

Nanoseeded Desupersaturation and Dissolution Tests for Elucidating Supersaturation Maintenance in Amorphous Solid Dispersions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Nanoseed Preparation via Wet Stirred Media Milling
2.2.2. Spray Drying
2.2.3. Particle Size and Morphology
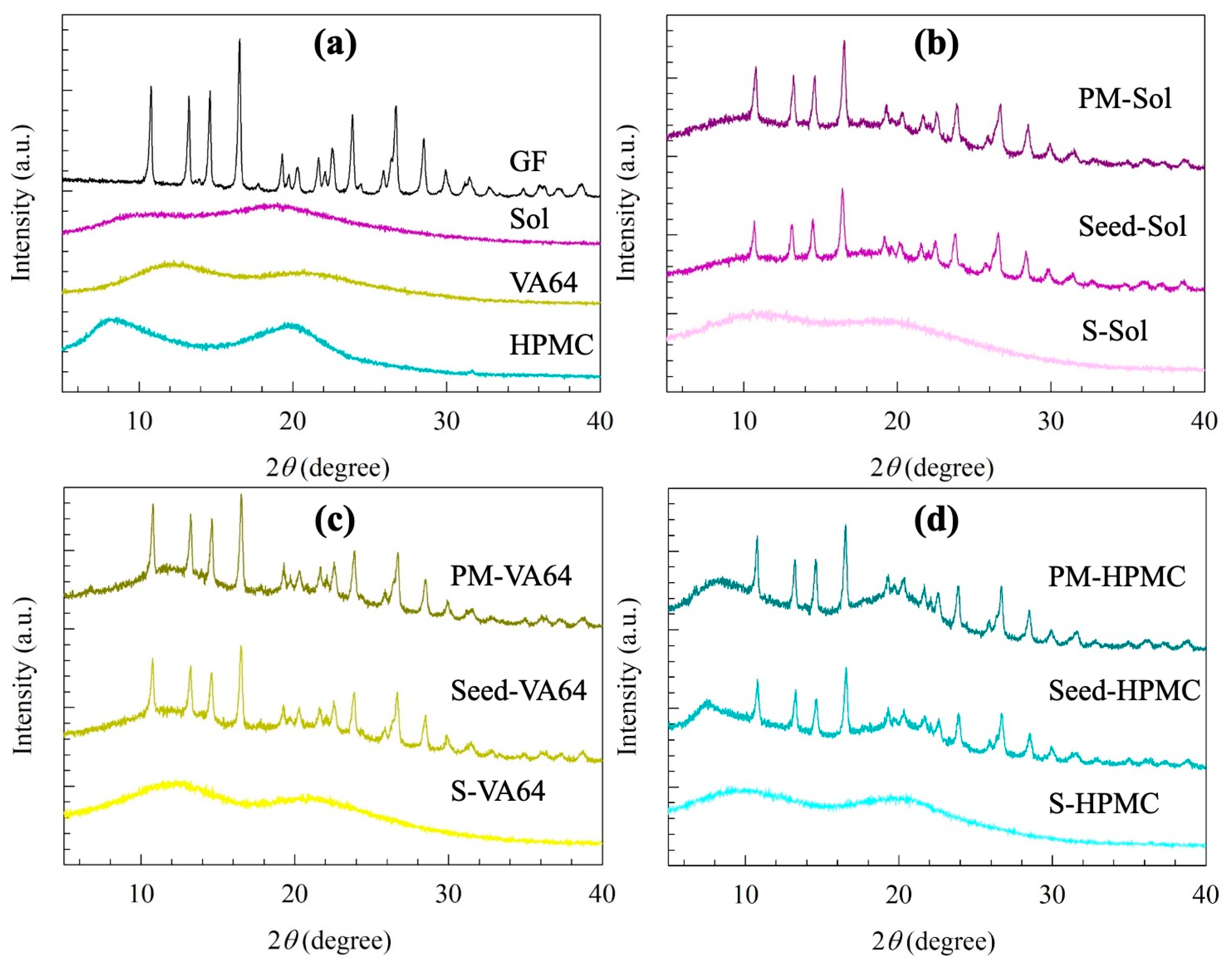
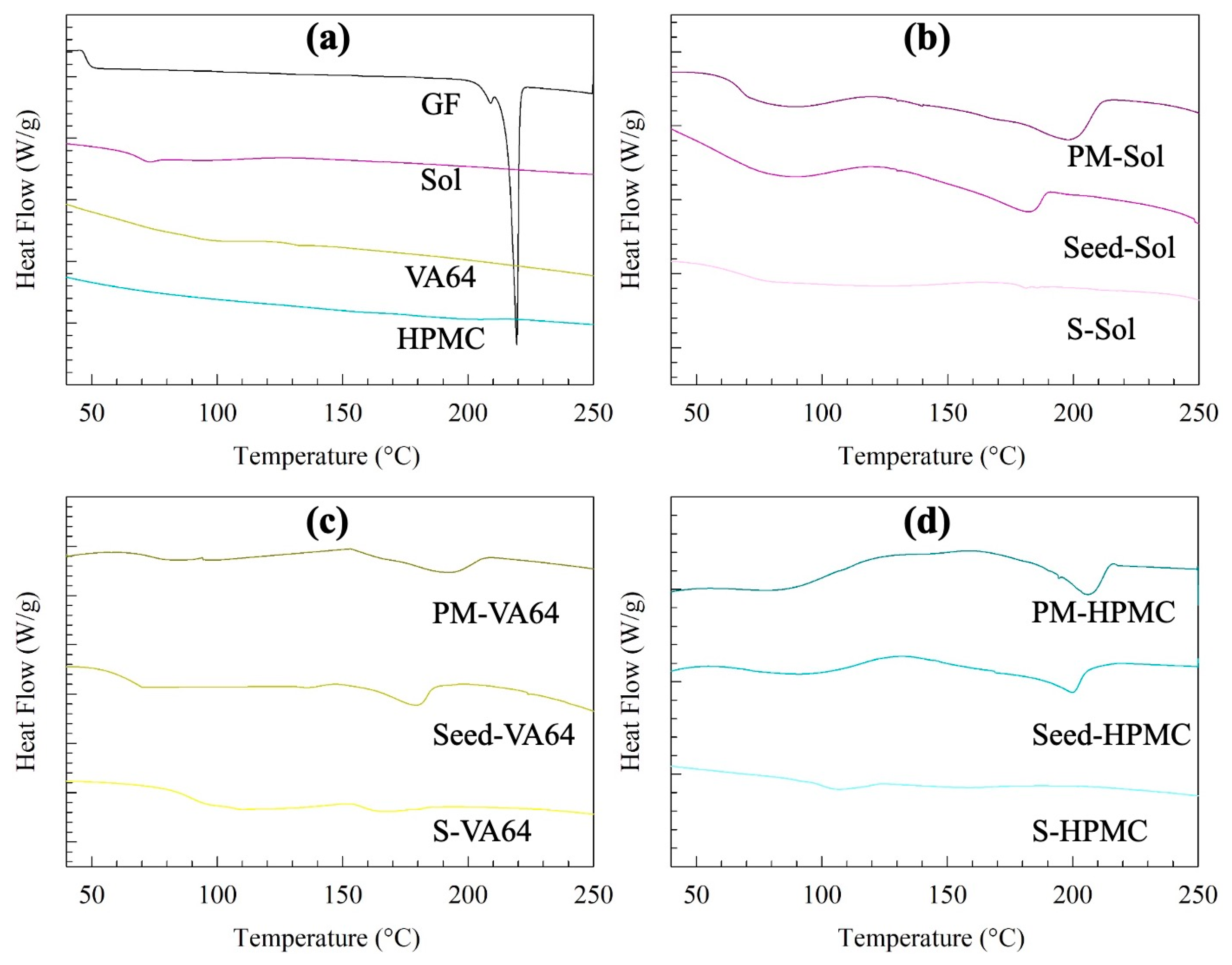
2.2.4. Solid-State Characterization
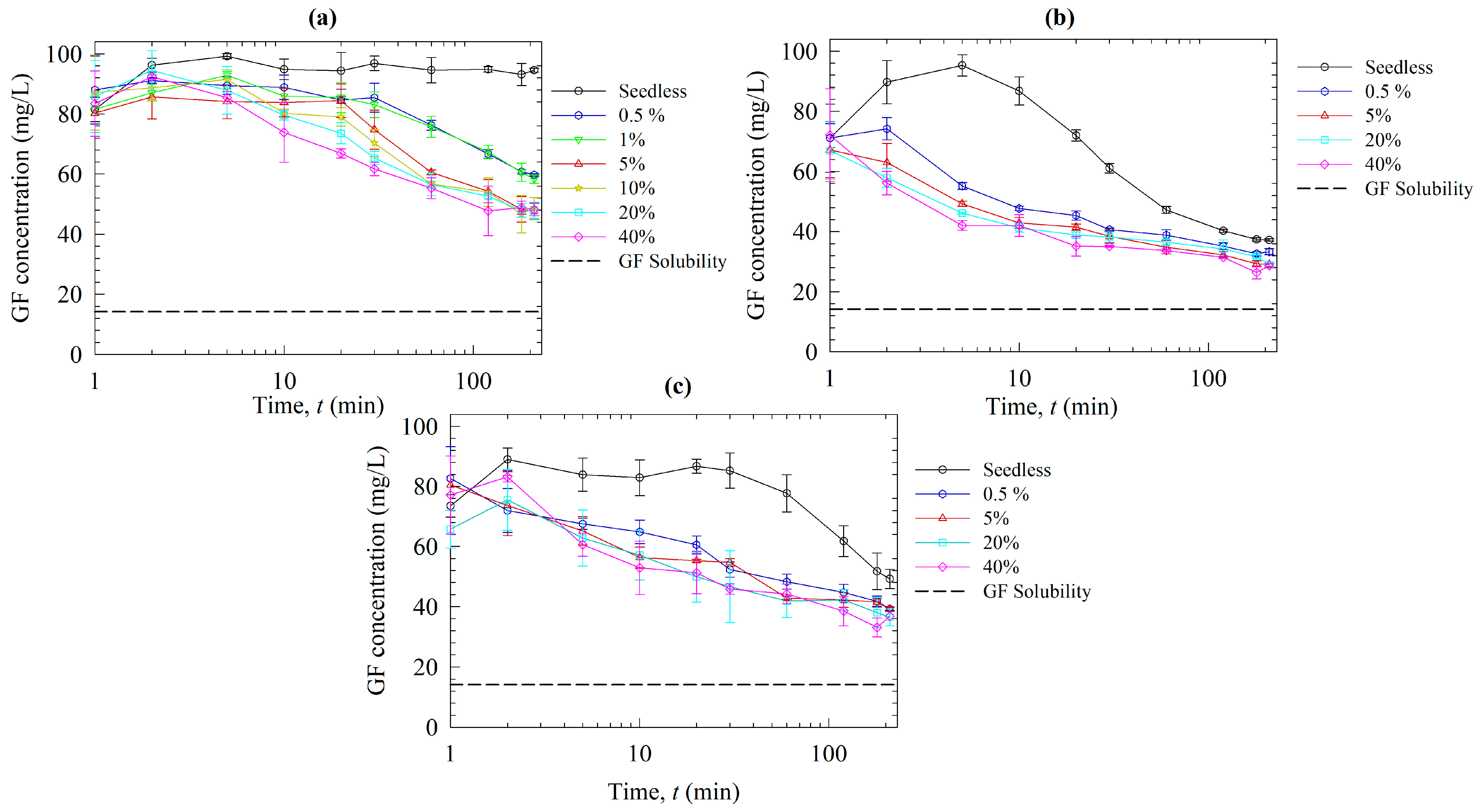
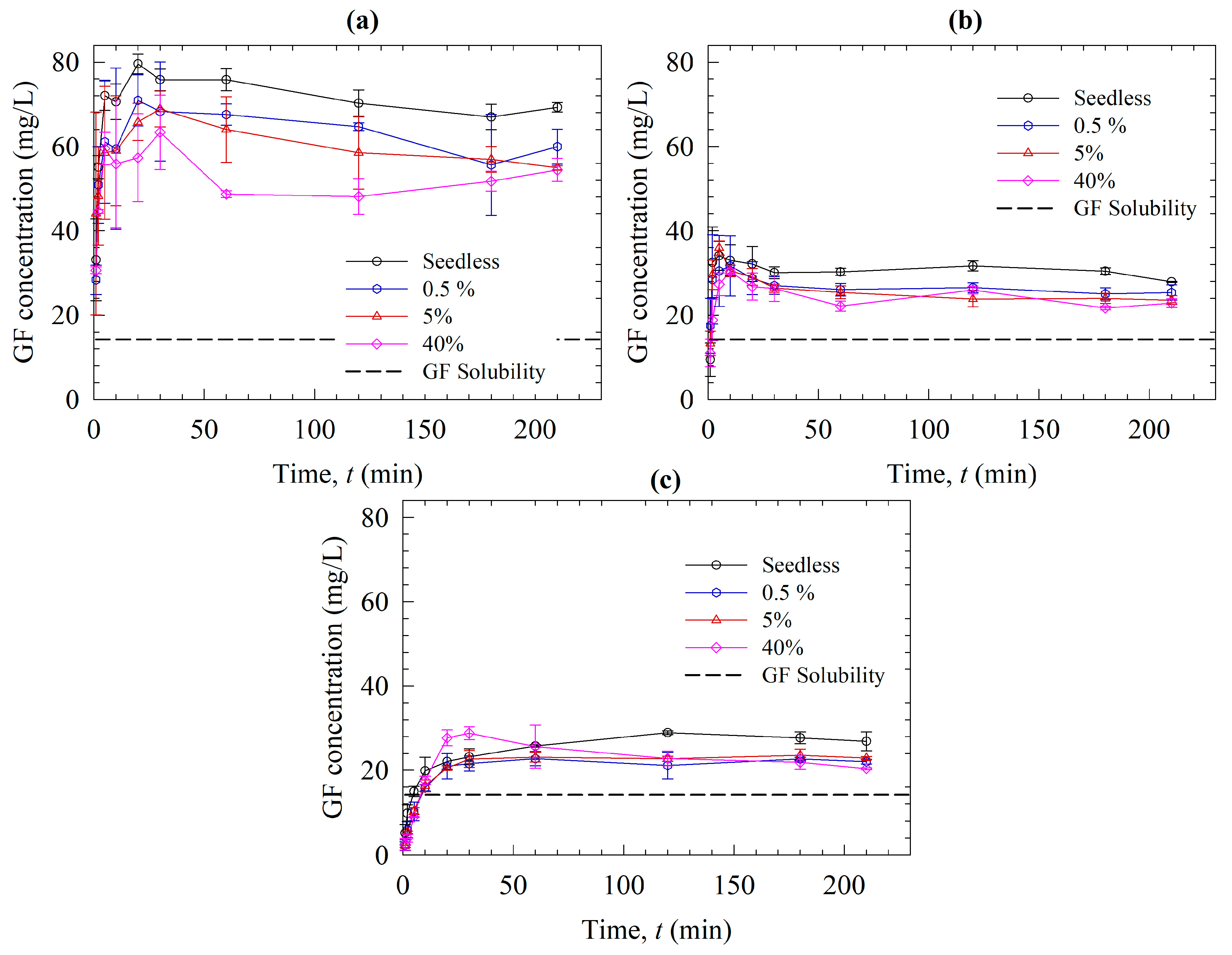
2.2.5. Seeded Desupersaturation and Dissolution Experiments
3. Results
3.1. Properties of the Seeds and Spray-Dried Powders
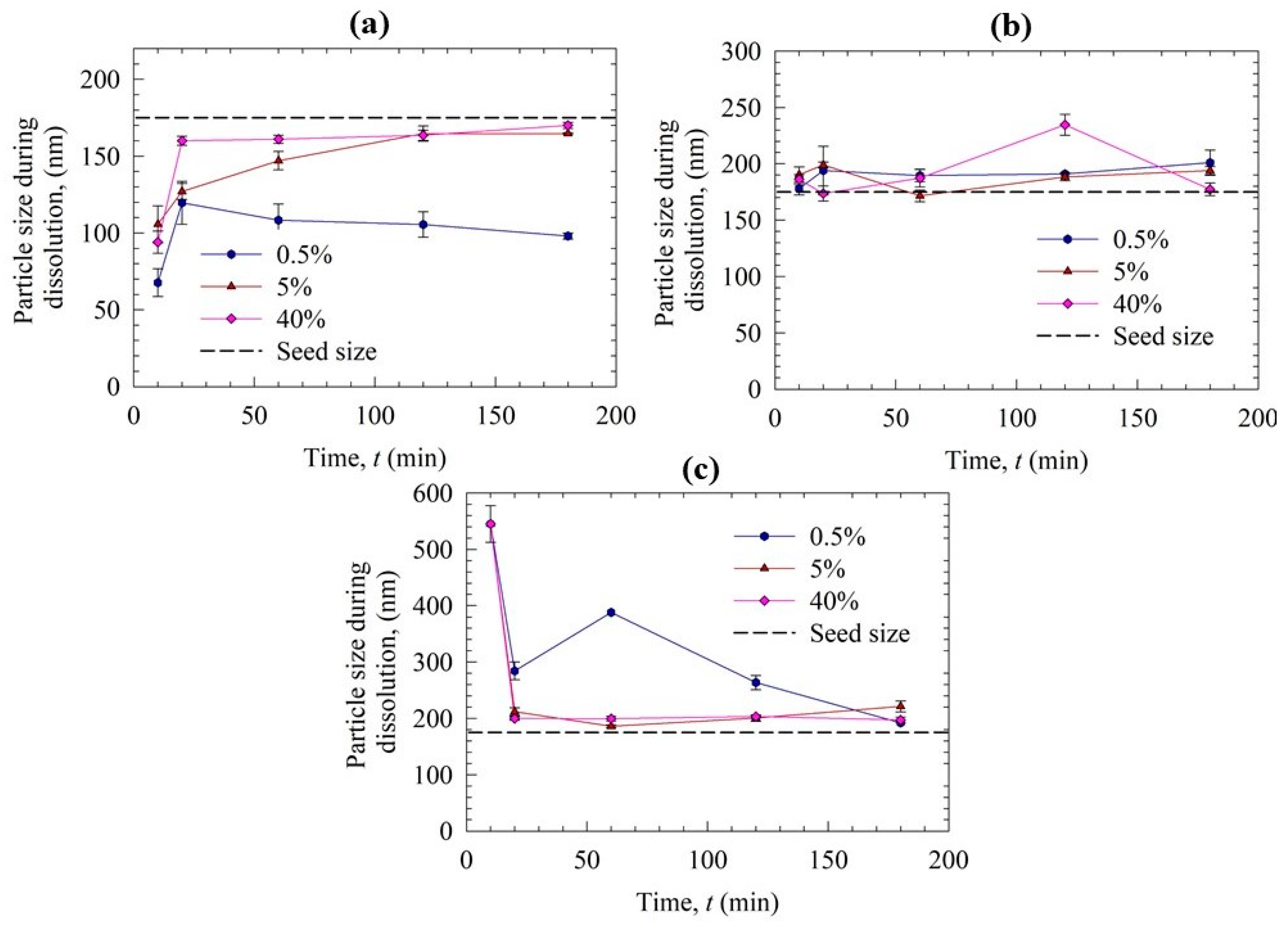
3.2. Solution-Mediated Precipitation of GF in the Solvent-Shift and Dissolution Tests
3.3. Roles of Polymers on Recrystallization Inhibition in the Presence of Nanoseeds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benet, L.Z.; Wu, C.-Y.; Custodio, J.M. Predicting drug absorption and the effects of food on oral bioavailability. Bull. Tech. Gattefosse 2006, 99, 9–16. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Wu, Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008, 10, 516–525. [Google Scholar] [CrossRef]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Malick, A.W. Amorphous Solid Dispersions; Springer: Berlin, Germany, 2014. [Google Scholar]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Gao, Y.; Zhou, D.; Mo, H.; Zhang, G.G.; Taylor, L.S. Dissolution and precipitation behavior of amorphous solid dispersions. J. Pharm. Sci. 2011, 100, 3316–3331. [Google Scholar] [CrossRef]
- Bhugra, C.; Pikal, M.J. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. J. Pharm. Sci. 2008, 97, 1329–1349. [Google Scholar] [CrossRef]
- Kawakami, K. Supersaturation and crystallization: Non-equilibrium dynamics of amorphous solid dispersions for oral drug delivery. Expert Opin. Drug Deliv. 2017, 14, 735–743. [Google Scholar] [CrossRef]
- Pui, Y.; Chen, Y.; Chen, H.; Wang, S.; Liu, C.; Tonnis, W.; Chen, L.; Serno, P.; Bracht, S.; Qian, F. Maintaining supersaturation of nimodipine by PVP with or without the presence of sodium lauryl sulfate and sodium taurocholate. Mol. Pharm. 2018, 15, 2754–2763. [Google Scholar] [CrossRef]
