
Potential Oral Anticancer Therapeutic Agents of Hexahydrocurcumin-Encapsulated Chitosan Nanoparticles against MDA-MB-231 Breast Cancer Cells
 , ,
, ,  and
and
Abstract
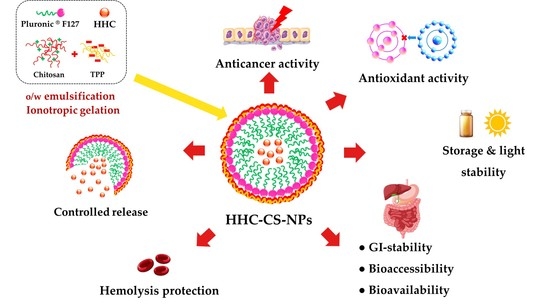
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Design
2.3. Fabrication of HHC-CS-NPs
2.4. Design and Optimization of HHC-CS-NPs
2.5. Characterization
2.5.1. Particle Size, Zeta Potential, Morphology, and Drug–Excipients Interaction
2.5.2. Determination of EE
2.6. In Vitro Drug Release Study
2.6.1. Simulated Gastrointestinal (GI) Conditions
2.6.2. In Simulated Body Fluid Condition
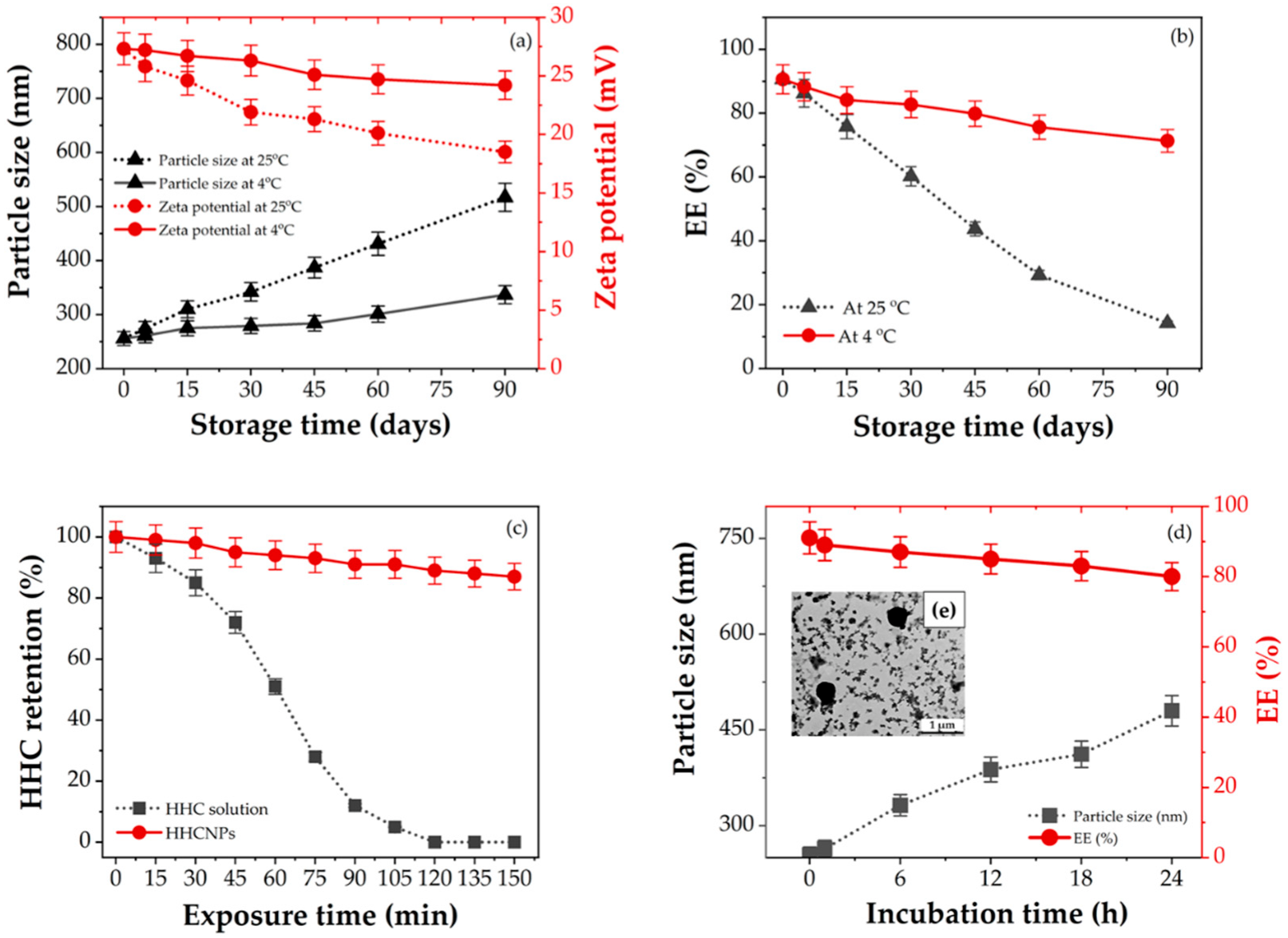
2.7. Determination of Physicochemical and Biological Stability
2.7.1. Storage Stability
2.7.2. Light Stability
2.7.3. In Vitro Protein Stability
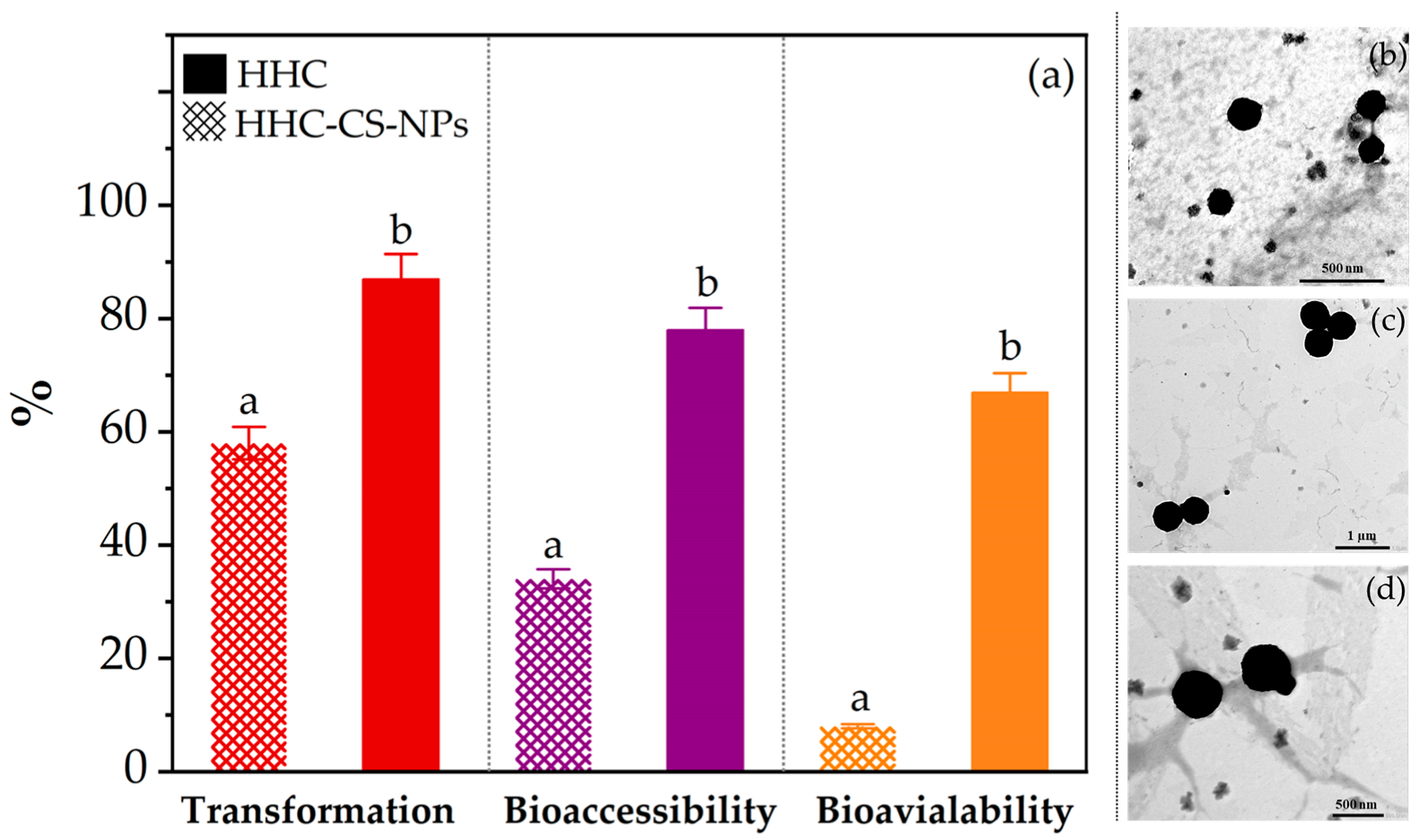
2.8. In Vitro Bioaccessibility and Bioavailability
2.9. In Vitro Antioxidant Capacity
2.9.1. DDPH Scavenging Activity
2.9.2. Hydroxyl (OH•) Scavenging Assay
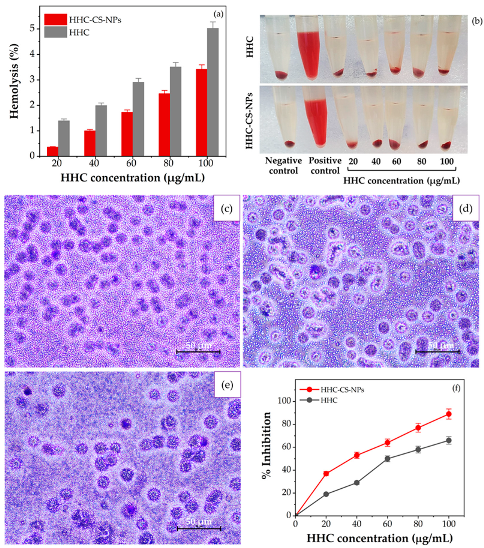
2.10. In Vitro Anti-Inflammatory Activity
2.10.1. Red Blood Cells Membrane Stabilization
2.10.2. Protein Denaturation Determination
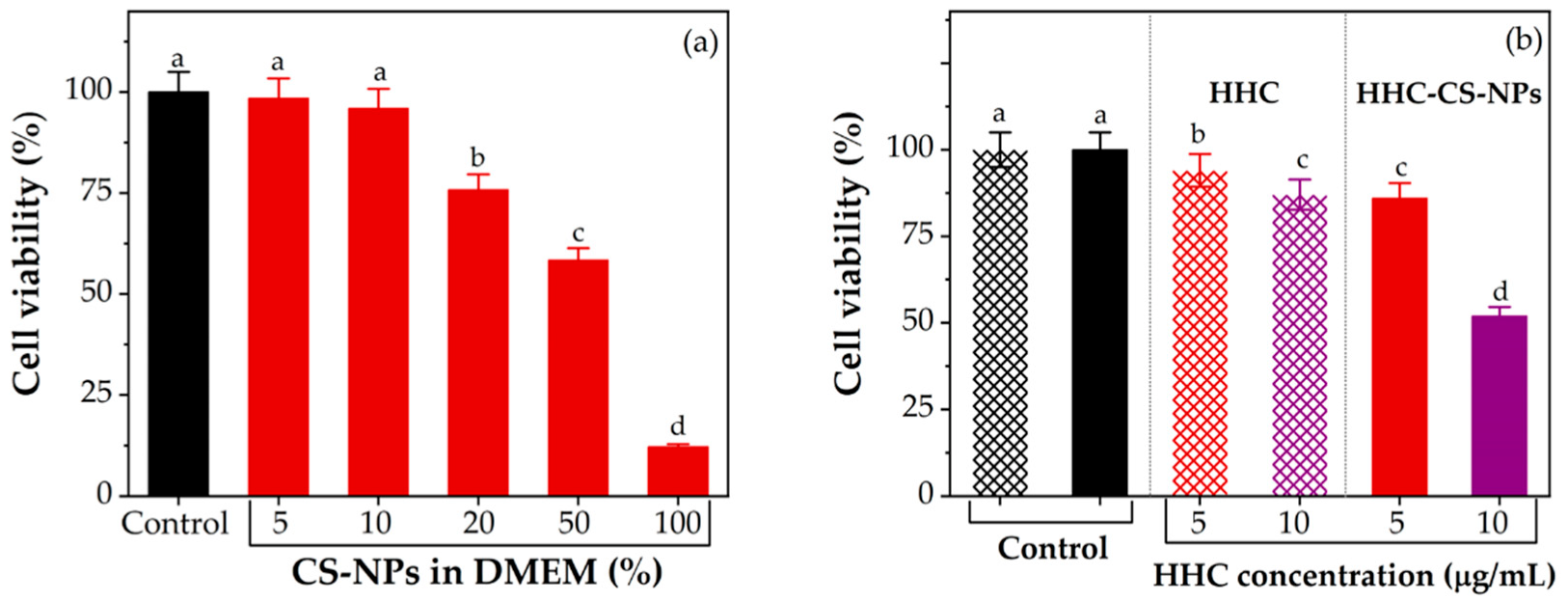
2.11. In Vitro Cytotoxicity
2.11.1. Evaluation of Cytotoxicity of CS-NPs
2.11.2. Evaluation of Cytotoxicity of CS-NPs
2.11.3. Evaluation of Cytotoxicity of HHCNPs
2.12. Western Blot Analysis
2.13. Caspase-3 and -9 Activities Analysis
2.14. Statistical Analysis
3. Results and Discussion
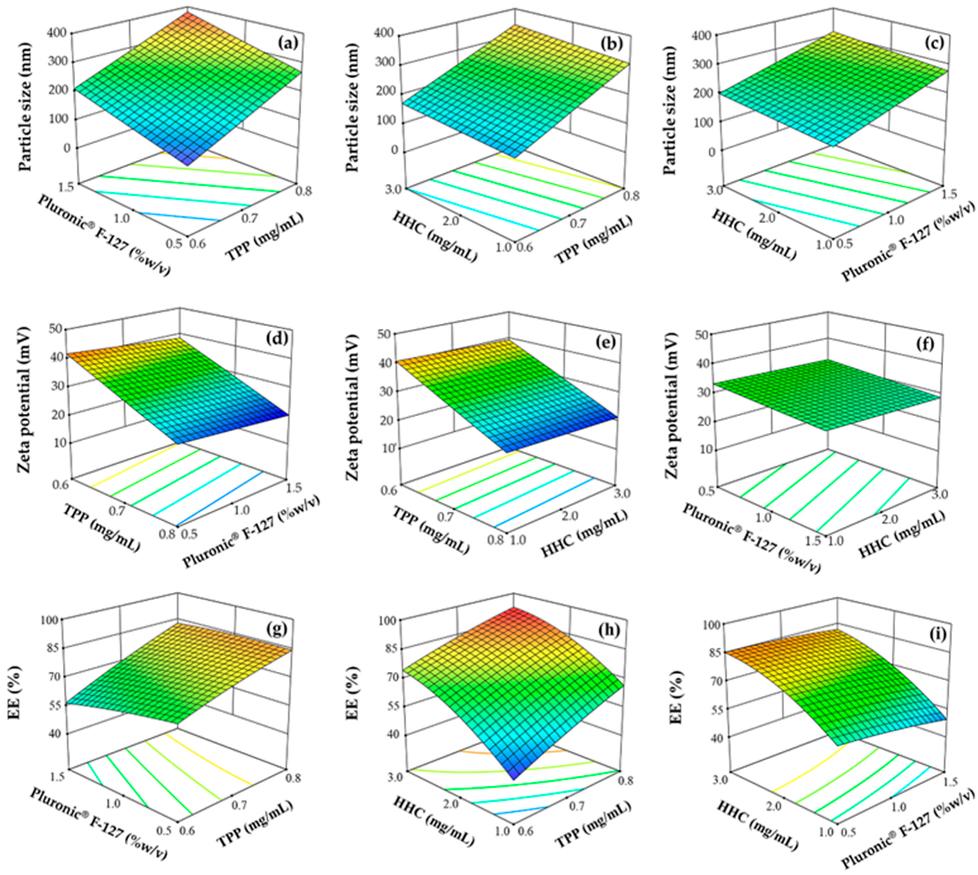
3.1. Statistical Analysis of the BBD
3.1.1. Effect of Factors on Particle Size
3.1.2. Effect of Factors on Zeta Potential
3.1.3. Effect of Factors on EE
3.2. Optimization and Model Validation
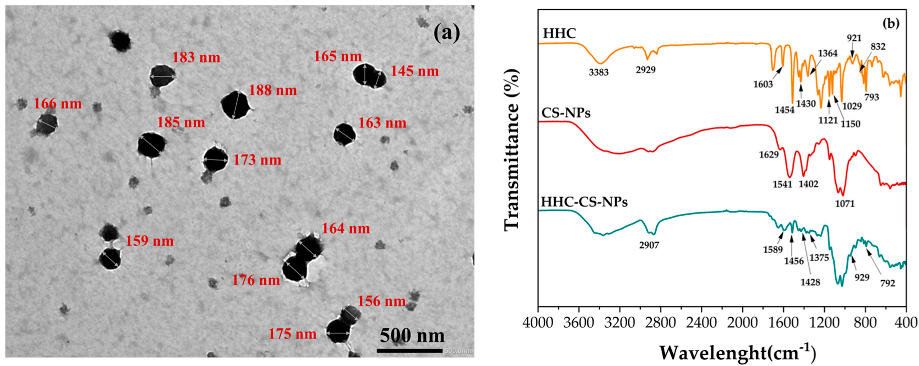
3.3. Characterizations
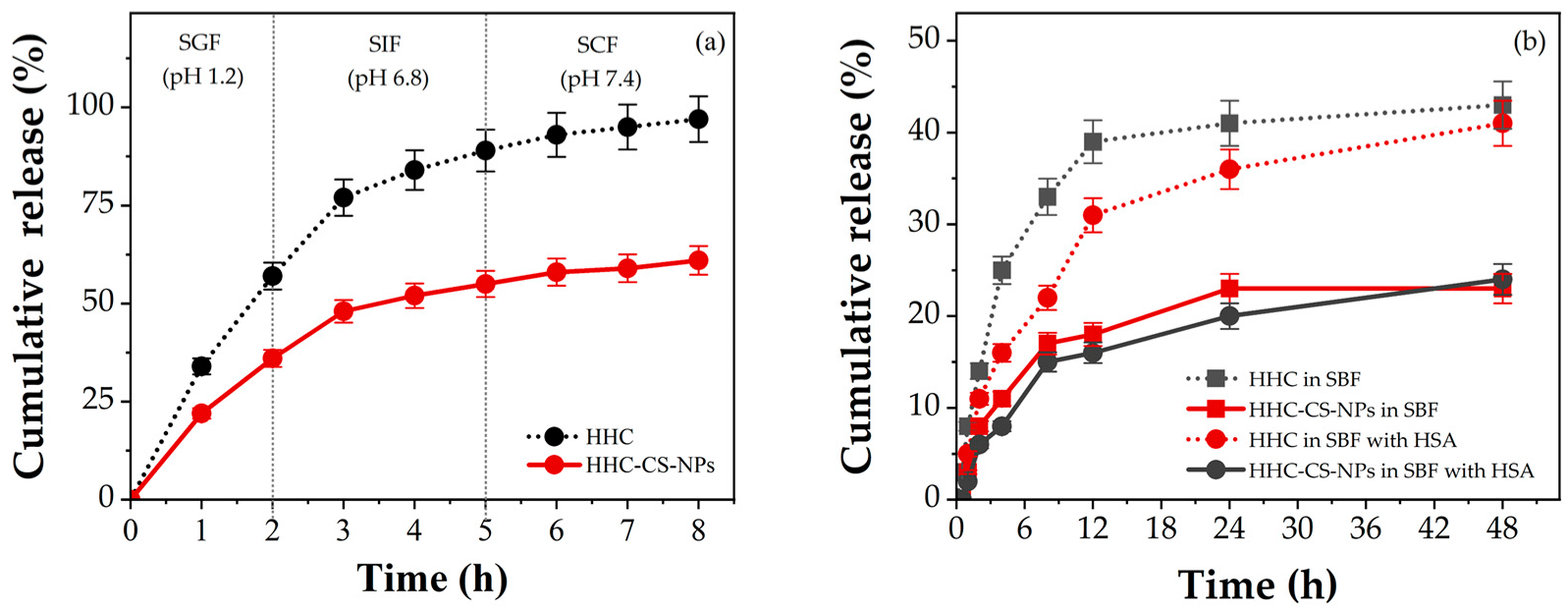
3.4. In Vitro Release Studies
3.5. Stability Studies
3.6. In Vitro Bioaccessibility and Bioavailability
3.7. In Vitro Antioxidant Activity
3.8. In Vitro Anti-Inflammatory Activity
3.9. In Vitro Cytotoxicity
3.10. Western Blot Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 15, 2021. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, A.; Gonsalves, A.; Menon, J.U. Pharmaceutics Current State of Breast Cancer Diagnosis, Treatment, and Theranostics. Pharmaceutics 2021, 13, 723. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.; Andre, F.; Tordai, A.; Mejia, J.; Symmans, W.; Gonzalez-Angulo, A.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. Am. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast Cancer: Biology, Biomarkers, and Treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Almansour, N.M. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef]
- Arunachalam, S.S.; Shetty, A.P.; Panniyadi, N.; Meena, C.; Kumari, J.; Rani, B.; Das, P.; Kumari, S. Study on Knowledge of Chemotherapy’s Adverse Effects and Their Self-Care Ability to Manage—The Cancer Survivors Impact. Clin. Epidemiol. Glob. Health 2021, 11, 100765. [Google Scholar] [CrossRef]
- El-Magboub, A.; Rojsitthisak, P.; Muangnoi, C.; Wichitnithad, W.; Romero, R.M.; Haworth, I.S. Biological Targets and Pharmacology of Curcumin. In Curcumin: Synthesis, Emerging Role in Pain Management and Health Implications; Pouliquen, D.L., Ed.; Nova Science Pub Inc.: Hauppauge, NY, USA, 2014; pp. 103–113. [Google Scholar]
- Truong, T.H.; Alcantara, K.P.; Bulatao, B.P.I.; Sorasitthiyanukarn, F.N.; Muangnoi, C.; Nalinratana, N.; Vajragupta, O.; Rojsitthisak, P.; Rojsitthisak, P. Chitosan-Coated Nanostructured Lipid Carriers for Transdermal Delivery of Tetrahydrocurcumin for Breast Cancer Therapy. Carbohydr. Polym. 2022, 288, 119401. [Google Scholar] [CrossRef]
