
Interplay of Breast Cancer Resistance Protein (Bcrp/Abcg2), Sex, and Fed State in Oral Pharmacokinetic Variability of Furosemide in Rats
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents, and Software
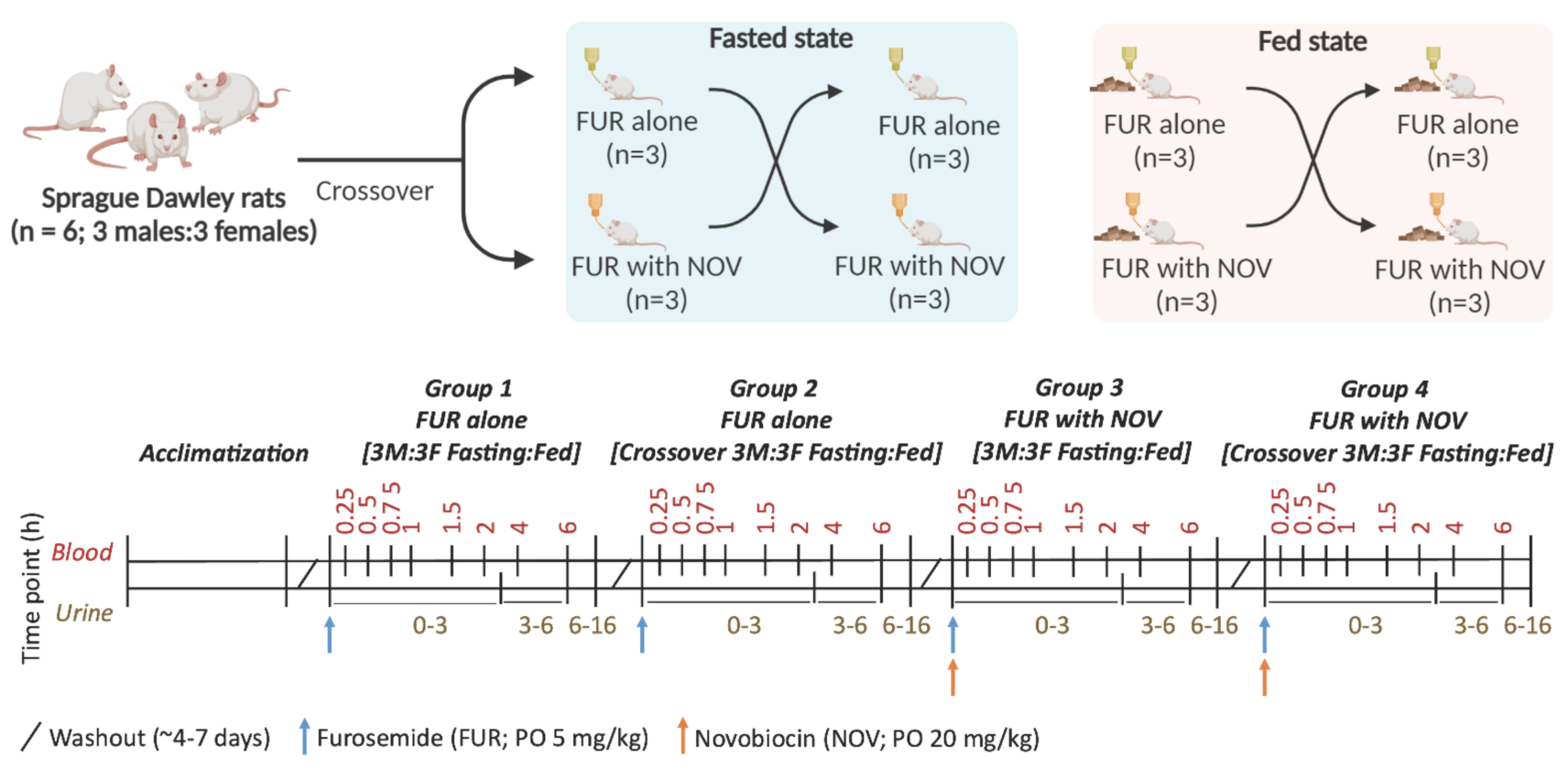
2.2. Rat Pharmacokinetic Study
2.3. Analysis of Blood and Urine Samples
2.4. Quantification of Bcrp Protein Abundance in Rat Tissues
3. Results
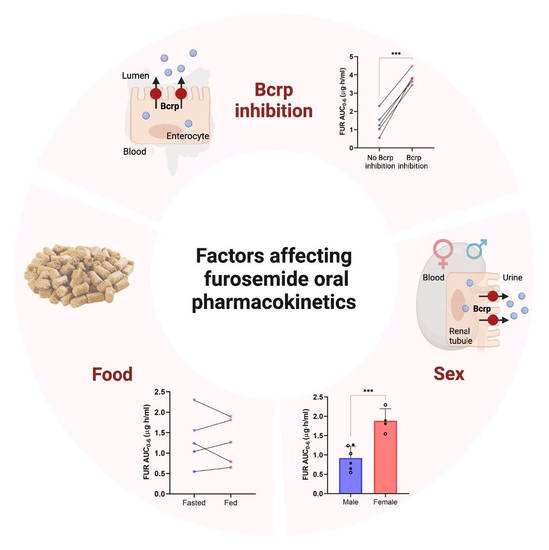
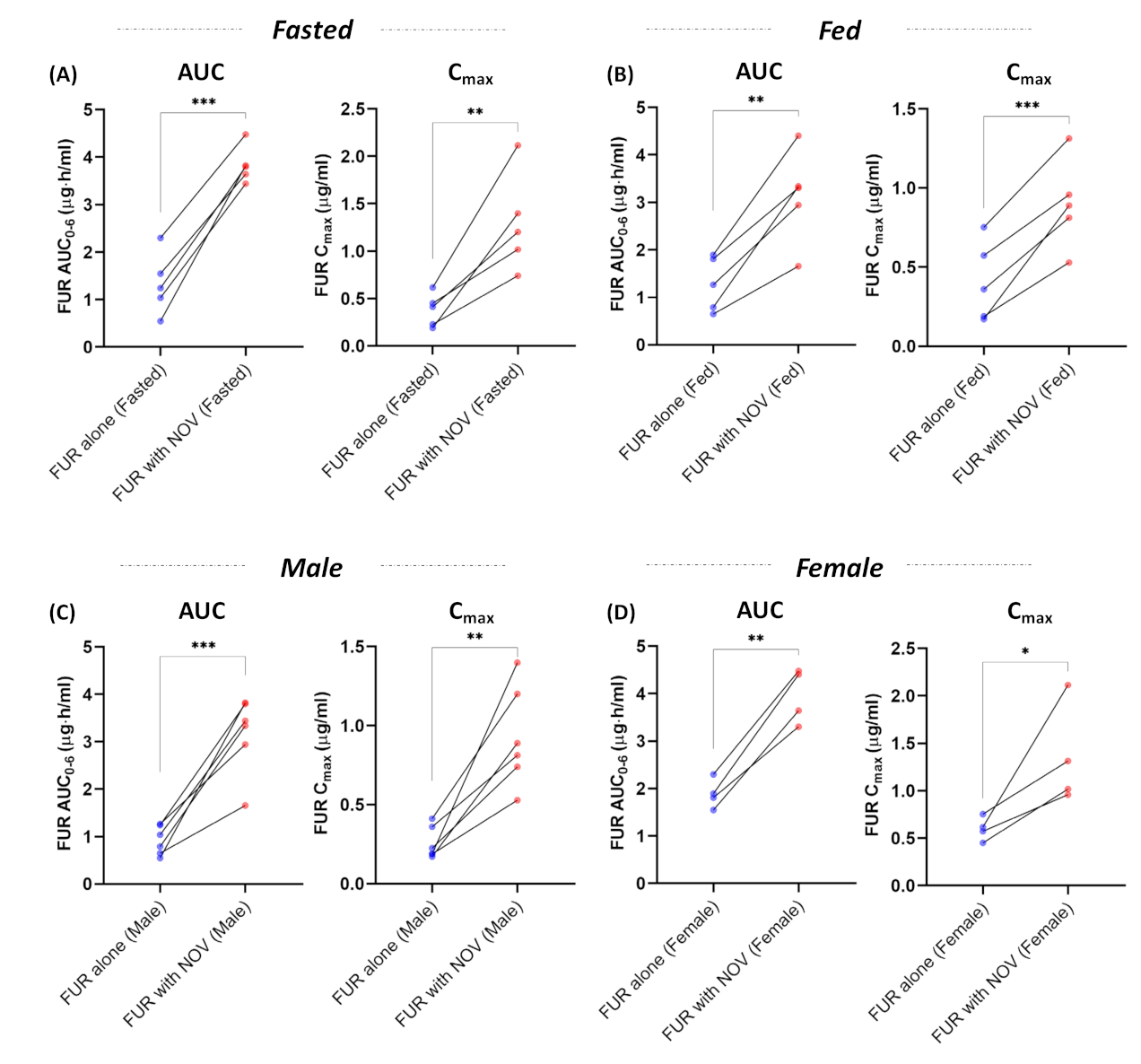
3.1. Effect of Bcrp Inhibition on Pharmacokinetics of Furosemide
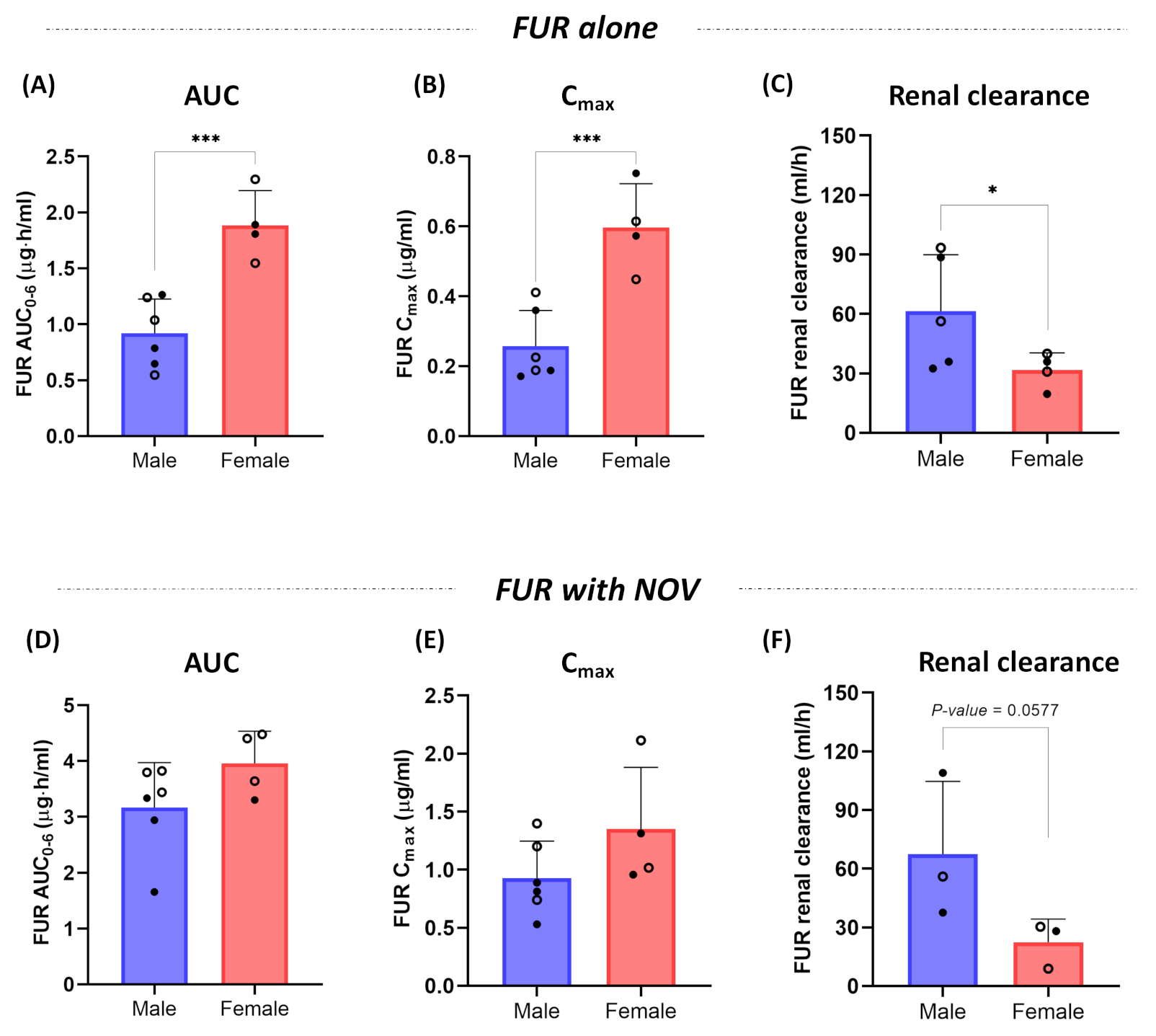
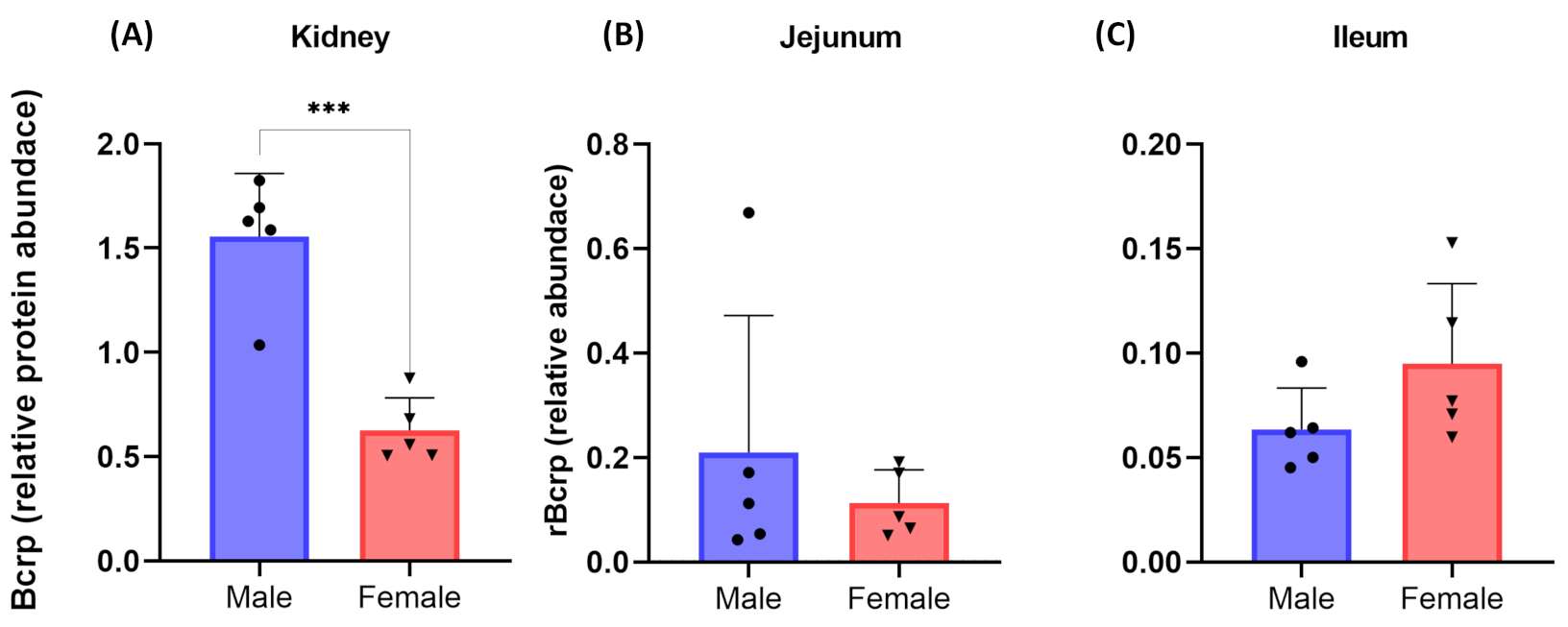
3.2. Effect of Sex on Pharmacokinetics of Furosemide and Novobiocin
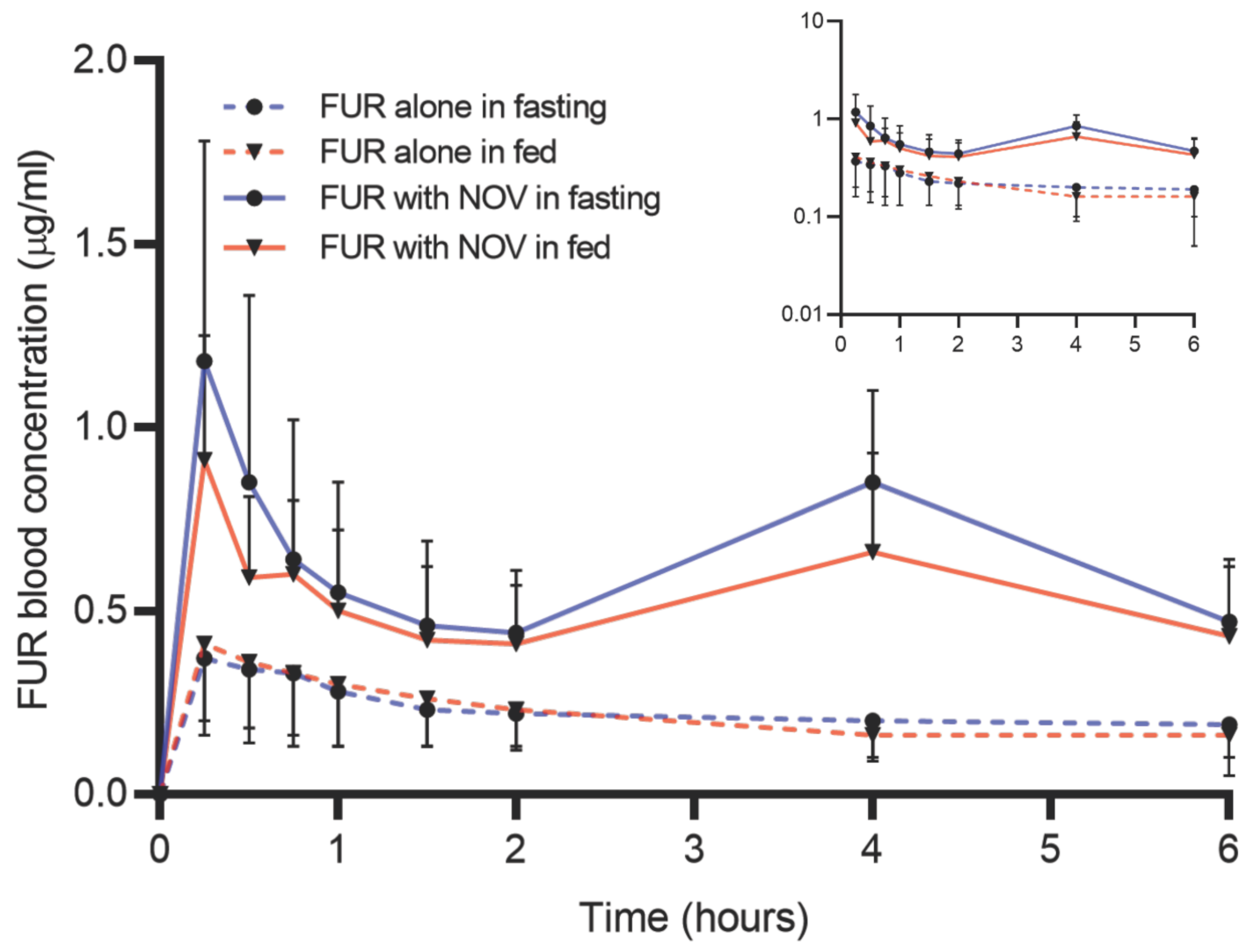
3.3. Cumulative Effect of Bcrp Inhibition, Sex, and Fed State on Pharmacokinetics of Furosemide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | area under the blood concentration-time profile curve |
| Bcrp | breast cancer resistance protein |
| CLR | renal clearance |
| Cmax | peak blood concentration |
| DDI | drug-drug interactions |
| FUR | Furosemide |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| NOV | Novobiocin |
| OAT | organic anion transporters |
| PK | Pharmacokinetics |
References
- Dixon, D.W.; Barwolf-Gohlke, C.; Gunnar, R.M. Comparative Efficacy and Safety of Bumetanide and Furosemide in Long-Term Treatment of Edema Due to Congestive Heart Failure. J. Clin. Pharmacol. 1981, 21, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Qavi, A.H.; Kamal, R.; Schrier, R.W. Clinical Use of Diuretics in Heart Failure, Cirrhosis, and Nephrotic Syndrome. Int. J. Nephrol. 2015, 2015, 975934. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H. Clinical Pharmacology in Diuretic Use. Clin. J. Am. Soc. Nephrol. 2019, 14, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.S.; Brater, D.C. Loop Diuretics: From the Na-K-2Cl Transporter to Clinical Use. Am. J. Physiol. Renal Physiol. 2003, 284, F11–F21. [Google Scholar] [CrossRef]
- Lennernäs, H. Human Intestinal Permeability. J. Pharm. Sci. 1998, 87, 403–410. [Google Scholar] [CrossRef]
- Wisher, D. Martindale: The Complete Drug Reference. 37th Ed. J. Med. Libr. Assoc. 2012, 100, 75–76. [Google Scholar] [CrossRef]
