
Mechanisms of a Mycobacterium tuberculosis Active Peptide
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptide
2.2. Bacteria
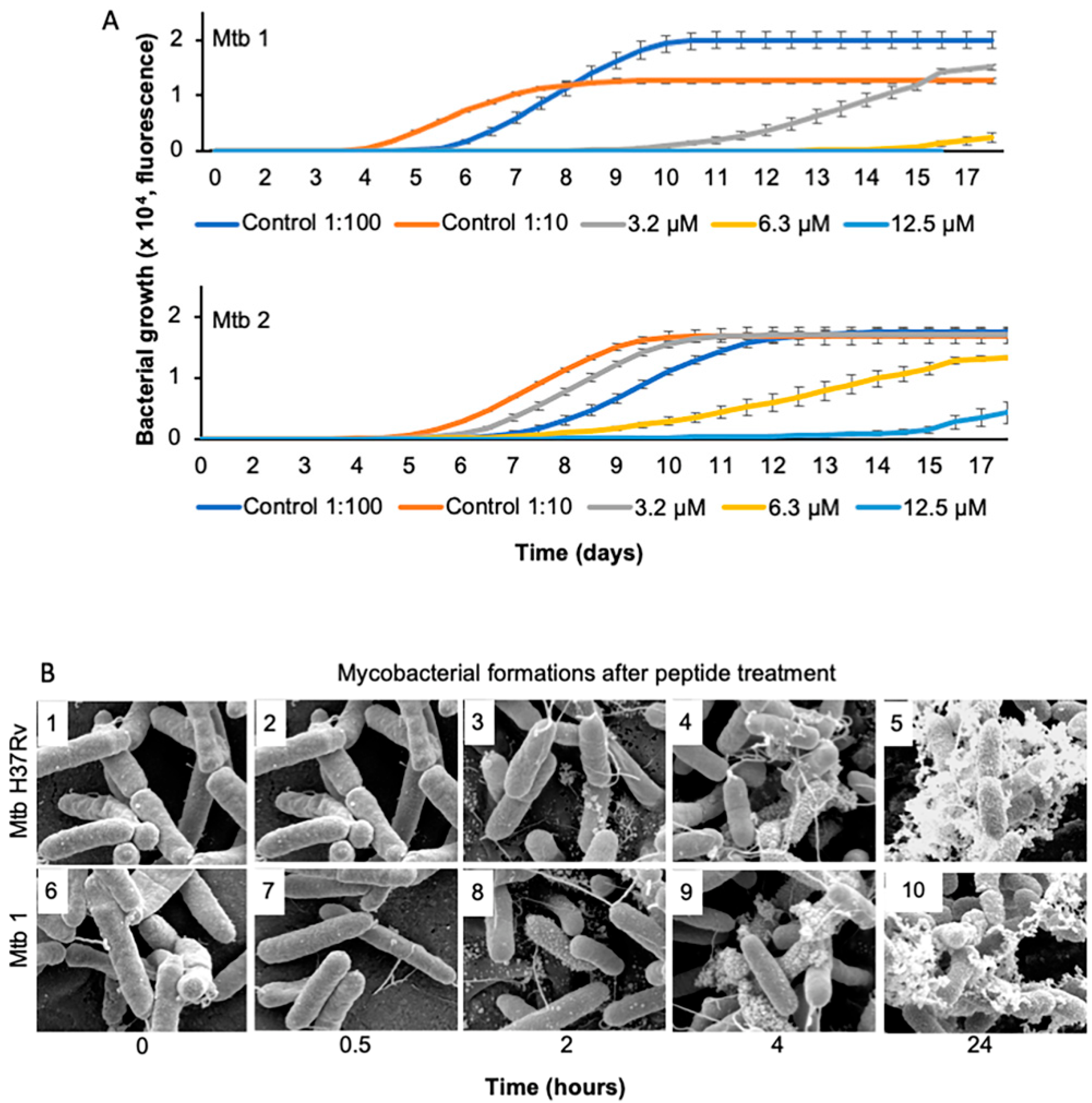
2.3. Growth Kinetics
2.4. Scanning Electron Microscopy
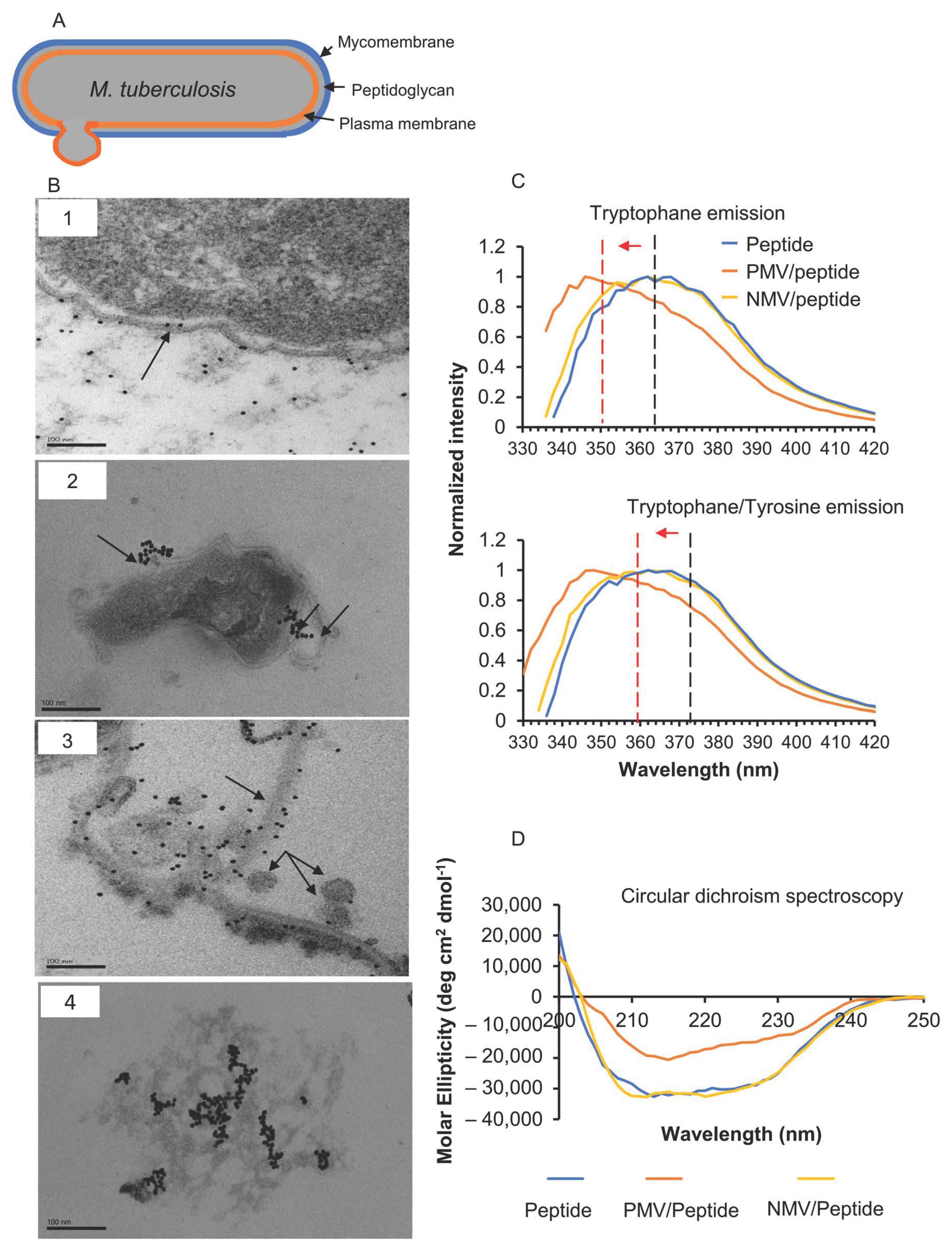
2.5. Transmission Electron Microscopy
2.6. Preparation of Large Unilamellar Vesicles
2.7. Circular Dichroism Spectroscopy