- Gomez, C.; Muangnoi, C.; Sorasitthiyanukarn, F.N.; Wongpiyabovorn, J.; Rojsitthisak, P.; Rojsitthisak, P. Synergistic Effects of Photo-Irradiation and Curcumin-Chitosan/Alginate Nanoparticles on Tumor Necrosis Factor-Alpha-Induced Psoriasis-like Proliferation of Keratinocytes. Molecules 2019, 24, 1388. [Google Scholar] [CrossRef] [Green Version]
- Goenka, S.; Simon, S.R. Comparative Study of Curcumin and Its Hydrogenated Metabolites, Tetrahydrocurcumin, Hexahydrocurcumin, and Octahydrocurcumin, on Melanogenesis in B16F10 and MNT-1 Cells. Cosmetics 2021, 8, 4. [Google Scholar] [CrossRef]
- Kou, S.G.; Peters, L.; Mucalo, M. Chitosan: A Review of Molecular Structure, Bioactivities and Interactions with the Human Body and Micro-Organisms. Carbohydr. Polym. 2022, 282, 119132. [Google Scholar] [CrossRef]
- Parhi, R. Drug Delivery Applications of Chitin and Chitosan: A Review. Environ. Chem. Lett. 2020, 18, 577–594. [Google Scholar] [CrossRef]
- Sorasitthiyanukarn, F.N.; Muangnoi, C.; Thaweesest, W.; Rojsitthisak, P.; Rojsitthisak, P. Enhanced Cytotoxic, Antioxidant and Anti-Inflammatory Activities of Curcumin Diethyl Disuccinate Using Chitosan-Tripolyphosphate Nanoparticles. J. Drug Deliv. Sci. Technol. 2019, 53, 101118. [Google Scholar] [CrossRef]
- Srimuangwong, K.; Tocharus, C.; Chintana, P.Y.; Suksamrarn, A.; Tocharus, J. Hexahydrocurcumin Enhances Inhibitory Effect of 5-Fluorouracil on HT-29 Human Colon Cancer Cells. World J. Gastroenterol. 2012, 18, 2383–2389. [Google Scholar] [CrossRef]
- Calvo, P.; Remuñán-López, C.; Vila-Jato, J.L.; Alonso, M.J. Novel Hydrophilic Chitosan—Polyethylene Oxide Nanoparticles as Protein Carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Rasband, W. ImageJ—Image Processing and Analysis in Java; National Institute of Health: Bethesda, MD, USA, 2011. Available online: http://imagej.nih.gov/ij (accessed on 14 January 2023).
- Omer, A.M.; Ahmed, M.S.; El-Subruiti, G.M.; Khalifa, R.E.; Eltaweil, A.S. Ph-Sensitive Alginate/Carboxymethyl Chitosan/Aminated Chitosan Microcapsules for Efficient Encapsulation and Delivery of Diclofenac Sodium. Pharmaceutics 2021, 13, 338. [Google Scholar] [CrossRef]
- Riekes, M.K.; Kuminek, G.; Rauber, G.S.; Cuffini, S.L.; Stulzer, H.K. Development and Validation of an Intrinsic Dissolution Method for Nimodipine Polymorphs. Cent Eur. J. Chem. 2014, 12, 549–556. [Google Scholar] [CrossRef]
- Suvarna, M.; Dyawanapelly, S.; Kansara, B.; Dandekar, P.; Jain, R. Understanding the Stability of Nanoparticle–Protein Interactions: Effect of Particle Size on Adsorption, Conformation and Thermodynamic Properties of Serum Albumin Proteins. ACS Appl. Nano Mater. 2018, 1, 5524–5535. [Google Scholar] [CrossRef]
- Price, M.E.; Cornelius, R.M.; Brash, J.L. Protein Adsorption to Polyethylene Glycol Modified Liposomes from Fibrinogen Solution and from Plasma. Biochim. Biophys. Acta 2001, 1512, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An Add-in Program for Modeling and Comparison of Drug Dissolution Profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- ICH Q1A(R2) International Conference on Harmonization (ICH). Guidance for Industry: Q1A(R2) Stability Testing of New Drug Substances and Products. Ich Harmon. Tripart. Guidel. 2003, 4, 24. [Google Scholar]
- Li, M.; Zahi, M.R.; Yuan, Q.; Tian, F.; Liang, H. Preparation and Stability of Astaxanthin Solid Lipid Nanoparticles Based on Stearic Acid. Eur. J. Lipid Sci. Technol. 2016, 118, 592–602. [Google Scholar] [CrossRef]