- Ebner, T.; Ishiguro, N.; Taub, M.E. The Use of Transporter Probe Drug Cocktails for the Assessment of Transporter-Based Drug-Drug Interactions in a Clinical Setting-Proposal of a Four Component Transporter Cocktail. J. Pharm. Sci. 2015, 104, 3220–3228. [Google Scholar] [CrossRef]
- Ponto, L.L.; Schoenwald, R.D. Furosemide (Frusemide) A Pharmacokinetic/Pharmacodynamic Review (Part I). Clin. Pharm. 1990, 18, 381–408. [Google Scholar] [CrossRef]
- Kerdpin, O.; Knights, K.M.; Elliot, D.J.; Miners, J.O. In Vitro Characterisation of Human Renal and Hepatic Frusemide Glucuronidation and Identification of the UDP-Glucuronosyltransferase Enzymes Involved in This Pathway. Biochem. Pharmacol. 2008, 76, 249–257. [Google Scholar] [CrossRef]
- Odlind, B.; Beermann, B. Renal Tubular Secretion and Effects of Furosemide. Clin. Pharmacol. Ther. 1980, 27, 784–790. [Google Scholar] [CrossRef]
- Smith, D.E.; Gee, W.L.; Brater, D.C.; Lin, E.T.; Benet, L.Z. Preliminary Evaluation of Furosemide-Probenecid Interaction in Humans. J. Pharm. Sci. 1980, 69, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Holenarsipur, V.K.; Mariappan, T.T.; Drexler, D.M.; Cantone, J.L.; Rajanna, P.; Singh Gautam, S.; Zhang, Y.; Gan, J.; Shipkova, P.A.; et al. Evidence for the Validity of Pyridoxic Acid (PDA) as a Plasma-Based Endogenous Probe for OAT1 and OAT3 Function in Healthy Subjects. J. Pharmacol. Exp. Ther. 2019, 368, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Skolnik, S.; Chen, X.; Wang, J. Attenuation of Intestinal Absorption by Major Efflux Transporters: Quantitative Tools and Strategies Using a Caco-2 Model. Drug Metab. Dispos. 2011, 39, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Chapa, R.; Li, C.Y.; Basit, A.; Thakur, A.; Ladumor, M.K.; Sharma, S.; Singh, S.; Selen, A.; Prasad, B. Contribution of Uptake and Efflux Transporters to Oral Pharmacokinetics of Furosemide. ACS Omega 2020, 5, 32939–32950. [Google Scholar] [CrossRef] [PubMed]
- Fabiano, V.; Carnovale, C.; Gentili, M.; Radice, S.; Zuccotti, G.V.; Clementi, E.; Perrotta, C.; Mameli, C. Enalapril Associated with Furosemide Induced Acute Kidney Injury in an Infant with Heart Failure. A Case Report, a Revision of the Literature and a Pharmacovigilance Database Analysis. Pharmacology 2016, 97, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Schröter, M.; Neubeck, M.; Ritz, E. Coadministration of Thiazides Increases the Efficacy of Loop Diuretics Even in Patients with Advanced Renal Failure. Kidney Int. 1994, 46, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kusuhara, H.; Adachi, M.