
Biodistribution of Intratracheal, Intranasal, and Intravenous Injections of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles in a Mouse Model for Drug Delivery Studies

 , , , , , , , ,
, , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Extracellular Vesicles
2.2. Tunable Resistive Pulse Sensing (tRPS)
2.3. Transmission Electron Microscopy
2.4. Immunophenotyping of MSC-EVs
2.5. Labeling and Cellular Uptake of MSC-EVs
2.6. In Vivo Administration
2.7. IVIS Imaging of In-Vivo-Administered MSC-EVs
2.8. Organ Biodistribution of Labeled DiR-EVs
2.9. Immunofluorescence Staining
2.10. Statistics
3. Results
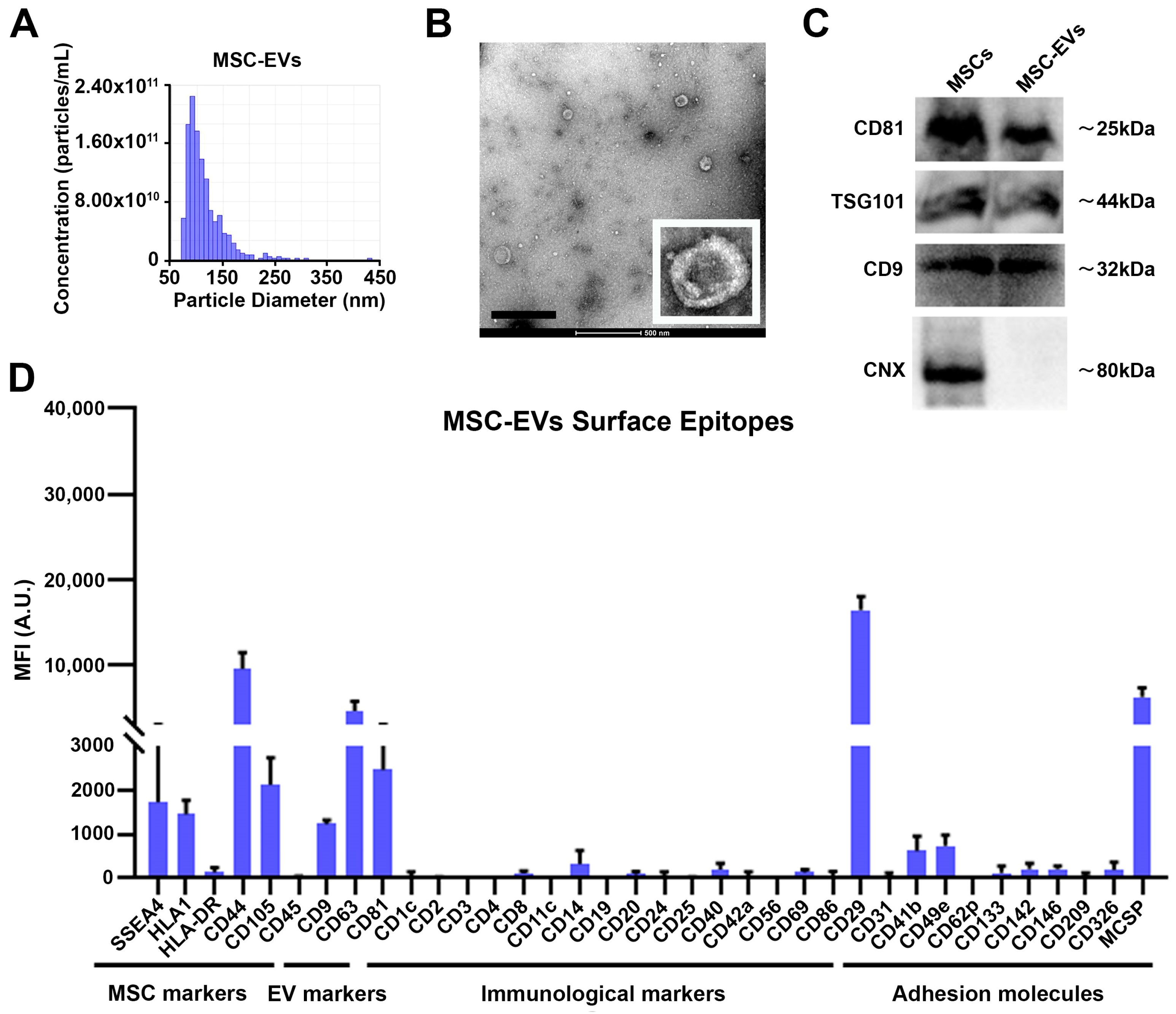
3.1. Characterization of MSC-EVs
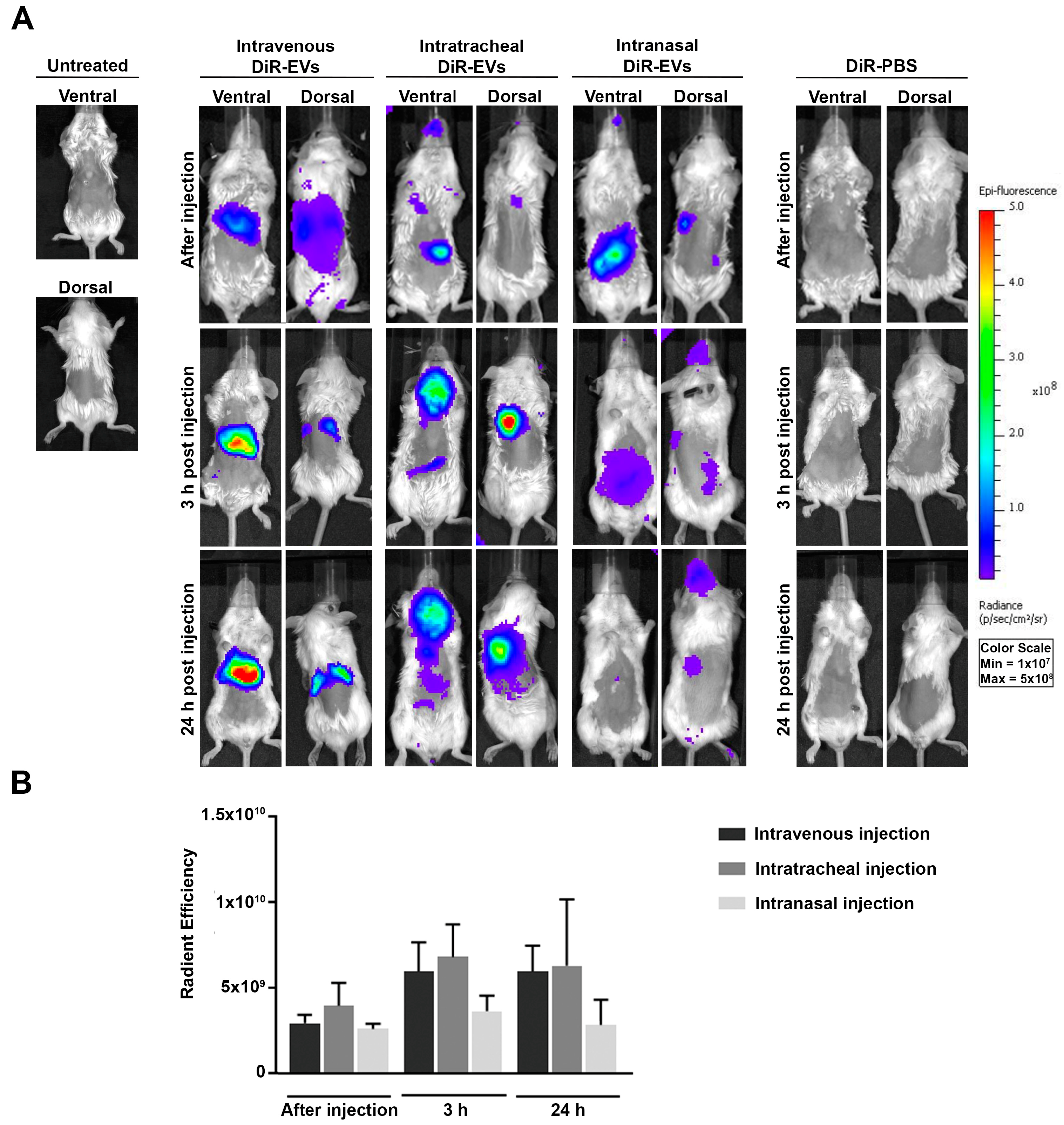
3.2. In Vivo Biodistribution of DiR-Labeled MSC-EVs in BALB/c Mice
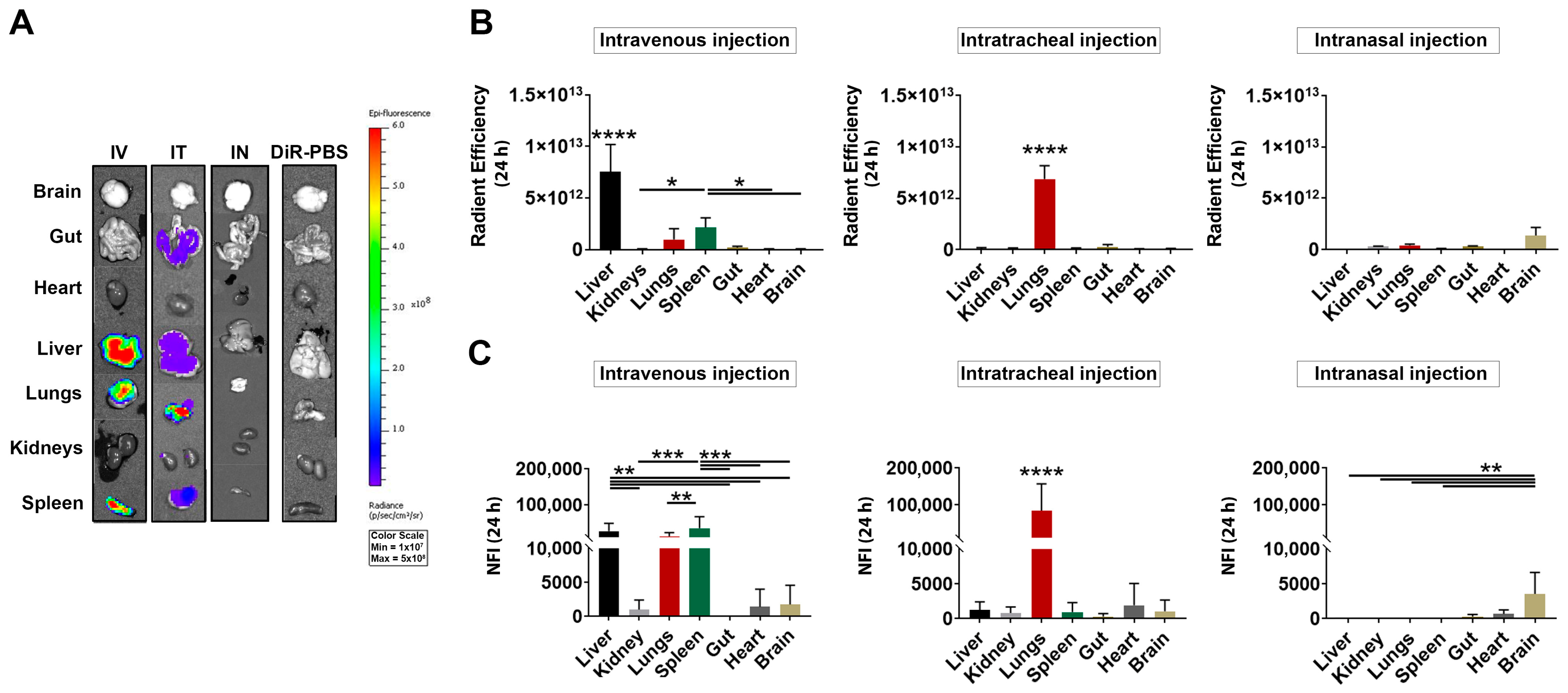
3.3. Ex Vivo Detection of DiR-Labeled MSC-EVs after IV, IT and IN Injection
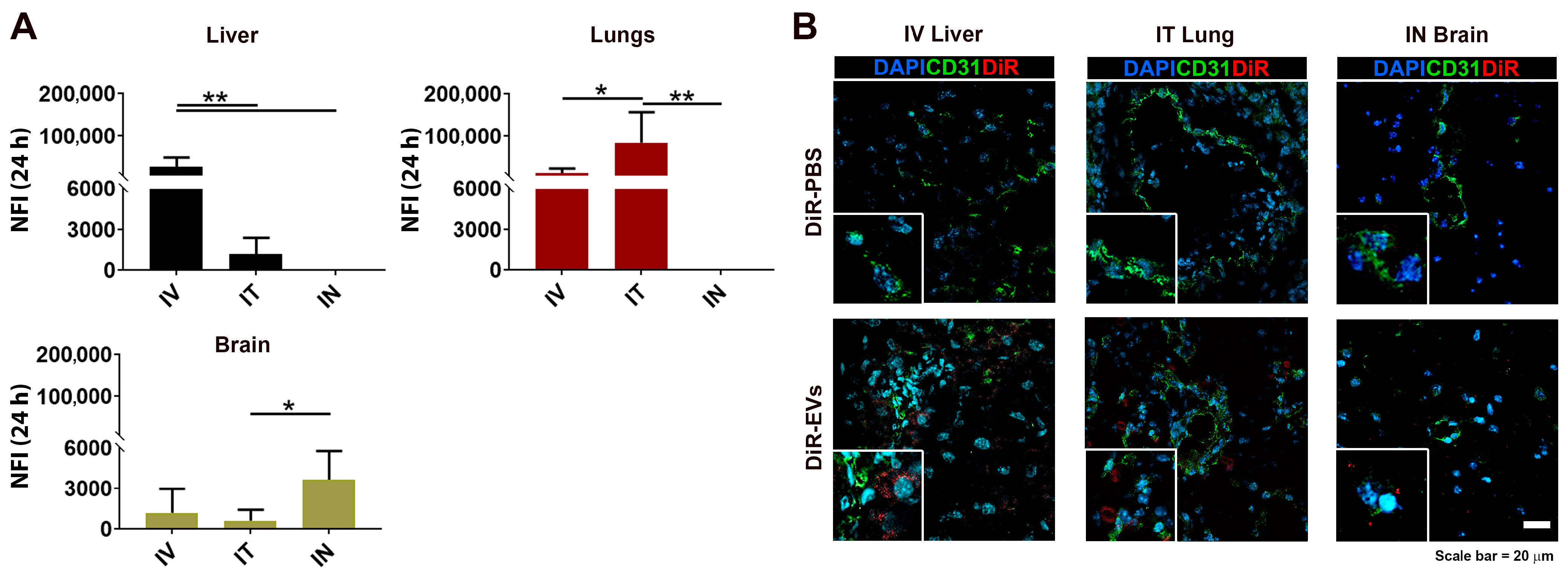
3.4. DiR-Labeled MSC-EV Tissue Detection after IV, IT and IN Administration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic Stem Cell-Derived Microvesicles Reprogram Hematopoietic Progenitors: Evidence for Horizontal Transfer of MRNA and Protein Delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-Mediated Transfer of MRNAs and MicroRNAs Is a Novel Mechanism of Genetic Exchange between Cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The Transport Mechanism of Extracellular Vesicles at the Blood-Brain Barrier. Curr. Pharm. Des. 2018, 23, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Deng, S.; Han, L.; Ren, Y.; Gu, J.; He, L.; Liu, T.; Yuan, Z.X. Mesenchymal Stem Cells, Exosomes and Exosome-Mimics as Smart Drug Carriers for Targeted Cancer Therapy. Colloids Surf. B Biointerfaces 2022, 209, 112163. [Google Scholar] [CrossRef] [PubMed]