- Shah, B.R.; Zhang, C.; Li, Y.; Li, B. Bioaccessibility and Antioxidant Activity of Curcumin after Encapsulated by Nano and Pickering Emulsion Based on Chitosan-Tripolyphosphate Nanoparticles. Food Res. Int. 2016, 89, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, F.; Perugini, L.; Marconi, E.; Messia, M.C.; Lopez, F. Enhanced Curcumin Bioavailability through Nonionic Surfactant/Caseinate Mixed Nanoemulsions. J. Food Sci. 2019, 84, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Chen, X.D. Validation of in vitro Bioaccessibility Assays—A Key Aspect in the Rational Design of Functional Foods towards Tailored Bioavailability. Curr. Opin. Food Sci. 2021, 39, 160–170. [Google Scholar] [CrossRef]
- Etcheverry, P.; Grusak, M.A.; Fleige, L.E. Application of in vitro Bioaccessibility and Bioavailability Methods for Calcium, Carotenoids, Folate, Iron, Magnesium, Polyphenols, Zinc, and Vitamins B6, B12, D, and E. Front. Physiol. 2012, 3, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Brake, K.; Gumireddy, A.; Tiwari, A.; Chauhan, H.; Kumari, D. In vivo Studies for Drug Development via Oral Delivery: Challenges, Animal Models and Techniques. Pharm. Anal. Acta 2017, 08, 560. [Google Scholar] [CrossRef]
- Dima, C.; Assadpour, E.; Dima, S.; Jafari, S.M. Nutraceutical Nanodelivery; an Insight into the Bioaccessibility/Bioavailability of Different Bioactive Compounds Loaded within Nanocarriers. Crit. Rev. Food Sci. Nutr. 2020, 61, 3031–3065. [Google Scholar] [CrossRef]
- Navarro-Hoyos, M.; Alvarado-Corella, D.; Moreira-Gonzalez, I.; Arnaez-Serrano, E.; Monagas-Juan, M. Polyphenolic Composition and Antioxidant Activity of Aqueous and Ethanolic Extracts from Uncaria Tomentosa Bark and Leaves. Antioxidants 2018, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Bertin, R.; Froldi, G. EC50 Estimation of Antioxidant Activity in DPPH* Assay Using Several Statistical Programs. Food Chem. 2013, 138, 414–420. [Google Scholar] [CrossRef]
- Lei, K.; Wei, W.; Liu, S.; Zhou, M.; Lin, X.; Cao, X. In Vitro Antioxidant Activity of the Anthocyanins in Sageretia Theezans Brongn Fruit. Int. J. Food Prop. 2016, 19, 210–221. [Google Scholar] [CrossRef]
- Jiang, L.; Yu, Y.; Li, Y.; Yu, Y.; Duan, J.; Zou, Y.; Li, Q.; Sun, Z. Oxidative Damage and Energy Metabolism Disorder Contribute to the Hemolytic Effect of Amorphous Silica Nanoparticles. Nanoscale Res. Lett. 2016, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Gunathilake, K.D.D.P.; Ranaweera, K.K.D.S.; Vasantha Rupasinghe, H.P. In vitro Anti-Inflammatory Properties of Selected Green Leafy Vegetables. Biomedicines 2018, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Muangnoi, C.; Ratnatilaka Na Bhuket, P.; Jithavech, P.; Supasena, W.; Paraoan, L.; Patumraj, S.; Rojsitthisak, P. Curcumin Diethyl Disuccinate, a Prodrug of Curcumin, Enhances Anti-Proliferative Effect of Curcumin against HepG2 Cells via Apoptosis Induction. Sci. Rep. 2019, 9, 11718. [Google Scholar] [CrossRef] [Green Version]
- Subhan, A.; Siva, S.; Yalamarty, K.; Filipczak, N.; Parveen, F.; Torchilin, V.P. Recent Advances in Tumor Targeting via EPR Effect for Cancer Treatment. J. Pers. Med. 2021, 11, 571. [Google Scholar] [CrossRef]
- Delan, W.K.; Zakaria, M.; Elsaadany, B.; ElMeshad, A.N.; Mamdouh, W.; Fares, A.R. Formulation of Simvastatin Chitosan Nanoparticles for Controlled Delivery in Bone Regeneration: Optimization Using Box-Behnken Design, Stability and in vivo Study. Int. J. Pharm 2020, 577, 119038. [Google Scholar] [CrossRef]
- García-Couce, J.; Tomás, M.; Fuentes, G.; Que, I.; Almirall, A.; Cruz, L.J. Chitosan/Pluronic F127 Thermosensitive Hydrogel as an Injectable Dexamethasone Delivery Carrier. Gels 2022, 8, 44. [Google Scholar] [CrossRef]
- Honary, S.; Zahir, F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems—A Review (Part 2). Trop J. Pharm. Res. 2013, 12, 265–273. [Google Scholar]
