
The Role of Solute Carrier Transporters in Efficient Anticancer Drug Delivery and Therapy

Abstract
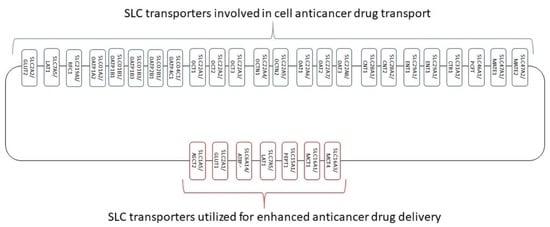
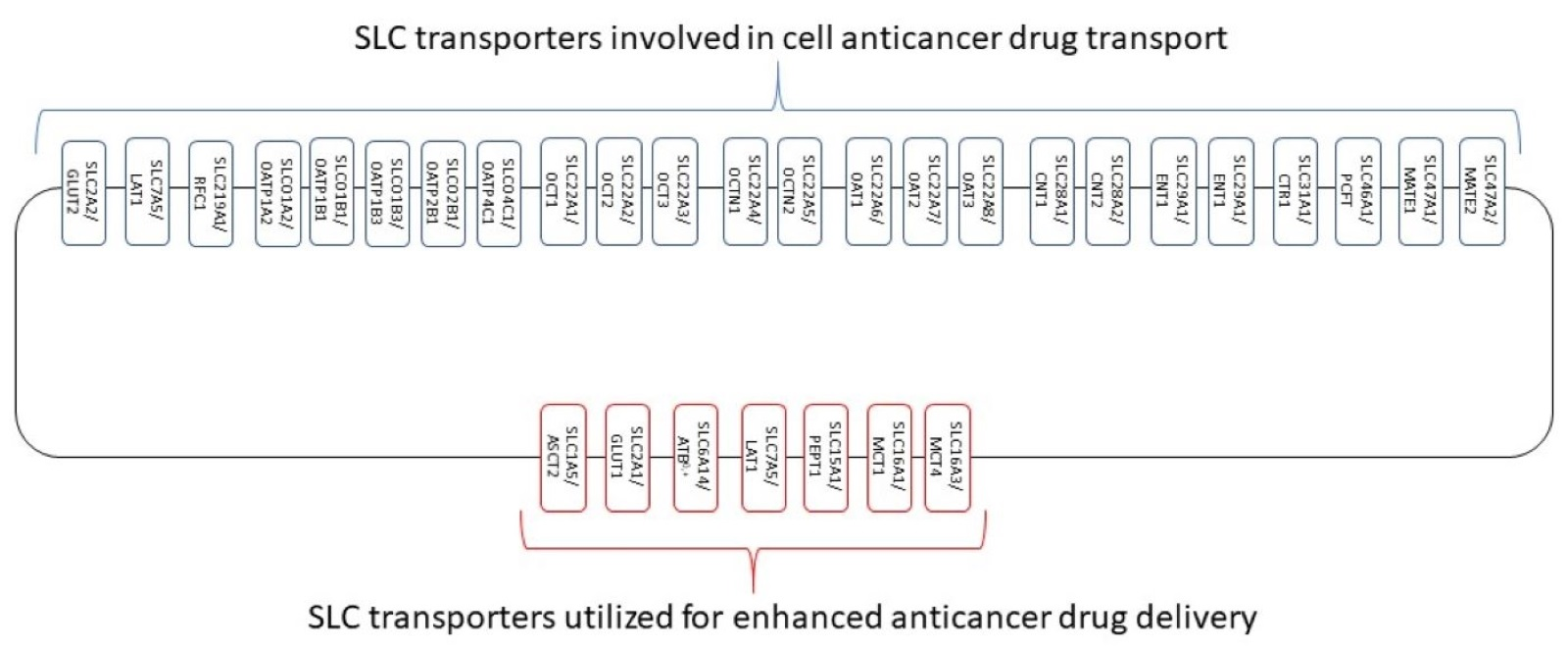
:
1. Introduction
2. Anticancer Drug Resistance
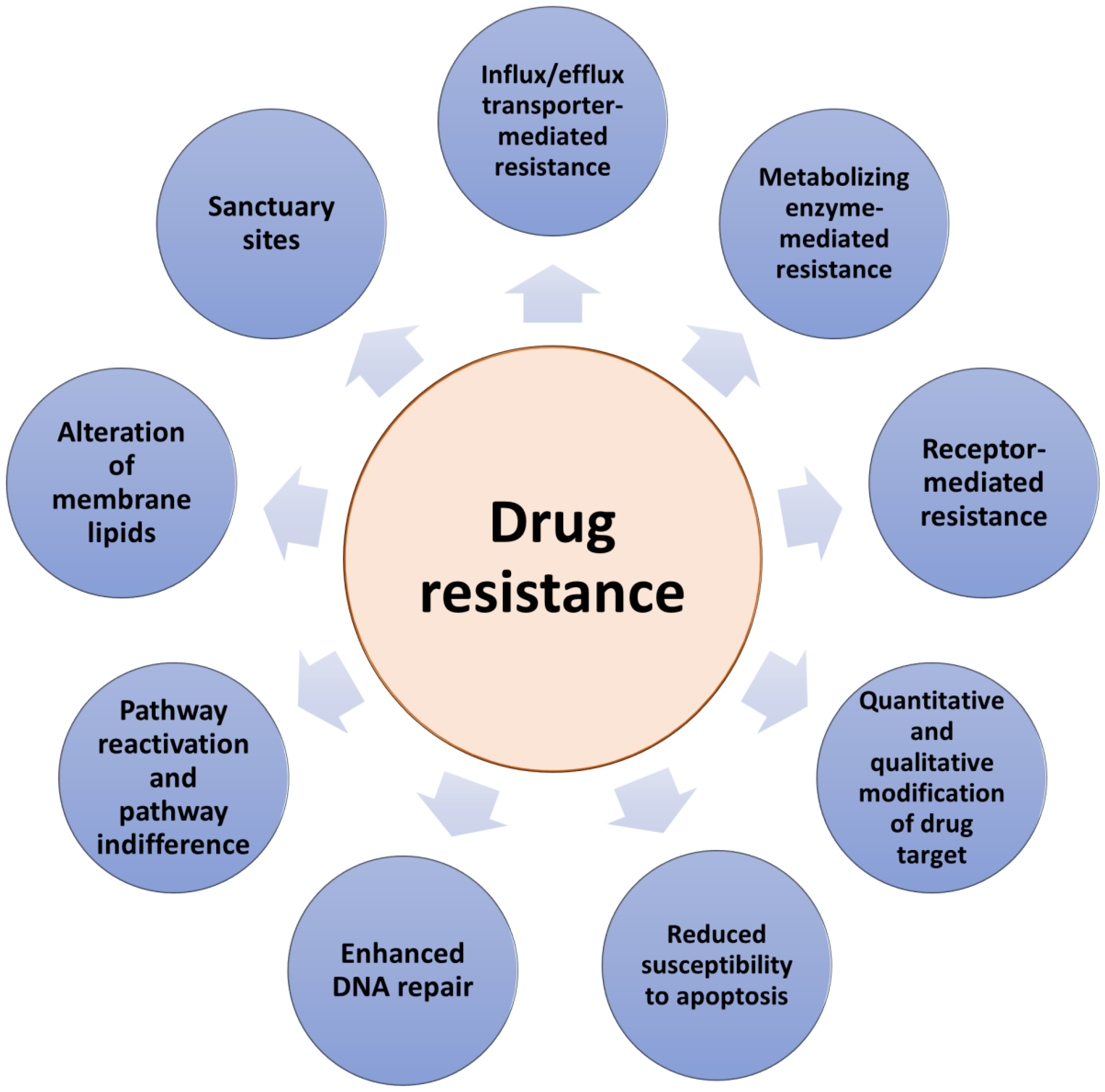
3. Mechanisms of Anticancer Drug Resistance
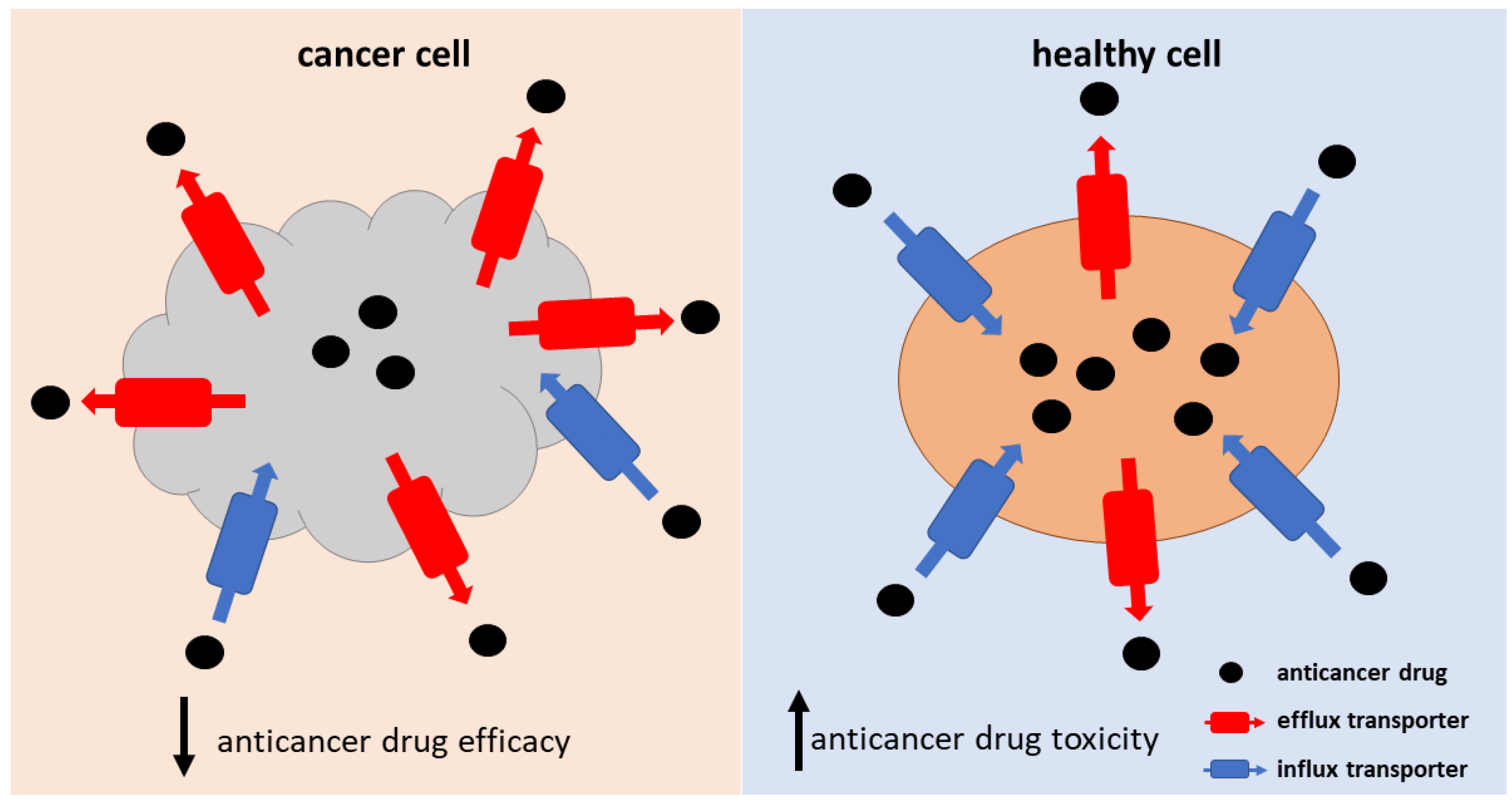
4. Transporter-Mediated Anticancer Drug Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Natural Substrates | Anticancer Drug Substrates | Tissue Expression | Expression in Cancer Compared to Normal Tissues * | References |
|---|---|---|---|---|---|---|
| SLC2A2 | GLUT2 | Glucose, glucosamine | Streptozotocin | Liver, pancreatic beta-cells, intestinal and renal epithelial cells | High expression: hepatocellular carcinoma c, invasive ductal carcinoma c, invasive colon tubular carcinoma c, pancreatic adenocarcinoma c, lung mesothelioma c, | [28,29,30,31,32] |
| SLC7A5 | LAT1 | Phenylalanine, leucine, tryptophan | Melphalan, acivicin | Brain (endothelial cells), testis, retina, esophagus, testis, placenta and bone marrow | High expression: colorectal cancer a, gliomablastoma b, triple-negative breast cancer and HER2-positive breast cancers and MYC driver ER-positive breast cancer c,d | [33,34,35,36,37,38,39,40,41,42,43] |
| SLC19A1 | RFC1 | Reduced folates, antifolates | Methotrexate, pemetrexed | Ubiquitous | High expression: non-small cell lung carcinoma and squamous cell carcinoma c, MYCN- amplified neuroblastoma, colorectal carcinoma d, urothelial bladder carcinomas d Low expression: ovarian cancers c | [44,45,46,47,48,49,50,51,52,53] |