- Mersmann, A. Crystallization Technology Handbook; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Rahman, M.; Coelho, A.; Tarabokija, J.; Ahmad, S.; Radgman, K.; Bilgili, E. Synergistic and antagonistic effects of various amphiphilic polymer combinations in enhancing griseofulvin release from ternary amorphous solid dispersions. Eur. J. Pharm. Sci. 2020, 150, 105354. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, H.; Hui-Gu, C.; Atef, E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J. Pharm. Sci. 2013, 102, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Schram, C.J.; Beaudoin, S.P.; Taylor, L.S. Polymer inhibition of crystal growth by surface poisoning. Cryst. Growth Des. 2016, 16, 2094–2103. [Google Scholar] [CrossRef]
- Ivanisevic, I. Physical stability studies of miscible amorphous solid dispersions. J. Pharm. Sci. 2010, 99, 4005–4012. [Google Scholar] [CrossRef]
- Moseson, D.E.; Mugheirbi, N.A.; Stewart, A.A.; Taylor, L.S. Nanometer-scale residual crystals in a hot melt extruded amorphous solid dispersion: Characterization by transmission electron microscopy. Cryst. Growth Des. 2018, 18, 7633–7640. [Google Scholar] [CrossRef]
- Paudel, A.; Van den Mooter, G. Influence of solvent composition on the miscibility and physical stability of naproxen/PVP K 25 solid dispersions prepared by cosolvent spray-drying. Pharm. Res. 2012, 29, 251–270. [Google Scholar] [CrossRef]
- Haser, A.; Cao, T.; Lubach, J.; Listro, T.; Acquarulo, L.; Zhang, F. Melt extrusion vs. spray drying: The effect of processing methods on crystalline content of naproxen-povidone formulations. Eur. J. Pharm. Sci. 2017, 102, 115–125. [Google Scholar] [CrossRef]
- Dedroog, S.; Pas, T.; Vergauwen, B.; Huygens, C.; Van den Mooter, G. Solid-state analysis of amorphous solid dispersions: Why DSC and XRPD may not be regarded as stand-alone techniques. J. Pharm. Biomed. Anal. 2020, 178, 112937. [Google Scholar] [CrossRef]