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Multidrug Resistance-Associated Protein 4 Is Involved in the Urinary Excretion of Hydrochlorothiazide and Furosemide. J. Am. Soc. Nephrol. 2007, 18, 37–45. [Google Scholar] [CrossRef]
- Jentzer, J.C.; DeWald, T.A.; Hernandez, A.F. Combination of Loop Diuretics with Thiazide-Type Diuretics in Heart Failure. J. Am. Coll. Cardiol. 2010, 56, 1527–1534. [Google Scholar] [CrossRef]
- Prasad, B.; Lai, Y.; Lin, Y.; Unadkat, J.D. Interindividual Variability in the Hepatic Expression of the Human Breast Cancer Resistance Protein (BCRP/ABCG2): Effect of Age, Sex, and Genotype. J. Pharm. Sci. 2013, 102, 787–793. [Google Scholar] [CrossRef]
- Billington, S.; Salphati, L.; Hop, C.E.C.A.; Chu, X.; Evers, R.; Burdette, D.; Rowbottom, C.; Lai, Y.; Xiao, G.; Humphreys, W.G.; et al. Interindividual and Regional Variability in Drug Transporter Abundance at the Human Blood-Brain Barrier Measured by Quantitative Targeted Proteomics. Clin. Pharmacol. Ther. 2019, 106, 228–237. [Google Scholar] [CrossRef]
- Storelli, F.; Yin, M.; Kumar, A.R.; Ladumor, M.K.; Evers, R.; Chothe, P.P.; Enogieru, O.J.; Liang, X.; Lai, Y.; Unadkat, J.D. The next Frontier in ADME Science: Predicting Transporter-Based Drug Disposition, Tissue Concentrations and Drug-Drug Interactions in Humans. Pharmacol. Ther. 2022, 238, 108271. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Prasad, B. Meta-Analysis of Food Effect on Oral Absorption of Efflux Transporter Substrate Drugs: Does Delayed Gastric Emptying Influence Drug Transport Kinetics? Pharmaceutics 2021, 13, 1035. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.; Radi, Z.; Vaidya, V.S.; Karasu, M.; Prasad, B. Kidney Cortical Transporter Expression across Species Using Quantitative Proteomics. Drug Metab. Dispos. 2019, 47, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Tucker, G.T. Measurement of the Renal Clearance of Drugs. Br. J. Clin. Pharmacol. 1981, 12, 761–770. [Google Scholar] [CrossRef]