- Kathe, N.; Henriksen, B.; Chauhan, H. Physicochemical Characterization Techniques for Solid Lipid Nanoparticles: Principles and Limitations. Drug Dev. Ind. Pharm. 2014, 40, 1565–1575. [Google Scholar] [CrossRef]
- Algharib, S.A.; Dawood, A.; Zhou, K.; Chen, D.; Li, C.; Meng, K.; Zhang, A.; Luo, W.; Ahmed, S.; Huang, L.; et al. Preparation of Chitosan Nanoparticles by Ionotropic Gelation Technique: Effects of Formulation Parameters and in vitro Characterization. J. Mol. Struct. 2022, 1252, 132129. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Le, D.V.; Do, D.H.; Le, Q.H. Development of Chitosan Graft Pluronic® F127 Copolymer Nanoparticles Containing DNA Aptamer for Paclitaxel Delivery to Treat Breast Cancer Cells. Adv. Nat. Sci. Nanosci. Nanotechnol. 2016, 7, 025018. [Google Scholar] [CrossRef]
- Deng, Q.Y.; Zhou, C.R.; Luo, B.H. Preparation and Characterization of Chitosan Nanoparticles Containing Lysozyme. Pharm. Biol. 2006, 44, 336–342. [Google Scholar] [CrossRef]
- Rosyada, A.; Sunarharum, W.B.; Waziiroh, E. Characterization of Chitosan Nanoparticles as An Edible Coating Material. IOP Conf. Ser. Earth Environ. Sci. 2019, 230, 012043. [Google Scholar] [CrossRef]
- Avadi, M.R.; Sadeghi, A.M.; Mohammadpour, N.; Abedin, S.; Atyabi, F.; Dinarvand, R.; Rafiee-Tehrani, M. Preparation and Characterization of Insulin Nanoparticles using Chitosan and Arabic Gum with Ionic Gelation Method. Nanomedicine 2010, 6, 58–63. [Google Scholar] [CrossRef]
- Lino, R.C.; de Carvalho, S.M.; Noronha, C.M.; Sganzerla, M.G.; da Rosa, C.G.; Nunes, M.R.; Barreto, P.L.M. Development and Characterization of Poly-ε-Caprolactone Nanocapsules Containing β-carotene Using the Nanoprecipitation Method and Optimized by Response Surface Methodology. Braz. Arch Biol. Technol. 2020, 63, e20190184. [Google Scholar] [CrossRef]
- Lim, J.; Yeap, S.P.; Che, H.X.; Low, S.C. Characterization of Magnetic Nanoparticle by Dynamic Light Scattering. Nanoscale Res. Lett. 2013, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yu, X.C.; Xu, S.F.; Xu, M. Paclitaxel and Etoposide co-Loaded Polymeric Nanoparticles for the Effective Combination Therapy Against Human Osteosarcoma. J. Nanobiotechnol. 2015, 22, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Zhang, Q.; Xu, H.; Yan, Z. Effects of Nanoparticle Size on Antitumor Activity of 10-Hydroxycamptothecin-Conjugated Gold Nanoparticles: In vitro and in vivo Studies. Int. J. Nanomed. 2016, 11, 929–940. [Google Scholar]
- Ching, Y.C.; Gunathilake, T.M.S.; Chuah, C.H.; Ching, K.Y.; Singh, R.; Liou, N.S. Curcumin/Tween 20-Incorporated Cellulose Nanoparticles with Enhanced Curcumin Solubility for Nano-Drug Delivery: Characterization and in vitro Evaluation. Cellulose 2019, 26, 5467–5481. [Google Scholar] [CrossRef]
- Qiao, Y.; Duan, L. Curcumin-Loaded Polyvinyl Butyral Film with Antibacterial Activity. E-Polymers 2020, 20, 673–681. [Google Scholar] [CrossRef]
- Lustriane, C.; Dwivany, F.M.; Suendo, V.; Reza, M. Effect of Chitosan and Chitosan-Nanoparticles on Post Harvest Quality of Banana Fruits. J. Plant Biotechnol. 2018, 45, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Kharia, A.A.; Singhai, A.K.; Verma, R. Formulation and Evaluation of Polymeric Nanoparticles of an Antiviral Drug for Gastroretention. Int. J. Pharm. Sci. Nanotechnol. 2012, 4, 1557–1562. [Google Scholar] [CrossRef]
- Bajpai, S.K.; Tankhiwale, R. Investigation of Water Uptake Behavior and Stability of Calcium Alginate/Chitosan Bi-Polymeric Beads: Part-1. React Funct. Polym. 2006, 66, 645–658. [Google Scholar] [CrossRef]
- Hoffman, A.S. The Origins and Evolution of “Controlled” Drug Delivery Systems. J. Control. Release 2008, 132, 153–163. [Google Scholar] [CrossRef]