| SLCO1A2 | OATP1A2 | Bile salts, organic anions and cations | Imatinib, methotrexate | Brain (endothelial cells), kidney, intestine, liver, eye | High expression: breast cancer, glioblastoma c Low expression: colorectal carcinoma liver metastases a, colorectal carcinoma d, | [35,54,55,56,57,58,59] |
| SLCO1B1 | OATP1B1 | Bile salts, organic anions | Cisplatin, carboplatin, Oxaliplatin, regorafenib, belzutifan, SN-38, etoposide, tamoxifen, sorafenib | Liver | High expression: ovarian d, colon d and pancreaticc cancers, castration resistant prostate cancer metastases d Low expression: hepatocellular carcinoma a, low in colorectal carcinoma liver metastases a, | [35,54,55,60,61,62,63] |
| SLCO1B3 | OATP1B3 | Bile salts, organic anions | Nilotinib, belzutifan, docetaxel, SN-38, oxaliplatin, carboplatin, cisplatin, imatinib, gefitinib, sorafenib, belzutifan | Liver | High expression: breast cancer c,d, colorectal carcinoma c,d, castration resistant prostate cancer metastases d Low expression: hepatocellular carcinomaa, colorectal carcinoma liver metastases a | [35,55,60,63] |
| SLCO2B1 | OATP2B1 | E-3-S, DHEAS | Etoposide, erlotinib | Liver, placenta, intestine, eye | High expression: prostate cancer with high Gleason score d and castration resistant prostate cancer metastases d Low expression: hepatocellular carcinoma a, low in colorectal carcinoma liver metastases a | [35,63,64] |
| SLCO4C1 | OATP4C1 | L-homoarginine | Methotrexate | Kidney | High expression: castration resistant prostate cancer metastases d | [35,63] |