- Marsac, P.J.; Rumondor, A.C.; Nivens, D.E.; Kestur, U.S.; Stanciu, L.; Taylor, L.S. Effect of temperature and moisture on the miscibility of amorphous dispersions of felodipine and poly (vinyl pyrrolidone). J. Pharm. Sci. 2010, 99, 169–185. [Google Scholar] [CrossRef]
- Knopp, M.M.; Wendelboe, J.; Holm, R.; Rades, T. Effect of amorphous phase separation and crystallization on the in vitro and in vivo performance of an amorphous solid dispersion. Eur. J. Pharm. Biopharm. 2018, 130, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent advances in understanding the micro-and nanoscale phenomena of amorphous solid dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef] [PubMed]
- Priemel, P.; Grohganz, H.; Gordon, K.; Rades, T.; Strachan, C. The impact of surface-and nano-crystallisation on the detected amorphous content and the dissolution behaviour of amorphous indomethacin. Eur. J. Pharm. Biopharm. 2012, 82, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hermans, A.; Kesisoglou, F.; Xu, W.; Dewitt, K.; Marota, M.; Colace, T. Possibilities and limiting factors for the use of dissolution as a quality control tool to detect presence of crystallinity for amorphous solid dispersions: An experimental and modeling investigation. J. Pharm. Sci. 2019, 108, 3054–3062. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, R.G.; Lodge, T.P.; Hillmyer, M.A. Nanoscale concentration quantification of pharmaceutical actives in amorphous polymer matrices by electron energy-loss spectroscopy. Langmuir 2016, 32, 7411–7419. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, R.G.; Lodge, T.P.; Hillmyer, M.A. Detection of pharmaceutical drug crystallites in solid dispersions by transmission electron microscopy. Mol. Pharm. 2015, 12, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Taylor, L.S. Nanoscale infrared, thermal, and mechanical characterization of telaprevir–polymer miscibility in amorphous solid dispersions Prepared by Solvent Evaporation. Mol. Pharm. 2016, 13, 1123–1136. [Google Scholar] [CrossRef]
- S’ari, M.; Blade, H.; Cosgrove, S.; Drummond-Brydson, R.; Hondow, N.; Hughes, L.P.; Brown, A. Characterization of amorphous solid dispersions and identification of low levels of crystallinity by transmission electron microscopy. Mol. Pharm. 2021, 18, 1905–1919. [Google Scholar] [CrossRef]