- Fonseca-Santos, B.; Chorilli, M. An Overview of Carboxymethyl Derivatives of Chitosan: Their Use as Biomaterials and Drug Delivery Systems. Mater. Sci. Eng. C 2017, 77, 1349–1362. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Baek, Y.; Yoo, H.J.; Lee, J.S.; Lee, H.G. Chitosan-Tripolyphosphate Nanoparticles Prepared by Ionic Gelation Improve the Antioxidant Activities of Astaxanthin in the in vitro and in vivo Model. Antioxidants 2022, 11, 479. [Google Scholar] [CrossRef]
- Hung, C.F.; Chen, J.K.; Liao, M.H.; Lo, H.M.; Fang, J.Y. Development and Evaluation of Emulsion-Liposome Blends for Resveratrol Delivery. J. Nanosci. Nanotechnol. 2006, 6, 2950–2958. [Google Scholar] [CrossRef]
- Jannin, B.; Menzel, M.; Berlot, J.P.; Delmas, D.; Lançon, A.; Latruffe, N. Transport of Resveratrol, a Cancer Chemopreventive Agent, to Cellular Targets: Plasmatic Protein Binding and Cell Uptake. Biochem. Pharmacol. 2004, 68, 1113–1118. [Google Scholar] [CrossRef]
- Phillips, D.J.; Pygall, S.R.; Cooper, V.B.; Mann, J.C. Overcoming Sink Limitations in Dissolution Testing: A Review of Traditional Methods and the Potential Utility of Biphasic Systems. J. Pharm. Pharmacol. 2012, 64, 1549–1559. [Google Scholar] [CrossRef]
- Fang, M.; Jin, Y.; Bao, W.; Gao, H.; Xu, M.; Wang, D.; Wang, X.; Yao, P.; Liu, L. In vitro Characterization and in vivo Evaluation of Nanostructured Lipid Curcumin Carriers for Intragastric Administration. Int. J. Nanomed. 2012, 7, 5395–5404. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, L.; Meng, L.; Wang, J.; Zhai, G. Design and Evaluation of a Self-Microemulsifying Drug Delivery System for Apigenin. Drug Dev. Ind. Pharm. 2013, 39, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Nasra, M.M.A.; Khiri, H.M.; Hazzah, H.A.; Abdallah, O.Y. Formulation, in vitro Characterization and Clinical Evaluation of Curcumin in-situ Gel for Treatment of Periodontitis. Drug Deliv. 2017, 24, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bini, R.A.; Silva, M.F.; Varanda, L.C.; da Silva, M.; Dreiss, C.A. Soft Nanocomposites of Gelatin and Poly(3-hydroxybutyrate) Nanoparticles for Dual Drug Release. Colloids Surf. B Biointerfaces 2017, 157, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fu, Y.; Niu, F.; Li, Z.; Ba, C.; Jin, B.; Chen, G.; Li, X. Enhanced Antioxidant Activity and in vitro Release of Propolis by Acid-Induced Aggregation Using Heat-Denatured Zein and Carboxymethyl Chitosan. Food Hydrocoll. 2018, 81, 104–112. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, P.; Wu, S.; Yang, T.; Chen, Y.; Zhang, X.; He, C.; Zheng, C.; Li, K.; Ma, X.; et al. Cisplatin and Curcumin co-Loaded Nano-Liposomes for the Treatment of Hepatocellular Carcinoma. Int. J. Pharm. 2018, 545, 261–273. [Google Scholar] [CrossRef]
- Doosti, M.; Dorraji, M.S.S.; Mousavi, S.N.; Rasoulifard, M.H.; Hosseini, S.H. Enhancing Quercetin Bioavailability by Superparamagnetic Starch-Based Hydrogel Grafted with Fumaric Acid: An in vitro and in vivo study. Colloids Surf. B Biointerfaces 2019, 183, 110487. [Google Scholar] [CrossRef]
- Okagu, O.D.; Verma, O.; McClements, D.J.; Udenigwea, C.C. Utilization of Insect Proteins to Formulate Nutraceutical Delivery Systems: Encapsulation and Release of Curcumin Using Mealworm Protein-Chitosan Nano-complexes. Int. J. Biol. Macromol. 2020, 151, 333–343. [Google Scholar] [CrossRef]
- Wang, X.; Peng, F.; Liu, F.; Xiao, Y.; Li, F.; Lei, H.; Wang, J.; Li, M.; Xu, H. Zein-Pectin Composite Nanoparticles as An Efficient Hyperoside Delivery System: Fabrication, Characterization, and in vitro Release Property. LWT–Food Sci. Technol. 2020, 133, 109869. [Google Scholar] [CrossRef]