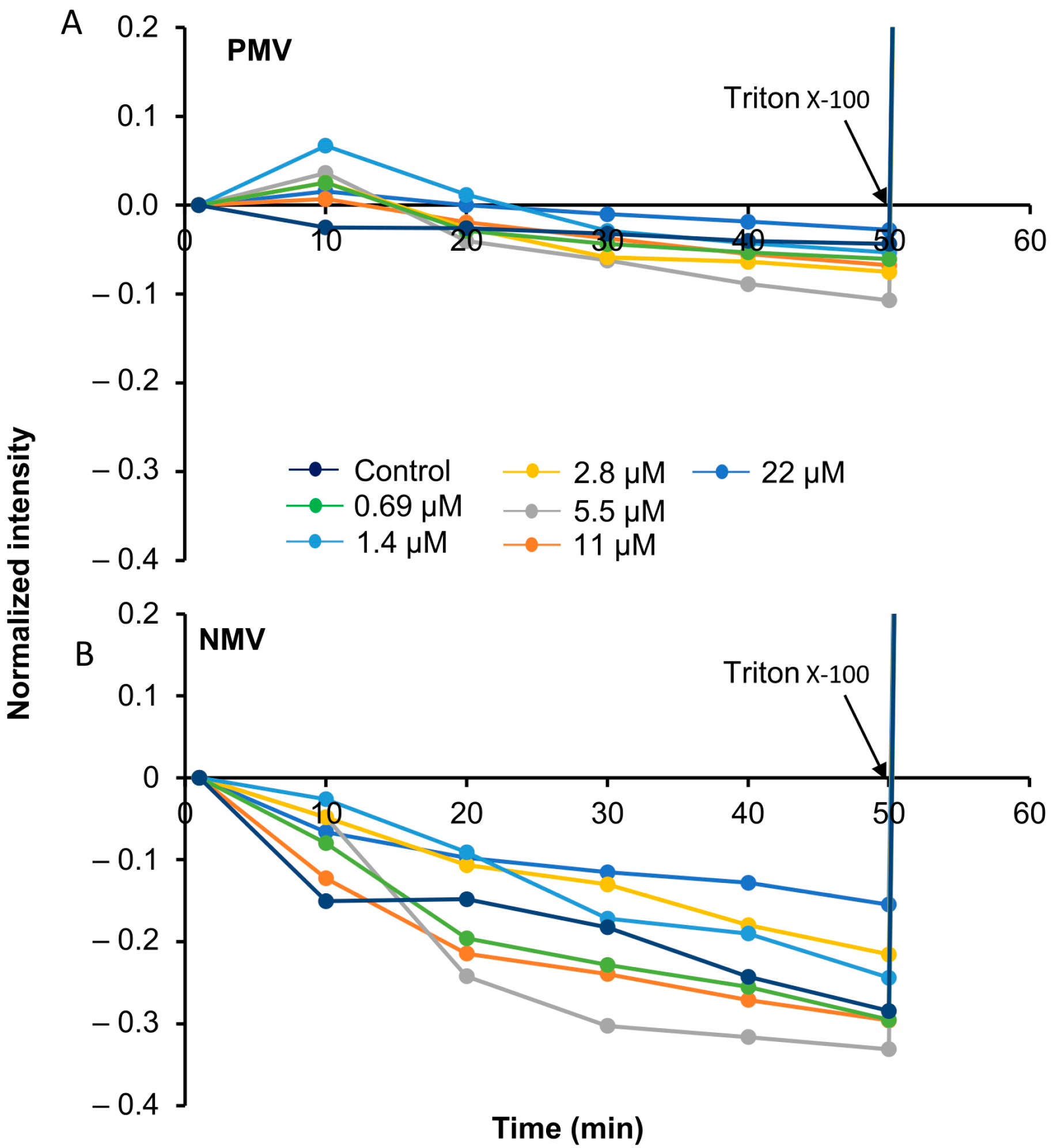
2.8. Calcein Experiment
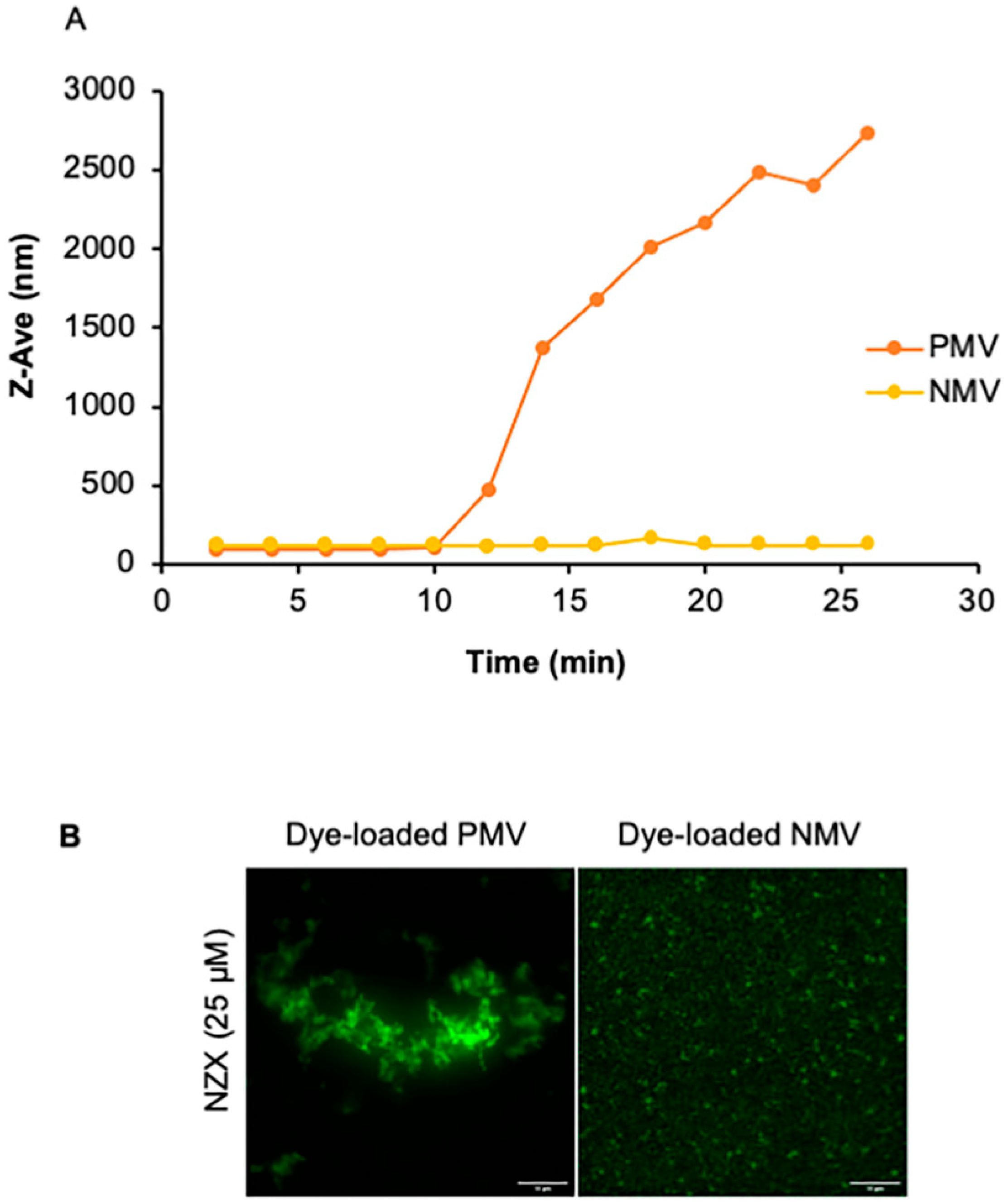
2.9. Dynamic Light Scattering
2.10. Intrinsic Tyrosine and Tryptophan Fluorescence Measurement
2.11. Labelling and Co-Immunoprecipitation of the Peptide
2.12. Sample Preparation for LC–MS/MS
2.13. Mass Spectrometry Acquisition
3. Results
3.1. The Peptide Interacts with M. tuberculosis Membrane
3.2. The Peptide Affects Mycobacterial Inner Membrane
3.3. Interfacial Binding of the Peptide to Lipid Membranes
3.4. Changes in Peptide Secondary Structure upon Interaction with Gram-Positive Membranes
3.5. Peptide Interactions Do Not Rupture the Membrane
3.6. The Peptide Causes Membrane Aggregations
3.7. The Peptide Targets Essential Proteins in Mycobacteria
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Boldrin, F.; Provvedi, R.; Cioetto Mazzabo, L.; Segafreddo, G.; Manganelli, R. Tolerance and Persistence to Drugs: A Main Challenge in the Fight Against Mycobacterium tuberculosis. Front. Microbiol. 2020, 11, 1924. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis (accessed on 4 January 2023).