- Moseson, D.E.; Taylor, L.S. The application of temperature-composition phase diagrams for hot melt extrusion processing of amorphous solid dispersions to prevent residual crystallinity. Int. J. Pharm. 2018, 553, 454–466. [Google Scholar] [CrossRef]
- Moseson, D.E.; Corum, I.D.; Lust, A.; Altman, K.J.; Hiew, T.N.; Eren, A.; Nagy, Z.K.; Taylor, L.S. Amorphous solid dispersions containing residual crystallinity: Competition between dissolution and matrix crystallization. AAPS J. 2021, 23, 69. [Google Scholar] [CrossRef]
- Moseson, D.E.; Parker, A.S.; Beaudoin, S.P.; Taylor, L.S. Amorphous solid dispersions containing residual crystallinity: Influence of seed properties and polymer adsorption on dissolution performance. Eur. J. Pharm. Sci. 2020, 146, 105276. [Google Scholar] [CrossRef]
- Paladino, E.; Doerr, F.J.S.; Bordos, E.; Onyemelukwe, I.I.; Lamprou, D.A.; Florence, A.J.; Gilmore, I.S.; Halbert, G.W. High spatial resolution ToF-SIMS imaging and image analysis strategies to monitor and quantify early phase separation in amorphous solid dispersions. Int. J. Pharm. 2022, 628, 122191. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, C.; Chen, H.; Zhu, A.; Qian, F. Impact of surfactants on polymer maintained nifedipine supersaturation in aqueous solution. Pharm. Res. 2020, 37, 113. [Google Scholar] [CrossRef] [PubMed]
- Que, C.; Gao, Y.; Raina, S.A.; Zhang, G.G.; Taylor, L.S. Paclitaxel crystal seeds with different intrinsic properties and their impact on dissolution of paclitaxel-HPMCAS amorphous solid dispersions. Cryst. Growth Des. 2018, 18, 1548–1559. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Raina, S.; Zhou, D.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Characterizing the impact of hydroxypropylmethyl cellulose on the growth and nucleation kinetics of felodipine from supersaturated solutions. Cryst. Growth Des. 2012, 12, 1538–1547. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Effect of binary additive combinations on solution crystal growth of the poorly water-soluble drug, Ritonavir. Cryst. Growth Des. 2012, 12, 6050–6060. [Google Scholar] [CrossRef]