- Su, Z.; Han, C.; Liu, E.; Zhang, F.; Liu, B.; Meng, X. Formation, Characterization and Application of Arginine-Modified Chitosan/γ-Poly Glutamic acid Nanoparticles as Carrier for Curcumin. Int. J. Biol. Macromol. 2021, 168, 215–222. [Google Scholar] [CrossRef]
- Cassano, R.; Serini, S.; Curcio, F.; Trombino, S.; Calviello, G. Preparation and Study of Solid Lipid Nanoparticles Based on Curcumin, Resveratrol and Capsaicin Containing Linolenic Acid. Pharmaceutics 2022, 14, 1593. [Google Scholar] [CrossRef]
- Tabboon, P.; Pongjanyakul, P.; Limpongsa, E.; Jaipakdee, N. In vitro Release, Mucosal Permeation and Deposition of Cannabidiol from Liquisolid Systems: The Influence of Liquid Vehicles. Pharmaceutics 2022, 14, 1787. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modelling of Drug Release from Delivery System Based on Hydroxypropyl Methylcellulose (HPMC). Adv. Drug Del. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for description of solute release III. Coupling of Diffusion and Relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Khushbu; Jindal, R. Thermal Stability and Optimization of Graphene Oxide Incorporated Chitosan and Sodium Alginate Based Nanocomposite Containing Inclusion Complexes of Paracetamol and β-Cyclodextrin for Prolonged Drug Delivery Systems. Polym. Bull. 2022, 80, 1751–1772. [Google Scholar] [CrossRef]
- Hu, K.; McClements, D.J. Fabrication of Biopolymer Nanoparticles by Antisolvent Precipitation and Electrostatic Deposition: Zein-Alginate Core/Shell Nanoparticles. Food Hydrocoll. 2015, 44, 101–108. [Google Scholar] [CrossRef]
- Almalik, A.; Benabdelkamel, H.; Masood, A.; Alanazi, I.O.; Alradwan, I.; Majrashi, M.A.; Alfadda, A.A.; Alghamdi, W.M.; Alrabiah, H.; Tirelli, N.; et al. Hyaluronic Acid Coated Chitosan Nanoparticles Reduced the Immunogenicity of the Formed Protein Corona. Sci. Rep. 2017, 7, 10542. [Google Scholar] [CrossRef] [Green Version]
- Leceta, I.; Guerrero, P.; Ibarburu, I.; Dueñas, M.T.; de La Caba, K. Characterization and Antimicrobial Analysis of Chitosan-Based Films. J. Food Eng. 2013, 116, 889–899. [Google Scholar] [CrossRef]
- Weissmann, G.; Spjlberg, I.; Krakauer, K. Arthritis Induced in Rabbits by Lysates of Granulocyte Lysosomes. Arthritis Rheum. 1969, 12, 103–116. [Google Scholar] [CrossRef]
- Hess, S.; Milonig, R. Inflammation. In Inflammation, Mechanism and Control; Lepow, L., Ward, P., Eds.; Academic Press: New York, NY, USA, 1972; pp. 1–2. [Google Scholar]
- Niza, E.; Nieto-Jiménez, C.; del Mar Noblejas-López, M.; Bravo, I.; Castro-Osma, A.; de La Cruz-Martínez, F.; Martínez De Sarasa Buchaca, M.; Posadas, I.; Canales-Vázquez, J.; Lara-Sanchez, A.; et al. Poly(Cyclohexene Phthalate) Nanoparticles for Controlled Dasatinib Delivery in Breast Cancer Therapy. Nanomaterials 2019, 9, 1208. [Google Scholar] [CrossRef] [Green Version]
- Malathy, S.; Priya, R.I. Naringin Loaded Chitosan Nanoparticle for Bone Regeneration: A Preliminary in vitro Study. J. Nanomed. Nanotechnol. 2018, 09, 507. [Google Scholar]
- El-Mekawy, A.; Hudson, H.; El-Baz, A.; Hamza, H.; El-Halafawy, K. Preparation of Chitosan Films Mixed with Superabsorbent Polymer and Evaluation of Its Haemostatic and Antibacterial Activities. J. Appl. Polym. Sci. 2010, 116, 3489–3496. [Google Scholar] [CrossRef]
- Umapathy, E.; Ndebia, E.J.; Meeme, A.; Adam, B.; Menziwa, P.; Nkeh-Chungag, B.N.; Iputo, J.E. An Experimental Evaluation of Albuca Setosa Aqueous Extract on Membrane Stabilization, Protein Denaturation and White Blood Cell Migration during Acute Inflammation. J. Med. Plant Res. 2010, 4, 789–795. [Google Scholar]