- Perets, N.; Betzer, O.; Shapira, R.; Brenstein, S.; Angel, A.; Sadan, T.; Ashery, U.; Popovtzer, R.; Offen, D. Golden Exosomes Selectively Target Brain Pathologies in Neurodegenerative and Neurodevelopmental Disorders. Nano Lett. 2019, 19, 3422–3431. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Betzer, O.; Elmaliach-Pnini, N.; Motiei, M.; Sadan, T.; Cohen-Berkman, M.; Dagan, O.; Popovtzer, A.; Yosepovich, A.; Barhom, H.; et al. ‘Golden’ Exosomes as Delivery Vehicles to Target Tumors and Overcome Intratumoral Barriers: In Vivo Tracking in a Model for Head and Neck Cancer. Biomater. Sci. 2021, 9, 2103–2114. [Google Scholar] [CrossRef]
- Rohban, R.; Pieber, T.R. Mesenchymal Stem and Progenitor Cells in Regeneration: Tissue Specificity and Regenerative Potential. Stem Cells Int. 2017, 2017, 5173732. [Google Scholar] [CrossRef]
- Psaraki, A.; Ntari, L.; Karakostas, C.; Korrou-Karava, D.; Roubelakis, M.G. Extracellular Vesicles Derived from Mesenchymal Stem/Stromal Cells: The Regenerative Impact in Liver Diseases. Hepatology 2022, 75, 1590–1603. [Google Scholar] [CrossRef]
- Zargar, M.J.; Kaviani, S.; Vasei, M.; Soufi Zomorrod, M.; Heidari Keshel, S.; Soleimani, M. Therapeutic Role of Mesenchymal Stem Cell-Derived Exosomes in Respiratory Disease. Stem Cell Res. Ther. 2022, 13, 194. [Google Scholar] [CrossRef]
- Vatsa, P.; Negi, R.; Ansari, U.A.; Khanna, V.K.; Pant, A.B. Insights of Extracellular Vesicles of Mesenchymal Stem Cells: A Prospective Cell-Free Regenerative Medicine for Neurodegenerative Disorders. Mol. Neurobiol. 2022, 59, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, M.; Merlotti, G.; Colombatto, A.; Bruno, S.; Stasi, A.; Franzin, R.; Castellano, G.; Grossini, E.; Fanelli, V.; Cantaluppi, V. Stem Cell-Derived Extracellular Vesicles as Potential Therapeutic Approach for Acute Kidney Injury. Front. Immunol. 2022, 13, 849891. [Google Scholar] [CrossRef] [PubMed]