- Zignol, M.; Dean, A.S.; Falzon, D.; van Gemert, W.; Wright, A.; van Deun, A.; Portaels, F.; Laszlo, A.; Espinal, M.A.; Pablos-Mendez, A.; et al. Twenty Years of Global Surveillance of Antituberculosis-Drug Resistance. N. Engl. J. Med. 2016, 375, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, G.V.; Keller, P.M.; Stucki, D.; Trauner, A.; Borrell, S.; Latshang, T.; Coscolla, M.; Rothe, T.; Homke, R.; Ritter, C.; et al. Acquired Resistance to Bedaquiline and Delamanid in Therapy for Tuberculosis. N. Engl. J. Med. 2015, 373, 1986–1988. [Google Scholar] [CrossRef]
- Tenland, E.; Krishnan, N.; Ronnholm, A.; Kalsum, S.; Puthia, M.; Morgelin, M.; Davoudi, M.; Otrocka, M.; Alaridah, N.; Glegola-Madejska, I.; et al. A novel derivative of the fungal antimicrobial peptide plectasin is active against Mycobacterium tuberculosis. Tuberculosis 2018, 113, 231–238. [Google Scholar] [CrossRef]
- Rao, K.U.; Henderson, D.I.; Krishnan, N.; Puthia, M.; Glegola-Madejska, I.; Brive, L.; Bjarnemark, F.; Millqvist Fureby, A.; Hjort, K.; Andersson, D.I.; et al. A broad spectrum anti-bacterial peptide with an adjunct potential for tuberculosis chemotherapy. Sci. Rep. 2021, 11, 4201. [Google Scholar] [CrossRef]
- Tenland, E.; Pochert, A.; Krishnan, N.; Umashankar Rao, K.; Kalsum, S.; Braun, K.; Glegola-Madejska, I.; Lerm, M.; Robertson, B.D.; Linden, M.; et al. Effective delivery of the anti-mycobacterial peptide NZX in mesoporous silica nanoparticles. PLoS ONE 2019, 14, e0212858. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sonksen, C.P.; Ludvigsen, S.; Raventos, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef]
- Schneider, T.; Kruse, T.; Wimmer, R.; Wiedemann, I.; Sass, V.; Pag, U.; Jansen, A.; Nielsen, A.K.; Mygind, P.H.; Raventos, D.S.; et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 2010, 328, 1168–1172. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.S.; Hansford, K.A.; Blaskovich, M.A.; Halai, R.; Cooper, M.A. Glycopeptide antibiotics: Back to the future. J. Antibiot. 2014, 67, 631–644. [Google Scholar] [CrossRef]
- Soetaert, K.; Rens, C.; Wang, X.M.; De Bruyn, J.; Laneelle, M.A.; Laval, F.; Lemassu, A.; Daffe, M.; Bifani, P.; Fontaine, V.; et al. Increased Vancomycin Susceptibility in Mycobacteria: A New Approach to Identify Synergistic Activity against Multidrug-Resistant Mycobacteria. Antimicrob. Agents Chemother. 2015, 59, 5057–5060. [Google Scholar] [CrossRef]
- Snewin, V.A.; Gares, M.P.; Gaora, P.O.; Hasan, Z.; Brown, I.N.; Young, D.B. Assessment of immunity to mycobacterial infection with luciferase reporter constructs. Infect. Immun. 1999, 67, 4586–4593. [Google Scholar] [CrossRef]
- Sturegard, E.; Angeby, K.A.; Werngren, J.; Jureen, P.; Kronvall, G.; Giske, C.G.; Kahlmeter, G.; Schon, T. Little difference between minimum inhibitory concentrations of Mycobacterium tuberculosis wild-type organisms determined with BACTEC MGIT 960 and Middlebrook 7H10. Clin. Microbiol. Infect. 2015, 21, 148.e5–148.e7. [Google Scholar] [CrossRef]