| SLC22A1 | OCT1 | Organic cations | Dasatanib, nintendanib | Liver, small intestine, kidney, lung, heart, skeletal muscle, brain (endothelial cells, neurons), adipose tissue, immune cells | Low expression: hepatocellular carcinoma a, colorectal carcinoma liver metastases a, cholangiocellular carcinoma c,d | [60,65,66,67,68,69,70] |
| SLC22A2 | OCT2 | Organic cations | Cisplatin, oxaliplatin | Kidney, small intestine, trachea and bronchi placenta, thymus, brain (neurons, endothelial cells), inner ear | High expression: clear cell renal carcinoma c,d Low expression: hepatocellular carcinoma c | [65,67,68,70,71,72,73,74,75] |
| SLC22A3 | OCT3 | Organic cations | Oxaliplatin | Heart, skeletal muscle, brain (neurons, glial cells, choroid plexus), small intestine, liver, lung, kidney, urinary bladder, mammary gland, skin blood vessels | High expression: head and neck squamous cell carcinoma c, colorectal carcinoma c Low expression: colorectal carcinoma liver metastases a, hepatocellular carcinoma c, cholangiocellular carcinoma c,d | [55,65,68,69,76,77,78,79,80,81,82,83,84] |
| SLC22A4 | OCTN1 | Ergothioneine, zwitterions, organic cations | Doxorubicin, mitoxantrone, imatinib, cytarabine | Kidney, intestine, spleen, heart, skeletal muscle, brain, mammary gland, thymus, prostate, airways, testis, eye, foetal liver, sperm, immune cells | Not found | [66,85,86,87,88,89,90] |
| SLC22A5 | OCTN2 | Zwitterions (L-carnitine), organic cations | Etoposide, imatinib | Skeletal muscle, kidney, prostate, lung, pancreas, heart, small intestine, adrenal gland, thyroid gland, liver | High expression: ER-positive breast cancers d, glioma | [66,91,92,93,94,95,96] |
