravenous iron: A review of their chemistry and key safety aspects-Hypersensitivity, hypophosphatemia, and cardiovascular safety. Expert Opin Drug Saf. 2021, 20, 757–769. [Google Scholar] [CrossRef]
- Feng, Q.; Liu, Y.; Huang, J.; Chen, K.; Huang, J.; Xiao, K. Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings. Sci. Rep. 2018, 8, 2082. [Google Scholar] [CrossRef]
- Patil, R.M.; Thorat, N.D.; Shete, P.B.; Bedge, P.A.; Gavde, S.; Joshi, M.G.; Bohara, R.A. Comprehensive cytotoxicity studies of superparamagnetic iron oxide nanoparticles. Biochem. Biophys. Rep. 2018, 13, 63–72. [Google Scholar] [CrossRef]
- Vakili-Ghartavol, R.; Momtazi-Borojeni, A.A.; Vakili-Ghartavol, Z.; Aiyelabegan, H.T.; Jaafari, M.R.; Rezayat, S.M.; Arbabi Bidgoli, S. Toxicity assessment of superparamagnetic iron oxide nanoparticles in different tissues. Artif. Cells. Nanomed. Biotechnol. 2020, 48, 443–451. [Google Scholar] [CrossRef]
- Saptarshi, S.R.; Duschl, A.; Lopata, A.L. Interaction of nanoparticles with proteins: Relation to bio-reactivity of the nanoparticle. J. Nanobiotechnol. 2013, 11, 26. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Millan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Simberg, D.; Park, J.-H.; Karmali, P.P.; Zhang, W.-M.; Merkulov, S.; McCrae, K.; Bhatia, S.N.; Sailor, M.; Ruoslahti, E. Differential proteomics analysis of the surface heterogeneity of dextran iron oxide nanoparticles and the implications for their in vivo clearance. Biomaterials 2009, 30, 3926–3933. [Google Scholar] [CrossRef]
- Rojas, J.M.; Sanz-Ortega, L.; Mulens-Arias, V.; Gutiérrez, L.; Pérez-Yagüe, S.; Barber, D.F. Superparamagnetic iron oxide nanoparticle uptake alters M2 macrophage phenotype, iron metabolism, migration and invasion. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1127–1138. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Barber, D.F. The Intrinsic Biological Identities of Iron Oxide Nanoparticles and Their Coatings: Unexplored Territory for Combinatorial Therapies. Nanomaterials 2020, 10, 837. [Google Scholar] [CrossRef]
- Virág, L.; Jaén, R.I.; Regdon, Z.; Boscá, L.; Prieto, P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol. 2019, 26, 101261. [Google Scholar] [CrossRef] [PubMed]
- Predoi, D. A Study on iron oxide nanoparticles coated with dextrin obtained by coprecipitation. Dig. J. Nanomater. Biostruct. 2007, 2, 169–173. [Google Scholar]
- Ciobanu, C.S.; Iconaru, S.L.; Gyorgy, E.; Radu, M.; Costache, M.; Dinischiotu, A.; Le Coustumer, P.; Lafdi, K.; Predoi, D. Biomedical properties and preparation of iron oxide-dextran nanostructures by MAPLE technique. Chem. Cent. J. 2012, 6, 17. [Google Scholar] [CrossRef]
- Butoi, B.; Ciobanu, C.S.; Iconaru, S.L.; Negrilă, C.C.; Badea, M.A.; Balas, M.; Dinischiotu, A.; Predoi, G.; Bita, B.; Groza, A.; et al. Iron-Oxide-Nanoparticles-Doped Polyaniline Composite Thin Films. Polymers 2022, 14, 1821. [Google Scholar] [CrossRef] [PubMed]
- Prodan, A.M.; Iconaru, S.L.; Chifiriuc, C.M.; Bleotu, C.; Ciobanu, C.S.; Motelica-Heino, M.; Sizaret, S.; Predoi, D. Magnetic Properties and Biological Activity Evaluation of Iron Oxide Nanoparticles. J. Nanomater. 2013, 2013, 893970. [Google Scholar] [CrossRef]