- Magarotto, F.; Sgrò, A.; Dorigo Hochuli, A.H.; Andreetta, M.; Grassi, M.; Saggioro, M.; Nogara, L.; Tolomeo, A.M.; Francescato, R.; Collino, F.; et al. Muscle Functional Recovery Is Driven by Extracellular Vesicles Combined with Muscle Extracellular Matrix in a Volumetric Muscle Loss Murine Model. Biomaterials 2021, 269, 120653. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, A.M.; Castagliuolo, I.; Piccoli, M.; Grassi, M.; Magarotto, F.; de Lazzari, G.; Malvicini, R.; Caicci, F.; Franzin, C.; Scarpa, M.; et al. Extracellular Vesicles Secreted by Mesenchymal Stromal Cells Exert Opposite Effects to Their Cells of Origin in Murine Sodium Dextran Sulfate-Induced Colitis. Front. Immunol. 2021, 12, 627605. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular Vesicle in Vivo Biodistribution Is Determined by Cell Source, Route of Administration and Targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Yoo, M.H.; Lee, A.R.; Moon, K.S. Characteristics of Extracellular Vesicles and Preclinical Testing Considerations Prior to Clinical Applications. Biomedicines 2022, 10, 869. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Z.; Liu, L.; Zhang, B.; Li, B. Exosomes Derived from Adipose Tissue, Bone Marrow, and Umbilical Cord Blood for Cardioprotection after Myocardial Infarction. J. Cell Biochem. 2020, 121, 2089–2102. [Google Scholar] [CrossRef]
- Collino, F.; Lopes, J.A.; Tapparo, M.; Tortelote, G.G.; Kasai-Brunswick, T.H.; Lopes, G.M.C.; Almeida, D.B.; Skovronova, R.; Wendt, C.H.C.; Miranda, K.R.d.; et al. Extracellular Vesicles Derived from Induced Pluripotent Stem Cells Promote Renoprotection in Acute Kidney Injury Model. Cells 2020, 9, 453. [Google Scholar] [CrossRef]
- Abreu, S.C.; Antunes, M.A.; Xisto, D.G.; Cruz, F.F.; Branco, V.C.; Bandeira, E.; Kitoko, J.Z.; de Araú Jo, A.F.; Dellatorre-Texeira, L.; Olsen, P.C.; et al. Bone Marrow, Adipose, and Lung Tissue-Derived Murine Mesenchymal Stromal Cells Release Different Mediators and Differentially Affect Airway and Lung Parenchyma in Experimental Asthma. Stem Cells Transl. Med. 2017, 6, 1557–1567. [Google Scholar] [CrossRef]