- Li, M.; Yaragudi, N.; Afolabi, A.; Dave, R.; Bilgili, E. Sub-100nm drug particle suspensions prepared via wet milling with low bead contamination through novel process intensification. Chem. Eng. Sci. 2015, 130, 207–220. [Google Scholar] [CrossRef]
- Rahman, M.; Arevalo, F.; Coelho, A.; Bilgili, E. Hybrid nanocrystal–amorphous solid dispersions (HyNASDs) as alternative to ASDs for enhanced release of BCS Class II drugs. Eur. J. Pharm. Biopharm. 2019, 145, 12–26. [Google Scholar] [CrossRef]
- Kataoka, M.; Masaoka, Y.; Yamazaki, Y.; Sakane, T.; Sezaki, H.; Yamashita, S. In vitro system to evaluate oral absorption of poorly water-soluble drugs: Simultaneous analysis on dissolution and permeation of drugs. Pharm. Res. 2003, 20, 1674–1680. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, G.G.; Law, D.; Grant, D.J.; Schmitt, E.A. Thermodynamics, molecular mobility and crystallization kinetics of amorphous griseofulvin. Mol. Pharm. 2008, 5, 927–936. [Google Scholar] [CrossRef]
- Terife, G.; Wang, P.; Faridi, N.; Gogos, C.G. Hot melt mixing and foaming of soluplus® and indomethacin. Polym. Eng. Sci. 2012, 52, 1629–1639. [Google Scholar] [CrossRef]
- Kolter, K.; Karl, M.; Gryczke, A.; Ludwigshafen am Rhein, B. Hot-melt Extrusion with BASF Pharma Polymers: Extrusion Compendium; BASF: Ludwigshafen, Germany, 2012. [Google Scholar]
- Chavan, R.B.; Thipparaboina, R.; Kumar, D.; Shastri, N.R. Evaluation of the inhibitory potential of HPMC, PVP and HPC polymers on nucleation and crystal growth. RSC Adv. 2016, 6, 77569–77576. [Google Scholar] [CrossRef]
- Rahman, M.; Ahmad, S.; Tarabokija, J.; Bilgili, E. Roles of surfactant and polymer in drug release from spray-dried hybrid nanocrystal-amorphous solid dispersions (HyNASDs). Powder Technol. 2020, 361, 663–678. [Google Scholar] [CrossRef]