- Xu, M.; Saxena, N.; Vrana, M.; Zhang, H.; Kumar, V.; Billington, S.; Khojasteh, C.; Heyward, S.; Unadkat, J.D.; Prasad, B. A Targeted LC-MS/MS Proteomics-Based Strategy to Characterize In Vitro Models Used in Drug Metabolism and Transport Studies. Anal. Chem. 2018, 90, 11873–11882. [Google Scholar] [CrossRef]
- Ahire, D.S.; Basit, A.; Karasu, M.; Prasad, B. Ultrasensitive Quantification of Drug-Metabolizing Enzymes and Transporters in Small Sample Volume by Microflow LC-MS/MS. J. Pharm. Sci. 2021, 110, 2833–2840. [Google Scholar] [CrossRef]
- Drozdzik, M.; Gröer, C.; Penski, J.; Lapczuk, J.; Ostrowski, M.; Lai, Y.; Prasad, B.; Unadkat, J.D.; Siegmund, W.; Oswald, S. Protein Abundance of Clinically Relevant Multidrug Transporters along the Entire Length of the Human Intestine. Mol. Pharm. 2014, 11, 3547–3555. [Google Scholar] [CrossRef]
- Al-Majdoub, Z.M.; Scotcher, D.; Achour, B.; Barber, J.; Galetin, A.; Rostami-Hodjegan, A. Quantitative Proteomic Map of Enzymes and Transporters in the Human Kidney: Stepping Closer to Mechanistic Kidney Models to Define Local Kinetics. Clin. Pharmacol. Ther. 2021, 110, 1389–1400. [Google Scholar] [CrossRef]
- Zou, W.; Shi, B.; Zeng, T.; Zhang, Y.; Huang, B.; Ouyang, B.; Cai, Z.; Liu, M. Drug Transporters in the Kidney: Perspectives on Species Differences, Disease Status, and Molecular Docking. Front. Pharmacol. 2021, 12, 746208. [Google Scholar] [CrossRef]
- Chow, M.S.; Schweizer, R. Estimation of Renal Creatinine Clearance in Patients with Unstable Serum Creatinine Concentrations: Comparison of Multiple Methods. Drug Intell. Clin. Pharm. 1985, 19, 385–390. [Google Scholar] [CrossRef]
- Piao, Y.; Liu, Y.; Xie, X. Change Trends of Organ Weight Background Data in Sprague Dawley Rats at Different Ages. J. Toxicol. Pathol. 2013, 26, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Busti, A.J.; Bain, A.M.; Hall, R.G.; Bedimo, R.G.; Leff, R.D.; Meek, C.; Mehvar, R. Effects of Atazanavir/Ritonavir or Fosamprenavir/Ritonavir on the Pharmacokinetics of Rosuvastatin. J. Cardiovasc. Pharmacol. 2008, 51, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Kiser, J.J.; Gerber, J.G.; Predhomme, J.A.; Wolfe, P.; Flynn, D.M.; Hoody, D.W. Drug/Drug Interaction between Lopinavir/Ritonavir and Rosuvastatin in Healthy Volunteers. J. Acquir. Immune Defic. Syndr. 