- Liu, D.; Cheng, F.; Pan, S.; Liu, Z. Stem Cells: A Potential Treatment Option for Kidney Diseases. Stem Cell Res. Ther. 2020, 11, 249. [Google Scholar] [CrossRef]
- Kang, M.; Jordan, V.; Blenkiron, C.; Chamley, L.W. Biodistribution of Extracellular Vesicles Following Administration into Animals: A Systematic Review. J. Extracell. Vesicles 2021, 10, e12085. [Google Scholar] [CrossRef]
- Li, J.; Komatsu, H.; Poku, E.K.; Olafsen, T.; Huang, K.X.; Huang, L.A.; Chea, J.; Bowles, N.; Chang, B.; Rawson, J.; et al. Biodistribution of Intra-Arterial and Intravenous Delivery of Human Umbilical Cord Mesenchymal Stem Cell-Derived Extracellular Vesicles in a Rat Model to Guide Delivery Strategies for Diabetes Therapies. Pharmaceuticals 2022, 15, 595. [Google Scholar] [CrossRef] [PubMed]
- Porzionato, A.; Zaramella, P.; Dedja, A.; Guidolin, D.; van Wemmel, K.; Macchi, V.; Jurga, M.; Perilongo, G.; de Caro, R.; Baraldi, E.; et al. Intratracheal Administration of Clinical-Grade Mesenchymal Stem Cell-Derived Extracellular Vesicles Reduces Lung Injury in a Rat Model of Bronchopulmonary Dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L6–L19. [Google Scholar] [CrossRef]
- Porzionato, A.; Zaramella, P.; Dedja, A.; Guidolin, D.; Bonadies, L.; Macchi, V.; Pozzobon, M.; Jurga, M.; Perilongo, G.; de Caro, R.; et al. Intratracheal Administration of Mesenchymal Stem Cell-Derived Extracellular Vesicles Reduces Lung Injuries in a Chronic Rat Model of Bronchopulmonary Dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L688–L704. [Google Scholar] [CrossRef] [PubMed]