- Iconaru, S.L.; Prodan, A.M.; Motelica-Heino, M.; Sizaret, S.; Predoi, D. Synthesis and characterization of polysaccharide-maghemite composite nanoparticles and their antibacterial properties. Nanoscale Res. Lett. 2012, 7, 576–584. [Google Scholar] [CrossRef]
- Radu, M.; Dinu, D.; Sima, C.; Burlacu, R.; Hermenean, A.; Ardelean, A.; Dinischiotu, A. Magnetite nanoparticles induced adaptive mechanisms counteract cell death in human pulmonary fibroblasts. Toxicol. In Vitro 2015, 29, 1492–1502. [Google Scholar] [CrossRef]
- Dinischiotu, A.; Stanca, L.; Gradinaru, D.; Petrache, S.N.; Radu, M.; Serban, A.I. Lipid peroxidation due to in vitro and in vivo exposure of biological samples to nanoparticles. Oxid. Stress Nanotechnol. 2013, 1028, 155–164. [Google Scholar]
- Paoletti, F.; Aldinucci, D.; Mocali, A.; Caparrini, A. A sensitive spectrophotometric method for the determination of superoxide dismutase activity in tissue extracts. Anal. Biochem. 1986, 154, 538–541. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. In Methods of Enzymatic Analysis; Bergmayer, H.U., Ed.; FRG: Weinheim, Germany, 1984; pp. 673–684. [Google Scholar]
- Beutler, E. Red cell metabolism. In A Manual of Biochemical Methods; Grune, S., Ed.; Elsevier: New York, NY, USA, 1971; pp. 71–73. [Google Scholar]
- Balas, M.; Ciobanu, C.S.; Burtea, C.; Stan, M.S.; Bezirtzoglou, E.; Predoi, D.; Dinischiotu, A. Synthesis, Characterization, and Toxicity Evaluation of Dextran-Coated Iron Oxide Nanoparticles. Metals 2017, 7, 63. [Google Scholar] [CrossRef]
- Predoi, S.-A.; Iconaru, S.L.; Predoi, D. In Vitro and In Vivo Biological Assays of Dextran Coated Iron Oxide Aqueous Magnetic Fluids. Pharmaceutics 2023, 15, 177. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, M.P.; Orlov, A.V.; Znoyko, S.L.; Bragina, V.A.; Gorshkov, B.G.; Ksenevich, T.I.; Cherkasov, V.R.; Nikitin, P.I. Multiplex biosensing with highly sensitive magnetic nanoparticle quantification method. J. Magn. Magn. Mater. 2018, 459, 260–264. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Jiwpanich, S.; Chacko, R.; Bickerton, S.; Thayumanavan, S. Surface-Functionalizable Polymer Nano-gels with Facile Hydrophobic Guest Encapsulation Capabilities. J. Am. Chem. Soc. 2010, 132, 8246–8247. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Chacko, R.T.; Jiwpanich, S.; Bickerton, S.; Babu, R.P.; Thayumanavan, S. Self-Cross-Linked Polymer Nanogels: A Versatile Nanoscopic Drug Delivery Platform. J. Am. Chem. Soc. 2010, 132, 17227–17235. [Google Scholar] [CrossRef] [PubMed]
- Aktan, B.; Chambre, L.; Sanyal, R.; Sanyal, A. ‘‘Clickable’’ Nanogels via Thermally Driven Self-Assembly of Polymers: Facile Access to Targeted Imaging Platforms using Thiol– Maleimide Conjugation. Biomacromolecules 2017, 18, 490–497. [Google Scholar] [CrossRef]
- Suganthi, K.S.; Rajan, K.S. Temperature induced changes in ZnO-water nanofluid: Zeta potential, size distribution and viscosity profiles. Int. J. Heat Mass Transf. 2012, 55, 7969–7980. [Google Scholar] [CrossRef]
- Nobbmann, U.L. Polydispersity–What Does It Mean for DLS and Chromatography. 2014. Available online: http://www.materials-talks.com/blog/2014/10/23/polydispersity-what-does-it-mean-for-dls-and-chromatography/ (accessed on 14 March 2018).