2008, 47, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.A.; la Porte, C.J.L.; Lee, L.S.; van Heeswijk, R.; Sabo, J.P.; Elgadi, M.M.; Piliero, P.J.; Barditch-Crovo, P.; Fuchs, E.; Flexner, C.; et al. Differential Effects of Tipranavir plus Ritonavir on Atorvastatin or Rosuvastatin Pharmacokinetics in Healthy Volunteers. Antimicrob. Agents Chemother. 2009, 53, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, H.; Furuie, H.; Inano, A.; Sunagawa, A.; Yamada, S.; Wu, C.; Fukizawa, S.; Morimoto, N.; Ieiri, I.; Morishita, M.; et al. Pharmacokinetic Interaction Study of Sulphasalazine in H.Healthy Subjects and the Impact of Curcumin as an in Vivo Inhibitor of BCRP. Br. J. Pharmacol. 2012, 166, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Maher, R.L.; Hanlon, J.T.; Hajjar, E.R. Clinical Consequences of Polypharmacy in Elderly. Expert Opin. Drug Saf. 2014, 13, 57–65. [Google Scholar] [CrossRef]
- Sönnerstam, E.; Sjölander, M.; Lövheim, H.; Gustafsson, M. Clinically Relevant Drug–Drug Interactions among Elderly People with Dementia. Eur. J. Clin. Pharmacol. 2018, 74, 1351–1360. [Google Scholar] [CrossRef]
- Zhang, Y.; Gupta, A.; Wang, H.; Zhou, L.; Vethanayagam, R.R.; Unadkat, J.D.; Mao, Q. BCRP Transports Dipyridamole and Is Inhibited by Calcium Channel Blockers. Pharm. Res. 2005, 22, 2023–2034. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the Breast Cancer Resistance Protein (BCRP/ABCG2) in Drug Transport--an Update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Savage, R.D.; Visentin, J.D.; Bronskill, S.E.; Wang, X.; Gruneir, A.; Giannakeas, V.; Guan, J.; Lam, K.; Luke, M.J.; Read, S.H.; et al. Evaluation of a Common Prescribing Cascade of Calcium Channel Blockers and Diuretics in Older Adults With Hypertension. JAMA Intern. Med. 2020, 180, 643–651. [Google Scholar] [CrossRef]
- Gradhand, U.; Kim, R.B. Pharmacogenomics of MRP Transporters (ABCC1-5) and BCRP (ABCG2). Drug Metab. Rev. 2008, 40, 317–354. [Google Scholar] [CrossRef]
- Urquhart, B.L.; Ware, J.A.; Tirona, R.G.; Ho, R.H.; Leake, B.F.; Schwarz, U.I.; Zaher, H.; Palandra, J.; Gregor, J.C.; Dresser, G.K.; et al. Breast Cancer Resistance Protein (ABCG2) and Drug Disposition: Intestinal Expression, Polymorphisms and Sulfasalazine as an in Vivo Probe. Pharm. Genom. 2008, 18, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-K.; Yoon, J.-H.; Woo, J.K.; Kang, J.-H.; Lee, K.-R.; Oh, S.H.; Chung, S.