- Svensson, L.; Baumgarten, M.; Morgelin, M.; Shannon, O. Platelet activation by Streptococcus pyogenes leads to entrapment in platelet aggregates, from which bacteria subsequently escape. Infect. Immun. 2014, 82, 4307–4314. [Google Scholar] [CrossRef]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta 2007, 1768, 2500–2509. [Google Scholar] [CrossRef]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 2012, 80, 374–381. [Google Scholar] [CrossRef]
- Governa, V.; Talbot, H.; Goncalves de Oliveira, K.; Cerezo-Magana, M.; Bang-Rudenstam, A.; Johansson, M.C.; Mansson, A.S.; Forsberg-Nilsson, K.; Marko-Varga, G.; Enriquez Perez, J.; et al. Landscape of surfaceome and endocytome in human glioma is divergent and depends on cellular spatial organization. Proc. Natl. Acad. Sci. USA 2022, 119, e2114456119. [Google Scholar] [CrossRef]
- Nagakubo, T.; Tahara, Y.O.; Miyata, M.; Nomura, N.; Toyofuku, M. Mycolic acid-containing bacteria trigger distinct types of membrane vesicles through different routes. iScience 2021, 24, 102015. [Google Scholar] [CrossRef]
- Prados-Rosales, R.; Baena, A.; Martinez, L.R.; Luque-Garcia, J.; Kalscheuer, R.; Veeraraghavan, U.; Camara, C.; Nosanchuk, J.D.; Besra, G.S.; Chen, B.; et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J. Clin. Invest. 2011, 121, 1471–1483. [Google Scholar] [CrossRef] [Green Version]
- Prados-Rosales, R.; Weinrick, B.C.; Pique, D.G.; Jacobs, W.R., Jr.; Casadevall, A.; Rodriguez, G.M. Role for Mycobacterium tuberculosis membrane vesicles in iron acquisition. J. Bacteriol. 2014, 196, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Batt, S.M.; Minnikin, D.E.; Besra, G.S. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem. J. 2020, 477, 1983–2006. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef]
- Modak, B.; Girkar, S.; Narayan, R.; Kapoor, S. Mycobacterial Membranes as Actionable Targets for Lipid-Centric Therapy in Tuberculosis. J. Med. Chem. 2022, 65, 3046–3065. [Google Scholar] [CrossRef]
- Vincent, A.T.; Nyongesa, S.; Morneau, I.; Reed, M.B.; Tocheva, E.I.; Veyrier, F.J. The Mycobacterial Cell Envelope: A Relict from the Past or the Result of Recent Evolution? Front. Microbiol. 2018, 9, 2341. [Google Scholar] [CrossRef]
- Roberts, K.E.; O’Keeffe, A.K.; Lloyd, C.J.; Clarke, D.J. Selective Dequenching by Photobleaching Increases Fluorescence Probe Visibility. J. Fluoresc. 2003, 13, 513–517. [Google Scholar] [CrossRef]
- Toyofuku, M.; Carcamo-Oyarce, G.; Yamamoto, T.; Eisenstein, F.; Hsiao, C.C.; Kurosawa, M.; Gademann, K.; Pilhofer, M.; Nomura, N.; Eberl, L. Prophage-triggered membrane vesicle formation through peptidoglycan damage in Bacillus subtilis. Nat. Commun. 2017, 8, 481. [Google Scholar] [CrossRef]
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.R.; Teixeira, C.; Sousa, C.F.; Bessa, L.J.; Gomes, P.; Gameiro, P. How Insertion of a Single Tryptophan in the N-Terminus of a Cecropin A-Melittin Hybrid Peptide Changes Its Antimicrobial and Biophysical Profile. Membranes 2021, 11, 48. [Google Scholar] [CrossRef]