- Li, M.; Lopez, N.; Bilgili, E. A study of the impact of polymer–surfactant in drug nanoparticle coated pharmatose composites on dissolution performance. Adv. Powder Technol. 2016, 27, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Guner, G.; Kannan, M.; Berrios, M.; Bilgili, E. Use of bead mixtures as a novel process optimization approach to nanomilling of drug suspensions. Pharm. Res. 2021, 38, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Lozano, A.; Yip, B.; Hanson, R.K. Acetone: A tracer for concentration measurements in gaseous flows by planar laser-induced fluorescence. Exp. Fluids 1992, 13, 369–376. [Google Scholar] [CrossRef]
- Sartory, D.P.; Grobbelaar, J.U. Extraction of chlorophyll a from freshwater phytoplankton for spectrophotometric analysis. Hydrobiologia 1984, 114, 177–187. [Google Scholar] [CrossRef]
- Mugheirbi, N.A.; Marsac, P.J.; Taylor, L.S. Insights into water-induced phase separation in itraconazole–hydroxypropylmethyl cellulose spin coated and spray dried dispersions. Mol. Pharm. 2017, 14, 4387–4402. [Google Scholar] [CrossRef]
- Krstić, M.; Đuriš, J.; Petrović, O.; Lazarević, N.; Cvijić, S.; Ibrić, S. Application of the melt granulation technique in development of self-emulsifying tablets with immediate release of carbamazepine. J. Drug Deliv. Sci. Technol. 2017, 39, 467–474. [Google Scholar] [CrossRef]
- Monteiro, A.; Afolabi, A.; Bilgili, E. Continuous production of drug nanoparticle suspensions via wet stirred media milling: A fresh look at the Rehbinder effect. Drug Dev. Ind. Pharm. 2013, 39, 266–283. [Google Scholar] [CrossRef]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Garside, J.; Mersmann, A.; Nývlt, J. Measurement of Crystal Growth and Nucleation Rates, 2nd ed.; IChemE: Rugby, UK, 2002. [Google Scholar]
- Li, M.; Furey, C.; Skros, J.; Xu, O.; Rahman, M.; Azad, M.; Dave, R.; Bilgili, E. Impact of matrix surface area on griseofulvin release from extrudates prepared via nanoextrusion. Pharmaceutics 2021, 13, 1036. [Google Scholar] [CrossRef] [PubMed]