-J.; Maeng, H.-J. Quercetin Is a Flavonoid Breast Cancer Resistance Protein Inhibitor with an Impact on the Oral Pharmacokinetics of Sulfasalazine in Rats. Pharmaceutics 2020, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Slitt, A.L.; Leazer, T.M.; Maher, J.M.; Klaassen, C.D. Tissue Distribution and Hormonal Regulation of the Breast Cancer Resistance Protein (Bcrp/Abcg2) in Rats and Mice. Biochem. Biophys. Res. Commun. 2005, 326, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Johnson, K.; Billington, S.; Lee, C.; Chung, G.W.; Brown, C.D.A.; Kelly, E.J.; Himmelfarb, J.; Unadkat, J.D. Abundance of Drug Transporters in the Human Kidney Cortex as Quantified by Quantitative Targeted Proteomics. Drug Metab. Dispos. 2016, 44, 1920–1924. [Google Scholar] [CrossRef]
- Brandoni, A.; Villar, S.R.; Torres, A.M. Gender-Related Differences in the Pharmacodynamics of Furosemide in Rats. Pharmacology 2004, 70, 107–112. [Google Scholar] [CrossRef]
- Magallanes, L.; Fagiolino, P.; Vázquez, M.; Fotaki, N.; Ibarra, M.; Lorier, M.; Bértola, V.; Barindelli, A. Influence of Food and Sex on the Pharmacokinetics and Pharmacodynamics of Furosemide. Curr. Top. Pharmacol. 2016, 20, 45–56. [Google Scholar]
- Loos, W.J.; Gelderblom, H.J.; Verweij, J.; Brouwer, E.; de Jonge, M.J.; Sparreboom, A. Gender-Dependent Pharmacokinetics of Topotecan in Adult Patients. Anticancer Drugs 2000, 11, 673–680. [Google Scholar] [CrossRef]
- Godfrey, C.; Sweeney, K.; Miller, K.; Hamilton, R.; Kremer, J. The Population Pharmacokinetics of Long-Term Methotrexate in Rheumatoid Arthritis. Br. J. Clin. Pharmacol. 1998, 46, 369–376. [Google Scholar] [CrossRef]
- Wall, A.M.; Gajjar, A.; Link, A.; Mahmoud, H.; Pui, C.H.; Relling, M.V. Individualized Methotrexate Dosing in Children with Relapsed Acute Lymphoblastic Leukemia. Leukemia 2000, 14, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbs, N.A.; Twelves, C.J.; Gillies, H.; James, C.A.; Harper, P.G.; Rubens, R.D. Gender Affects Doxorubicin Pharmacokinetics in Patients with Normal Liver Biochemistry. Cancer Chemother. Pharmacol. 1995, 36, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.R.; Kelman, A.W.; Kerr, D.J.; Robert, J.; Whiting, B. Variability in the Pharmacokinetics of Epirubicin: A Population Analysis. Cancer Chemother. Pharmacol. 1992, 29, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Cerrutti, J.A.; Brandoni, A.; Quaglia, N.B.; Torres, A.M. Sex Differences in P-Aminohippuric Acid Transport in Rat Kidney: Role of Membrane Fluidity and Expression of OAT1. Mol. Cell. Biochem. 2002, 233, 175–179. [Google Scholar] [CrossRef]