- Lázaro-Ibánez, E.; Al-Jamal, K.T.; Dekker, N.; Faruqu, F.N.; Saleh, A.F.; Silva, A.M.; Wang, J.T.W.; Rak, J. Selection of Fluorescent, Bioluminescent, and Radioactive Tracers to Accurately Reflect Extracellular Vesicle Biodistribution in Vivo. ACS Nano 2021, 15, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; Balaj, L.; Boulanger, C.M.; Carter, D.R.F.; Compeer, E.B.; D’Angelo, G.; el Andaloussi, S.; Goetz, J.G.; Gross, J.C.; Hyenne, V.; et al. The Power of Imaging to Understand Extracellular Vesicle Biology in Vivo. Nat. Methods 2021, 18, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, Functionality, and Re-Release of Extracellular Vesicle-Encapsulated Cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef]
- Arifin, D.R.; Witwer, K.W.; Bulte, J.W.M. Non-Invasive Imaging of Extracellular Vesicles: Quo Vaditis in Vivo? J. Extracell. Vesicles 2022, 11, e12241. [Google Scholar] [CrossRef]
- Cassidy, P.J.; Radda, G.K. Molecular Imaging Perspectives. J. R. Soc. Interface 2005, 2, 133–144. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Bruno, S.; Chatterjee, D.; Quesenberry, P.J.; Tetta, C.; Camussi, G. Biodistribution of Mesenchymal Stem Cell-Derived Extracellular Vesicles in a Model of Acute Kidney Injury Monitored by Optical Imaging. Int. J. Mol. Med. 2014, 33, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Jalabert, A.; Vial, G.; Guay, C.; Wiklander, O.P.B.; Nordin, J.Z.; Aswad, H.; Forterre, A.; Meugnier, E.; Pesenti, S.; Regazzi, R.; et al. Exosome-like Vesicles Released from Lipid-Induced Insulin-Resistant Muscles Modulate Gene Expression and Proliferation of Beta Recipient Cells in Mice. Diabetologia 2016, 59, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.B.; Hällbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with Size-Exclusion Liquid Chromatography for High Yield Isolation of Extracellular Vesicles Preserving Intact Biophysical and Functional Properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and Delivery Efficiency of Unmodified Tumor-Derived Exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.F.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef]