- Dian, L.; Yu, E.; Chen, X.; Wen, X.; Zhang, Z.; Qin, L.; Wang, Q.; Li, G.; Wu, C. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res. Lett. 2014, 9, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, S.; Kurokawa, T.; Sato, H.; Kadota, K.; Tozuka, Y. Evaluation of the micellization mechanism of an amphipathic graft copolymer with enhanced solubility of ipriflavone. Chem. Pharm. Bull. 2016, 64, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Understanding polymer properties important for crystal growth inhibition– Impact of chemically diverse polymers on solution crystal growth of ritonavir. Cryst. Growth Des. 2012, 12, 3133–3143. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining supersaturation in aqueous drug solutions: Impact of different polymers on induction times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.J.; Pouton, C.W. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef]
- Khan, J.; Rades, T.; Boyd, B. The precipitation behavior of poorly water-soluble drugs with an emphasis on the digestion of lipid based formulations. Pharm. Res. 2016, 33, 548–562. [Google Scholar] [CrossRef]
- Fedors, R.F. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14, 147–154. [Google Scholar] [CrossRef]
- Sakellariou, P.; Rowe, R.C.; White, E.F.T. The solubility parameters of some cellulose derivatives and polyethylene glycols used in tablet film coating. Int. J. Pharm. 1986, 31, 175–177. [Google Scholar] [CrossRef]
- Thakral, S.; Thakral, N.K. Prediction of drug–polymer miscibility through the use of solubility parameter based Flory–Huggins interaction parameter and the experimental validation: PEG as model polymer. J. Pharm. Sci. 2013, 102, 2254–2263. [Google Scholar] [CrossRef]
- Forster, A.; Hempenstall, J.; Rades, T. Characterization of glass solutions of poorly water-soluble drugs produced by melt extrusion with hydrophilic amorphous polymers. J. Pharm. Pharmacol. 2001, 53, 303–315. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | GF (% w/v) a | Polymer (% w/v) a | SDS (% w/v) a | Water (mL) | Acetone (mL) | Ethanol (mL) |
|---|---|---|---|---|---|---|
| Seed-Sol | 2.5 | 7.5 | 0.125 | 240 | 0 | 0 |
| Seed-VA64 | 2.5 | 7.5 | 0.125 | 240 | 0 | 0 |
| Seed-HPMC | 2.5 | 7.5 | 0.125 | 240 | 0 | 0 |
| S-Sol | 2.5 | 7.5 | 0.125 | 40 | 200 | 0 |
| S-VA64 | 2.5 | 7.5 | 0.125 | 40 | 140 | 60 |