- Kelly, M.R.; Cutler, R.E.; Forrey, A.W.; Kimpel, B.M. Pharmacokinetics of Orally Administered Furosemide. Clin. Pharmacol. Ther. 1974, 15, 178–186. [Google Scholar]
- Hammarlund, M.M.; Odlind, B.; Paalzow, L.K. Acute Tolerance to Furosemide Diuresis in Humans. Pharmacokinetic-Pharmacodynamic Modeling. J. Pharmacol. Exp. Ther. 1985, 233, 447–453. [Google Scholar]
- Ogata, H.; Kawatsu, Y.; Maruyama, Y.; Machida, K.; Haga, T. Bioavailability and Diuretic Effect of Furosemide during Long-Term Treatment of Chronic Respiratory Failure. Eur. J. Clin. Pharmacol. 1985, 28, 53–59. [Google Scholar] [CrossRef]
- Beermann, B.; Midskov, C. Reduced Bioavailability and Effect of Furosemide given with Food. Eur. J. Clin. Pharmacol. 1986, 29, 725–727. [Google Scholar] [CrossRef]
- Paintaud, G.; Alván, G.; Eckernäs, S.A.; Wakelkamp, M.; Grahnén, A. The Influence of Food Intake on the Effect of Two Controlled Release Formulations of Furosemide. Biopharm. Drug Dispos. 1995, 16, 221–232. [Google Scholar] [CrossRef]
- McCrindle, J.L.; Li Kam Wa, T.C.; Barron, W.; Prescott, L.F. Effect of Food on the Absorption of Frusemide and Bumetanide in Man. Br. J. Clin. Pharmacol. 1996, 42, 743–746. [Google Scholar] [CrossRef]
- Bard, R.L.; Bleske, B.E.; Nicklas, J.M. Food: An Unrecognized Source of Loop Diuretic Resistance. Pharmacotherapy 2004, 24, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Chungi, V.S.; Dittert, L.W.; Smith, R.B. Gastrointestinal Sites of Furosemide Absorption in Rats. Int. J. Pharm. 1979, 4, 27–38. [Google Scholar] [CrossRef]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of Rat and Mouse Gastrointestinal PH, Fluid and Lymphoid Tissue, and Implications for in-Vivo Experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, K.; Oka, M.; Soda, H.; Yoshikawa, M.; Ikegami, Y.; Tsurutani, J.; Nakatomi, K.; Nakamura, Y.; Doi, S.; Kitazaki, T.; et al. Reversal of Breast Cancer Resistance Protein (BCRP/ABCG2)-Mediated Drug Resistance by Novobiocin, a Coumermycin Antibiotic. Int. J. Cancer 2004, 108, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Hirano, H.; Nakagawa, H.; Fukami, T.; Oosumi, K.; Murakami, K.; Kimura, H.; Kouchi, T.; Konomi, M.; Tao, E.; et al. A New Strategy of High-Speed Screening and Quantitative Structure-Activity Relationship Analysis to Evaluate Human ATP-Binding Cassette Transporter ABCG2-Drug Interactions. J. Pharmacol. Exp. Ther. 2006, 317, 1114–1124. [Google Scholar] [CrossRef]
- Su, Y.; Hu, P.; Lee, S.-H.; Sinko, P.J. Using Novobiocin as a Specific Inhibitor of Breast Cancer Resistant Protein to Assess the Role of Transporter in the Absorption and Disposition of Topotecan. J. Pharm. Pharm. Sci. 2007, 10, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Elsby, R.; Martin, P.; Surry, D.; Sharma, P.; Fenner, K. Solitary Inhibition of the Breast Cancer Resistance Protein Efflux Transporter Results in a Clinically Significant Drug-Drug Interaction with Rosuvastatin by Causing up to a 2-Fold Increase in Statin Exposure. Drug Metab. Dispos. 2016, 44, 398–408. [Google Scholar] [CrossRef]
- Suzuki, K.; Taniyama, K.; Aoyama, T.; Watanabe, Y. Usefulness of Novobiocin as a Selective Inhibitor of Intestinal Breast Cancer Resistance Protein (Bcrp) in Rats. Xenobiotica 2020, 50, 1121–1127. [Google Scholar] [CrossRef]
- Duan, P.; You, G. Novobiocin Is a Potent Inhibitor for Human Organic Anion Transporters. Drug Metab. Dispos. 2009, 37, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm (accessed on 25 September 2022).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Mettu, V.S.; Prasad, B. Interplay of Breast Cancer Resistance Protein (Bcrp/Abcg2), Sex, and Fed State in Oral Pharmacokinetic Variability of Furosemide in Rats. Pharmaceutics 2023, 15, 542. https://doi.org/10.3390/pharmaceutics15020542
Sharma S, Mettu VS, Prasad B. Interplay of Breast Cancer Resistance Protein (Bcrp/Abcg2), Sex, and Fed State in Oral Pharmacokinetic Variability of Furosemide in Rats. Pharmaceutics. 2023; 15(2):542. https://doi.org/10.3390/pharmaceutics15020542
Chicago/Turabian StyleSharma, Sheena, Vijaya Saradhi Mettu, and Bhagwat Prasad. 2023. "Interplay of Breast Cancer Resistance Protein (Bcrp/Abcg2), Sex, and Fed State in Oral Pharmacokinetic Variability of Furosemide in Rats" Pharmaceutics 15, no. 2: 542. https://doi.org/10.3390/pharmaceutics15020542