- Bera, B. Nanoporous silicon prepared by vapor phase strain etch and sacrificial technique. In Proceedings of the International Conference on Microelectronic Circuit and System (Micro), Kolkata, India, 11–12 July 2015; pp. 42–45. [Google Scholar]
- Clarke, S. Development of Hierarchical Magnetic Nanocomposite Materials for Biomedical Applications. Ph.D. Thesis, Dublin City University, Northside, Dublin, 2013. [Google Scholar]
- Worldwide, M.I. Dynamic Light Scattering, Common Terms Defined; Inform White Paper; Malvern Instruments Limited: Malvern, UK, 2011; pp. 1–6. [Google Scholar]
- Saraswathy, A.; Nazeer, S.S.; Nimi, N.; Arumugam, S.; Shenoy, S.J.; Jayasree, R.S. Synthesis and characterization of dextran stabilized superparamagnetic iron oxide nano-particles for in vivo MR imaging of liver fibrosis. Carbohydr. Polym. 2014, 101, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Carp, O.; Patron, L.; Culita, D.; Budrugeac, P.; Feder, M.; Diamandescu, L. Thermal analysis of two types of dextran-coated magnetite. J. Therm. Anal. Calorim. 2010, 101, 181–187. [Google Scholar] [CrossRef]
- Can, H.K.; Kavlak, S.; ParviziKhosroshahi, S.; Güune, A. Preparation, characterization and dynamical mechanical properties ofdextran-coated iron oxide nanoparticles (DIONPs). Artif. Cells Nanomed. Biotechnol. 2018, 46, 421–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easo, S.L.; Mohanan, P.V. Dextran stabilized iron oxide nanoparticles:synthesis, characterization and in vitro studies. Carbohydr Polym. 2013, 92, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Bautista, M.C.; Miguel-Bomati, O.; Morales, M.P.; Serna, C.J. Veintemillas-Verdaguer, S. Surface characterization of dextran-coated iron oxide nanoparticles prepared bylaser pyrolysis and coprecipitation. J. Magn. Magn. Mater. 2005, 293, 20–27. [Google Scholar] [CrossRef]
- Shaterabadi, Z.; Nabiyouni, G.; Soleymani, M. High impact of in situ dextran coating on biocompatibility, stability and magnetic properties of iron oxide nanoparticles. Mater. Sci. Eng. C 2017, 75, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Merly, L.; Smith, S.L. Murine RAW264.7 cell line as an immune target: Are we missing something? Immunopharm Immunotox 2017, 39, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Reichell, D.; Tripathi, M.; Perez, J.M. Biological Effects of Nanoparticles on Macrophage Polarization in the Tumor Microenvironment. Nanotheranostics 2019, 3, 66–88. [Google Scholar] [CrossRef]
- Saha, K.; Rahimi, M.; Yazdani, M.; Kim, S.T.; Moyano, D.F.; Hou, S.; Das, R.; Mout, R.; Rezaee, F.; Mahmoudi, M.; et al. Regulation of Macrophage Recognition through the Interplay of Nanoparticle Surface Functionality and Protein Corona. ACS Nano 2016, 10, 4421–4430. [Google Scholar] [CrossRef]
- Petrat, F.; Paluch, S.; Dogruöz, E.; Dörfler, P.; Kirch, M.; Korth, H.-G.; Sustmann, R.; de Groot, H. Reduction of Fe (III) Ions Complexed to Physiological Ligands by Lipoyl Dehydrogenase and Other Flavoenzymes In Vitro. J. Biol. Chem. 2003, 278, 46403–46413. [Google Scholar] [CrossRef]
- Masoud, R.; Bizouarn, T.; Trepout, S.; Wien, F.; Baciou, L.; Marco, S.; Houée Levin, C. Titanium Dioxide Nanoparticles Increase Superoxide Anion Production by Acting on NADPH Oxidase. PLoS ONE 2015, 10, e0144829. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current Question. Expert. Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef]