| S-HPMC | 2.5 | 7.5 | 0.125 | 40 | 200 | 0 |
| Polymer in the Seed | Test | Seed (% w/w) |
|---|---|---|
| Sol | Desupersaturation | 0 |
| Dissolution | ||
| Desupersaturation | 0.5 | |
| Dissolution | ||
| Desupersaturation | 1 | |
| Desupersaturation | 5 | |
| Dissolution | ||
| Desupersaturation | 10 | |
| Desupersaturation | 20 | |
| Desupersaturation | 40 | |
| Dissolution | ||
| VA64 | Desupersaturation | 0 |
| Dissolution | ||
| Desupersaturation | 0.5 | |
| Dissolution | ||
| Desupersaturation | 5 | |
| Dissolution | ||
| Desupersaturation | 20 | |
| Desupersaturation | 40 | |
| Dissolution | ||
| HPMC | Desupersaturation | 0 |
| Dissolution | ||
| Desupersaturation | 0.5 | |
| Dissolution | ||
| Desupersaturation | 5 | |
| Dissolution | ||
| Desupersaturation | 20 | |
| Desupersaturation | 40 | |
| Dissolution |
| Formulation | Particle Size Statistics of the Spray-Dried Particles (µm) | Drug Content, Relative Standard Deviation (% w/w, %) | ||
|---|---|---|---|---|
| d10 ± SD | d50 ± SD | d90 ± SD | ||
| S-Sol | 1.06 ± 0.0 | 5.15 ± 0.1 | 13.17 ± 0.2 | 22.2, 0.51 |
| S-VA64 | 2.07 ± 0.0 | 8.81 ± 0.1 | 19.30 ± 0.3 | 20.9, 1.1 |
| S-HPMC | 1.85 ± 0.0 | 7.38 ± 0.2 | 22.57 ± 0.7 | 23.7, 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guner, G.; Amjad, A.; Berrios, M.; Kannan, M.; Bilgili, E. Nanoseeded Desupersaturation and Dissolution Tests for Elucidating Supersaturation Maintenance in Amorphous Solid Dispersions. Pharmaceutics 2023, 15, 450. https://doi.org/10.3390/pharmaceutics15020450
Guner G, Amjad A, Berrios M, Kannan M, Bilgili E. Nanoseeded Desupersaturation and Dissolution Tests for Elucidating Supersaturation Maintenance in Amorphous Solid Dispersions. Pharmaceutics. 2023; 15(2):450. https://doi.org/10.3390/pharmaceutics15020450
Chicago/Turabian StyleGuner, Gulenay, Ayesha Amjad, Matthew Berrios, Manisha Kannan, and Ecevit Bilgili. 2023. "Nanoseeded Desupersaturation and Dissolution Tests for Elucidating Supersaturation Maintenance in Amorphous Solid Dispersions" Pharmaceutics 15, no. 2: 450. https://doi.org/10.3390/pharmaceutics15020450