- Chawla, M.; Mishra, S.; Anand, K.; Parikh, P.; Mehta, M.; Vij, M.; Verma, T.; Singh, P.; Jakkala, K.; Verma, H.N.; et al. Redox-dependent condensation of the mycobacterial nucleoid by WhiB4. Redox Biol. 2018, 19, 116–133. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, X.; Teng, D.; Mao, R.; Hao, Y.; Yang, N.; Zong, L.; Wang, J. Mode of action of plectasin-derived peptides against gas gangrene-associated Clostridium perfringens type A. PLoS ONE 2017, 12, e0185215. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J.; Liang, D. Aggregation, fusion, and leakage of liposomes induced by peptides. Langmuir 2014, 30, 7334–7342. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 7439. [Google Scholar] [CrossRef]
- D’Este, F.; Benincasa, M.; Cannone, G.; Furlan, M.; Scarsini, M.; Volpatti, D.; Gennaro, R.; Tossi, A.; Skerlavaj, B.; Scocchi, M. Antimicrobial and host cell-directed activities of Gly/Ser-rich peptides from salmonid cathelicidins. Fish Shellfish Immunol. 2016, 59, 456–468. [Google Scholar] [CrossRef]
- Mardirossian, M.; Perebaskine, N.; Benincasa, M.; Gambato, S.; Hofmann, S.; Huter, P.; Muller, C.; Hilpert, K.; Innis, C.A.; Tossi, A.; et al. The Dolphin Proline-Rich Antimicrobial Peptide Tur1A Inhibits Protein Synthesis by Targeting the Bacterial Ribosome. Cell Chem. Biol. 2018, 25, 530–539.e537. [Google Scholar] [CrossRef]
- Mardirossian, M.; Sola, R.; Beckert, B.; Collis, D.W.P.; Di Stasi, A.; Armas, F.; Hilpert, K.; Wilson, D.N.; Scocchi, M. Proline-Rich Peptides with Improved Antimicrobial Activity against E. coli, K. pneumoniae, and A. baumannii. ChemMedChem 2019, 14, 2025–2033. [Google Scholar] [CrossRef]
- Kong, T.H.; Coates, A.R.; Butcher, P.D.; Hickman, C.J.; Shinnick, T.M. Mycobacterium tuberculosis expresses two chaperonin-60 homologs. Proc. Natl. Acad. Sci. USA 1993, 90, 2608–2612. [Google Scholar] [CrossRef]
- Lund, P.A. Microbial molecular chaperones. Adv. Microb. Physiol. 2001, 44, 93–140. [Google Scholar] [CrossRef]
- Ojha, A.; Anand, M.; Bhatt, A.; Kremer, L.; Jacobs, W.R., Jr.; Hatfull, G.F. GroEL1: A dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 2005, 123, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Henderson, B.; Lund, P.A.; Tormay, P.; Ahmed, M.T.; Gurcha, S.S.; Besra, G.S.; Coates, A.R. A Mycobacterium tuberculosis mutant lacking the groEL homologue cpn60.1 is viable but fails to induce an inflammatory response in animal models of infection. Infect. Immun. 2008, 76, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Lu, C.; Soetaert, K.; S’Heeren, C.; Peirs, P.; Laneelle, M.A.; Lefevre, P.; Bifani, P.; Content, J.; Daffe, M.; et al. Biochemical and immunological characterization of a cpn60.1 knockout mutant of Mycobacterium bovis BCG. Microbiology 2011, 157, 1205–1219. [Google Scholar] [CrossRef]