- Zhang, P.; Dong, B.; Zeng, E.; Wang, F.; Jiang, Y.; Li, D.; Liu, D. In Vivo Tracking of Multiple Tumor Exosomes Labeled by Phospholipid-Based Bioorthogonal Conjugation. Anal Chem. 2018, 90, 11273–11279. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of Distinct Nanoparticles and Subsets of Extracellular Vesicles by Asymmetric Flow Field-Flow Fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Wan, L.; Xia, T.; Du, Y.; Liu, J.; Xie, Y.; Zhang, Y.; Guan, F.; Wu, J.; Wang, X.; Shi, C. Exosomes from Activated Hepatic Stellate Cells Contain GLUT1 and PKM2: A Role for Exosomes in Metabolic Switch of Liver Nonparenchymal Cells. FASEB J. 2019, 33, 8530–8542. [Google Scholar] [CrossRef]
- Cai, J.; Wu, J.; Wang, J.; Li, Y.; Hu, X.; Luo, S.; Xiang, D. Extracellular Vesicles Derived from Different Sources of Mesenchymal Stem Cells: Therapeutic Effects and Translational Potential. Cell Biosci. 2020, 10, 69. [Google Scholar] [CrossRef]
- Hyvärinen, K.; Holopainen, M.; Skirdenko, V.; Ruhanen, H.; Lehenkari, P.; Korhonen, M.; Käkelä, R.; Laitinen, S.; Kerkelä, E. Mesenchymal Stromal Cells and Their Extracellular Vesicles Enhance the Anti-Inflammatory Phenotype of Regulatory Macrophages by Downregulating the Production of Interleukin (IL)-23 and IL-22. Front. Immunol. 2018, 9, 771. [Google Scholar] [CrossRef]
- Fujii, S.; Miura, Y.; Fujishiro, A.; Shindo, T.; Shimazu, Y.; Hirai, H.; Tahara, H.; Takaori-Kondo, A.; Ichinohe, T.; Maekawa, T. Graft-Versus-Host Disease Amelioration by Human Bone Marrow Mesenchymal Stromal/Stem Cell-Derived Extracellular Vesicles Is Associated with Peripheral Preservation of Naive T Cell Populations. Stem Cells 2018, 36, 434–445. [Google Scholar] [CrossRef]