- El-Ansary, A.; Al-Daihan, S. On the Toxicity of Therapeutically Used Nanoparticles: An Overview. J. Toxicol. 2009, 2009, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.L.; Qi, W.; Han, F.; Shao, J.Z.; Gao, J.Q. Toxicity Evaluation of Biodegradable Chitosan Nanoparticles Using a Zebrafish Embryo Model. Int. J. Nanomedicine 2011, 6, 3351–3359. [Google Scholar]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of Apoptosis by the BCL-2 Protein Family: Implications for Physiology and Therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-Cell Malignancies and Overcoming Therapeutic Resistance. Cell Death Dis. 2020, 11, 941. [Google Scholar] [CrossRef]
- Tomek, M.; Akiyama, T.; Dass, C.R. Role of Bcl-2 in Tumour Cell Survival and Implications for Pharmacotherapy. J. Pharm. Pharmacol. 2012, 64, 1695–1702. [Google Scholar] [CrossRef]
- Vivek, R.; Thangam, R.; Nipunbabu, V.; Ponraj, T.; Kannan, S. Oxaliplatin-Chitosan Nanoparticles Induced Intrinsic Apoptotic Signaling Pathway: A “Smart” Drug Delivery System to Breast Cancer Cell Therapy. Int. J. Biol. Macromol. 2014, 65, 289–297. [Google Scholar] [CrossRef]
- Wicha, P.; Tocharus, J.; Janyou, A.; Jittiwat, J.; Changtam, C.; Suksamrarn, A.; Tocharus, C. Hexahydrocurcumin Protects against Cerebral Ischemia/Reperfusion Injury, Attenuates Inflammation, and Improves Antioxidant Defenses in a Rat Stroke Model. PLoS ONE 2017, 12, e0189211. [Google Scholar] [CrossRef]
- Khodapasand, E.; Jafarzadeh, N.; Farrokhi, F.; Kamalidehghan, B.; Houshmand, M. Is Bax/Bcl-2 Ratio Considered as a Prognostic Marker with Age and Tumor Location in Colorectal Cancer? Iran. Biomed. J. 2015, 19, 69–75. [Google Scholar]
- Abdel-Hakeem, M.A.; Mongy, S.; Hassan, B.; Tantawi, O.I.; Badawy, I. Curcumin Loaded Chitosan-Protamine Nanoparticles Revealed Antitumor Activity via Suppression of NF-ΚB, Proinflammatory Cytokines and Bcl-2 Gene Expression in the Breast Cancer Cells. J. Pharm. Sci. 2021, 110, 3298–3305. [Google Scholar] [CrossRef]
- Sandhiutami, N.M.D.; Arozal, W.; Louisa, M.; Rahmat, D.; Wuyung, P.E. Curcumin Nanoparticle Enhances the Anticancer Effect of Cisplatin by Inhibiting PI3K/AKT and JAK/STAT3 Pathway in Rat Ovarian Carcinoma Induced by DMBA. Front. Pharmacol. 2021, 11, 603235. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Lin, Y.-J.; Huang, W.-T.; Hung, C.-C.; Lin, H.-Y.; Tu, Y.-C.; Liu, D.-M.; Lan, S.-J.; Sheu, M.-J. Molecules Demethoxycurcumin-Loaded Chitosan Nanoparticle Downregulates DNA Repair Pathway to Improve Cisplatin-Induced Apoptosis in Non-Small Cell Lung Cancer. Molecules 2018, 23, 3217. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.; Tao, X.; Xia, Z.; Hu, S.; Xue, J.; Zhu, Q.; Pan, X.; Zhang, Q.; Li, S. Pathogen Hijacks Programmed Cell Death Signaling by Arginine ADPR-Deacylization of Caspases. Mol. Cell 2022, 82, 1806–1820.e8. [Google Scholar] [CrossRef]
- Jiao, C.; Chen, W.; Tan, X.; Liang, H.; Li, J.; Yun, H.; He, C.; Chen, J.; Ma, X.; Xie, Y.; et al. Ganoderma Lucidum Spore Oil Induces Apoptosis of Breast Cancer Cells in vitro and in vivo by Activating Caspase-3 and Caspase-9. J. Ethnopharmacol. 2020, 247, 112256. [Google Scholar] [CrossRef]
- Motwani, S.K.; Chopra, S.; Kohli, T.K.; Ahmad, F.J.; Khar, R.K. Chitosan-Sodium Alginate Nanoparticles as Submicroscopic Reservoirs for Ocular Delivery: Formulation, Optimisation and in vitro Characterisation. Eur. J. Pharm. Biopharm. 2008, 68, 513–525. [Google Scholar] [CrossRef]
- Khuri, A.I.; Mukhopadhyay, S. Response Surface Methodology. WIREs Comp. Stat. 2010, 2, 128–149. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level Used | |||
|---|---|---|---|
| Low (−1) | Medium (0) | High (+1) | |
| Factors | |||
| X1 = TPP concentration (mg/mL) | 0.6 | 0.7 | 0.8 |
| X2 = Pluronic® F-127 concentration (% w/v) | 0.5 | 1 | 1.5 |
| X3 = HHC concentration (mg/mL) | 1 | 2 | 3 |