- Wu, M.; Li, M.; Yue, Y.; Xu, W. DNA vaccine with discontinuous T-cell epitope insertions into HSP65 scaffold as a potential means to improve immunogenicity of multi-epitope Mycobacterium tuberculosis vaccine. Microbiol. Immunol. 2016, 60, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Zimhony, O.; Schwarz, A.; Raitses-Gurevich, M.; Peleg, Y.; Dym, O.; Albeck, S.; Burstein, Y.; Shakked, Z. AcpM, the meromycolate extension acyl carrier protein of Mycobacterium tuberculosis, is activated by the 4′-phosphopantetheinyl transferase PptT, a potential target of the multistep mycolic acid biosynthesis. Biochemistry 2015, 54, 2360–2371. [Google Scholar] [CrossRef]
- Lewthwaite, J.C.; Coates, A.R.; Tormay, P.; Singh, M.; Mascagni, P.; Poole, S.; Roberts, M.; Sharp, L.; Henderson, B. Mycobacterium tuberculosis chaperonin 60.1 is a more potent cytokine stimulator than chaperonin 60.2 (Hsp 65) and contains a CD14-binding domain. Infect. Immun. 2001, 69, 7349–7355. [Google Scholar] [CrossRef]
- Cehovin, A.; Coates, A.R.; Hu, Y.; Riffo-Vasquez, Y.; Tormay, P.; Botanch, C.; Altare, F.; Henderson, B. Comparison of the moonlighting actions of the two highly homologous chaperonin 60 proteins of Mycobacterium tuberculosis. Infect. Immun. 2010, 78, 3196–3206. [Google Scholar] [CrossRef]
- Hickey, T.B.; Ziltener, H.J.; Speert, D.P.; Stokes, R.W. Mycobacterium tuberculosis employs Cpn60.2 as an adhesin that binds CD43 on the macrophage surface. Cell Microbiol. 2010, 12, 1634–1647. [Google Scholar] [CrossRef]
- Sajid, A.; Arora, G.; Gupta, M.; Singhal, A.; Chakraborty, K.; Nandicoori, V.K.; Singh, Y. Interaction of Mycobacterium tuberculosis elongation factor Tu with GTP is regulated by phosphorylation. J. Bacteriol. 2011, 193, 5347–5358. [Google Scholar] [CrossRef]
- Brotz-Oesterhelt, H.; Brunner, N.A. How many modes of action should an antibiotic have? Curr. Opin. Pharmacol. 2008, 8, 564–573. [Google Scholar] [CrossRef]
- Gray, D.A.; Wenzel, M. More Than a Pore: A Current Perspective on the In Vivo Mode of Action of the Lipopeptide Antibiotic Daptomycin. Antibiotics 2020, 9, 17. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Coverage % | # Proteins | # PSM | # Unique Peptides |
|---|---|---|---|---|
| Cpn 60.1 | 44 | 16 | 31 | 15 |
| EF-Tu | 48 | 12 | 28 | 11 |
| Cpn 60.2 | 31 | 11 | 18 | 9 |
| AcpM | 24 | 2 | 3 | 2 |
| HCP | 29 | 2 | 8 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, K.U.; Li, P.; Welinder, C.; Tenland, E.; Gourdon, P.; Sturegård, E.; Ho, J.C.S.; Godaly, G. Mechanisms of a Mycobacterium tuberculosis Active Peptide. Pharmaceutics 2023, 15, 540. https://doi.org/10.3390/pharmaceutics15020540
Rao KU, Li P, Welinder C, Tenland E, Gourdon P, Sturegård E, Ho JCS, Godaly G. Mechanisms of a Mycobacterium tuberculosis Active Peptide. Pharmaceutics. 2023; 15(2):540. https://doi.org/10.3390/pharmaceutics15020540
Chicago/Turabian StyleRao, Komal Umashankar, Ping Li, Charlotte Welinder, Erik Tenland, Pontus Gourdon, Erik Sturegård, James C. S. Ho, and Gabriela Godaly. 2023. "Mechanisms of a Mycobacterium tuberculosis Active Peptide" Pharmaceutics 15, no. 2: 540. https://doi.org/10.3390/pharmaceutics15020540