- Gupta, D.; Liang, X.; Pavlova, S.; Wiklander, O.P.B.; Corso, G.; Zhao, Y.; Saher, O.; Bost, J.; Zickler, A.M.; Piffko, A.; et al. Quantification of Extracellular Vesicles in Vitro and in Vivo Using Sensitive Bioluminescence Imaging. J. Extracell. Vesicles 2020, 9, 1800222. [Google Scholar] [CrossRef]
- Grange, C.; Tritta, S.; Tapparo, M.; Cedrino, M.; Tetta, C.; Camussi, G.; Brizzi, M.F. Stem Cell-Derived Extracellular Vesicles Inhibit and Revert Fibrosis Progression in a Mouse Model of Diabetic Nephropathy. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Collino, F.; Lopes, J.A.; Corrêa, S.; Abdelhay, E.; Takiya, C.M.; Wendt, C.H.C.; de Miranda, K.R.; Vieyra, A.; Lindoso, R.S. Adipose-Derived Mesenchymal Stromal Cells Under Hypoxia: Changes in Extracellular Vesicles Secretion and Improvement of Renal Recovery after Ischemic Injury. Cell Physiol. Biochem. 2019, 52, 1463–1483. [Google Scholar] [CrossRef]
- Yang, S.; Liu, P.; Gao, T.; Song, D.; Zhao, X.; Li, Y.; Wu, J.; Wang, L.; Wang, Z.; Hao, J.; et al. Every Road Leads to Rome: Therapeutic Effect and Mechanism of the Extracellular Vesicles of Human Embryonic Stem Cell-Derived Immune and Matrix Regulatory Cells Administered to Mouse Models of Pulmonary Fibrosis through Different Routes. Stem Cell Res. Ther. 2022, 13, 163. [Google Scholar] [CrossRef]
- Pacienza, N.; Lee, R.H.; Bae, E.H.; Kim, D.K.; Liu, Q.; Prockop, D.J.; Yannarelli, G. In Vitro Macrophage Assay Predicts the In Vivo Anti-Inflammatory Potential of Exosomes from Human Mesenchymal Stromal Cells. Mol. Ther. Methods Clin. Dev. 2019, 13, 67–76. [Google Scholar] [CrossRef]
- Malvicini, R.; Santa-Cruz, D.; de Lazzari, G.; Tolomeo, A.M.; Sanmartin, C.; Muraca, M.; Yannarelli, G.; Pacienza, N. Macrophage Bioassay Standardization to Assess the Anti-Inflammatory Activity of Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles. Cytotherapy 2022, 24, 999–1012. [Google Scholar] [CrossRef]