- Möller, M.N.; Cuevasanta, E.; Orrico, F.; Lopez, A.C.; Thomson, L.; Denicola, A. Diffusion and Transport of Reactive Species Across Cell Membrane. In Bioactive Lipids in Health and Disease; Trostchansky, A., Rubbo, H., Eds.; (AEMB, 1127); Springer: Berlin/Heidelberg, Germany, 2019; pp. 3–19. [Google Scholar]
- Lynch, R.E.; Fridovich, I. Permeation of the erythrocyte stroma by superoxide radical. J. Biol. Chem. 1978, 253, 4697–4699. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell. Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Gottfredsen, R.H.; Larsen, U.G.; Enghild, J.J.; Petersen, S.V. Hydrogen peroxide induce modifications of human extracellular superoxide dismutase that results in enzyme inhibition. Redox Biol. 2013, 1, 24–31. [Google Scholar] [CrossRef]
- Tokarz, P.; Płoszaj, T.; Regdon, Z.; Virag, L.; Robaszkiewicz, A. PARP1-LSD1 functional interplay controls transcription of SOD2 that protects human pro-inflammatory macrophages from death under an oxidative condition. Free Radic. Biol. Med. 2019, 131, 218–224. [Google Scholar] [CrossRef]
- Ganini, D.; Santos, J.H.; Bonini, M.G.; Mason, R.P. Switch of Mitochondrial Superoxide Dismutase into a Prooxidant Peroxidase in Manganese-Deficient Cells and Mice. Cell. Chem. Biol. 2018, 25, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; Clair, D.K. Manganese Superoxide Dismutase: Guardian of the Powerhouse. Int. J. Molec. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, T.A.F.; Hernández Navarro, B.C.; Pérez, J.A.M. Endogenous Antioxidants: A Review of their Role in Oxidative Stress. In A Master Regulator of Oxidative Stress-The Transcription Factor Nrf2; Morales-Gonzalez, J.A., Morales-Gonzalez, A., Madrigal-Santillan, E.O., Eds.; IntechOpen: London, UK, 2016. [Google Scholar]
- Röhrdanz, E.; Schmuck, G.; Ohler, S.; Kahl, R. The influence of oxidative stress on catalase and Mn SOD gene transcription in astrocytes. Brain. Res. 2001, 900, 128–136. [Google Scholar] [CrossRef]
- Makino, N.; Mochizuki, Y.; Bannai, S.; Sugita, Y. Kinetic studies on the removal of extracellular hydrogen peroxide by cultured fibroblasts. J. Biol. Chem. 1994, 269, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015; p. 753. [Google Scholar]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awathi, S. Antioxidant role of glutathione-S-transferase: 4-Hydroxynonenal, a key molecule in stress-mediated signals. Tox. Appl. Pharm. 2015, 289, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Easo, S.L.; Mohanan, P.V. Toxicological evaluation of dextran stabilized iron oxide nanoparticles in human peripheral blood lymphocytes. Biointerfases 2016, 16, 058601. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balas, M.; Iconaru, S.L.; Dinischiotu, A.; Buton, N.; Predoi, D. Response of the Endogenous Antioxidant Defense System Induced in RAW 264.7 Macrophages upon Exposure to Dextran-Coated Iron Oxide Nanoparticles. Pharmaceutics 2023, 15, 552. https://doi.org/10.3390/pharmaceutics15020552
Balas M, Iconaru SL, Dinischiotu A, Buton N, Predoi D. Response of the Endogenous Antioxidant Defense System Induced in RAW 264.7 Macrophages upon Exposure to Dextran-Coated Iron Oxide Nanoparticles. Pharmaceutics. 2023; 15(2):552. https://doi.org/10.3390/pharmaceutics15020552
Chicago/Turabian StyleBalas, Mihaela, Simona Liliana Iconaru, Anca Dinischiotu, Nicolas Buton, and Daniela Predoi. 2023. "Response of the Endogenous Antioxidant Defense System Induced in RAW 264.7 Macrophages upon Exposure to Dextran-Coated Iron Oxide Nanoparticles" Pharmaceutics 15, no. 2: 552. https://doi.org/10.3390/pharmaceutics15020552