| Responses | Constraints | ||
| Y1 = Particle size (nm) | Minimize | ||
| Y2 = Zeta potential (mV) | Y2 ≥ 20 | ||
| Y3 = Encapsulation efficiency (%) | Maximize | ||
| Runs | Factors | Observed Responses | ||||
|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 | Y3 | |
| 1 | 0.6 | 0.5 | 2 | 102 ± 15 | 43.8 ± 0.2 | 67.5 ± 1.3 |
| 2 | 0.8 | 0.5 | 2 | 269 ± 11 | 24.5 ± 0.5 | 85.5 ± 2.4 |
| 3 | 0.6 | 1.5 | 2 | 187 ± 17 | 35.9 ± 0.4 | 55.8 ± 1.9 |
| 4 | 0.8 | 1.5 | 2 | 387 ± 21 | 22.2 ± 0.8 | 83.4 ± 2.2 |
| 5 | 0.6 | 1.0 | 1 | 154 ± 14 | 41.7 ± 0.3 | 43.4 ± 1.8 |
| 6 | 0.8 | 1.0 | 1 | 326 ± 18 | 21.9 ± 0.7 | 67.3 ± 1.5 |
| 7 | 0.6 | 1.0 | 3 | 198 ± 10 | 39.7 ± 1.1 | 74.1 ± 2.3 |
| 8 | 0.8 | 1.0 | 3 | 338 ± 26 | 20.7 ± 0.4 | 91.2 ± 0.8 |
| 9 | 0.7 | 0.5 | 1 | 168 ± 12 | 30.7 ± 0.2 | 57.5 ± 2.4 |
| 10 | 0.7 | 1.5 | 1 | 275 ± 19 | 29.6 ± 0.8 | 49.2 ± 1.1 |
| 11 | 0.7 | 0.5 | 3 | 192 ± 11 | 30.1 ± 1.2 | 86.5 ± 2.6 |
| 12 | 0.7 | 1.5 | 3 | 331 ± 22 | 28.5 ± 0.5 | 82.8 ± 1.7 |
| 13 a | 0.7 | 1.0 | 2 | 216 ± 24 | 31.5 ± 1.6 | 76.6 ± 1.3 |
| 14 a | 0.7 | 1.0 | 2 | 230 ± 12 | 30.4 ± 0.3 | 75.4 ± 2.8 |
| 15 a | 0.7 | 1.0 | 2 | 225 ± 17 | 31.6 ± 0.8 | 73.4 ± 1.9 |
| Factor | Optimum | Response | Predicted | Observed | % Error |
|---|---|---|---|---|---|
| TPP (mg/mL) | 0.75 | Y1 | 250 | 256 ± 14 | 2.34 |
| Pluronic® F-127 (% w/v) | 0.50 | Y2 | 26.8 | 27.3 ± 0.7 | 1.83 |
| HHC (mg/mL) | 3.00 | Y3 | 88.7 | 90.6 ± 1.7 | 2.09 |
| Model | Evaluation Criteria | ||||||
|---|---|---|---|---|---|---|---|
| Media | R2adjusted | AIC | MSC | kn | n | m | |
| Zero-order (F = k0·t) | SBF | 0.292 | 57.668 | −0.357 | 0.667 | - | - |
| SBF + 1% HSA | 0.552 | 52.946 | 0.196 | 0.640 | - | - | |
| First-order () | SBF | 0.401 | 56.171 | −0.191 | 0.008 | - | - |
| SBF + 1% HSA | 0.631 | 51.197 | 0.391 | 0.008 | - | - | |
| Hixson–Crowell (F = 100 [1 − (1 − kHC·t)3]) | SBF | 0.365 | 56.698 | −0.249 | 0.003 | - | - |
| SBF + 1% HSA | 0.605 | 51.811 | 0.322 | 0.002 | - | - | |
| Korsmeyer–Peppas (F = kKP·tn) | SBF | 0.885 | 42.153 | 1.367 | 6.201 | 0.376 | - |
| SBF + 1% HSA | 0.931 | 36.875 | 1.981 | 4.635 | 0.446 | - | |
| Higuchi (F = kH·t0.5) | SBF | 0.850 | 43.664 | 1.199 | 4.208 | - | - |
| SBF + 1% HSA | 0.932 | 35.926 | 2.087 | 3.905 | - | - | |
| Peppas–Sahlin (F = k1·tm + k2·tm) | SBF | 0.970 | 30.696 | 2.639 | k1 = 4.601 k2 = −0.213 | - | 0.679 |
| SBF + 1% HSA | 0.969 | 30.088 | 2.736 | k1 = 3.522 k2 = −0.130 | - | 0.699 | |
| Hopfenberg (F = 100 [1 − (1 − kHB·t)n]) | SBF | 0.315 | 58.172 | −0.413 | 0.000 | 719.798 | - |
| SBF + 1% HSA | 0.578 | 53.199 | 0.168 | 0.002 | 767.388 | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorasitthiyanukarn, F.N.; Muangnoi, C.; Gomez, C.B.; Suksamrarn, A.; Rojsitthisak, P.; Rojsitthisak, P. Potential Oral Anticancer Therapeutic Agents of Hexahydrocurcumin-Encapsulated Chitosan Nanoparticles against MDA-MB-231 Breast Cancer Cells. Pharmaceutics 2023, 15, 472. https://doi.org/10.3390/pharmaceutics15020472
Sorasitthiyanukarn FN, Muangnoi C, Gomez CB, Suksamrarn A, Rojsitthisak P, Rojsitthisak P. Potential Oral Anticancer Therapeutic Agents of Hexahydrocurcumin-Encapsulated Chitosan Nanoparticles against MDA-MB-231 Breast Cancer Cells. Pharmaceutics. 2023; 15(2):472. https://doi.org/10.3390/pharmaceutics15020472
Chicago/Turabian StyleSorasitthiyanukarn, Feuangthit N., Chawanphat Muangnoi, Clinton B. Gomez, Apichart Suksamrarn, Pranee Rojsitthisak, and Pornchai Rojsitthisak. 2023. "Potential Oral Anticancer Therapeutic Agents of Hexahydrocurcumin-Encapsulated Chitosan Nanoparticles against MDA-MB-231 Breast Cancer Cells" Pharmaceutics 15, no. 2: 472. https://doi.org/10.3390/pharmaceutics15020472