- Ramos-Zaldívar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalán, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular Vesicles through the Blood–Brain Barrier: A Review. Fluids Barriers CNS 2022, 19, 60. [Google Scholar] [CrossRef]
- Driedonks, T.; Jiang, L.; Carlson, B.; Han, Z.; Liu, G.; Queen, S.E.; Shirk, E.N.; Gololobova, O.; Liao, Z.; Nyberg, L.H.; et al. Pharmacokinetics and Biodistribution of Extracellular Vesicles Administered Intravenously and Intranasally to Macaca Nemestrina. J. Extracell. Biol. 2022, 1, e59. [Google Scholar] [CrossRef]
- Türker, S.; Onur, E.; Ózer, Y. Nasal Route and Drug Delivery Systems. Pharm. World Sci. 2004, 26, 137–142. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolomeo, A.M.; Zuccolotto, G.; Malvicini, R.; De Lazzari, G.; Penna, A.; Franco, C.; Caicci, F.; Magarotto, F.; Quarta, S.; Pozzobon, M.; et al. Biodistribution of Intratracheal, Intranasal, and Intravenous Injections of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles in a Mouse Model for Drug Delivery Studies. Pharmaceutics 2023, 15, 548. https://doi.org/10.3390/pharmaceutics15020548
Tolomeo AM, Zuccolotto G, Malvicini R, De Lazzari G, Penna A, Franco C, Caicci F, Magarotto F, Quarta S, Pozzobon M, et al. Biodistribution of Intratracheal, Intranasal, and Intravenous Injections of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles in a Mouse Model for Drug Delivery Studies. Pharmaceutics. 2023; 15(2):548. https://doi.org/10.3390/pharmaceutics15020548
Chicago/Turabian StyleTolomeo, Anna Maria, Gaia Zuccolotto, Ricardo Malvicini, Giada De Lazzari, Alessandro Penna, Chiara Franco, Federico Caicci, Fabio Magarotto, Santina Quarta, Michela Pozzobon, and et al. 2023. "Biodistribution of Intratracheal, Intranasal, and Intravenous Injections of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles in a Mouse Model for Drug Delivery Studies" Pharmaceutics 15, no. 2: 548. https://doi.org/10.3390/pharmaceutics15020548