| SLC22A6 | OAT1 | Organic anions | Methotrexate, bleomycin | Kidney, placenta, choroid plexus | Low expression: kidney renal clear cell carcinoma and kidney renal papillary cell carcinoma d | [97,98,99,100,101,102] |
| SLC22A7 | OAT2 | Organic anions | Irinotecan, docetaxel, 5-fluorouracil | Liver, kidney, eye | Low expression: colorectal carcinoma liver metastases a, kidney renal clear cell carcinoma and kidney renal papillary cell carcinoma d | [55,97,102,103,104,105,106,107] |
| SLC22A8 | OAT3 | Organic anions | Methotrexate | Kidney, choroid plexus | Low expression: kidney renal clear cell carcinoma and kidney renal papillary cell carcinoma d | [97,98,99,100,101,102] |
| SLC28A1 | CNT1 | Pyrimidine nucleosides and adenosine | Gemcitabine | Kidney, liver, small intestine, bone marrow macrophages and the brain | High expression: serous, mucinous and endometroid ovarian carcinomas c, uterine cervix carcinomas c Low expression: clear cell ovarian carcinomas c, pancreatic ductal adenocarcinoma d | [108,109,110,111,112,113,114,115] |
| SLC28A2 | CNT2 | Purine nucleosides and uridine | Gemcitabine,5-fluorouridine, 5-fluoro-2′-deoxyuridine | Heart, skeletal muscle, liver, kidney, intestine, pancreas, placenta, brain, spleen, rectum, colon, immune system | High expression: lung, ovary, uterus and prostate cancers d Low expression: hepatocellular carcinoma, colorectal carcinoma, colorectal carcinoma liver metastases d, kidney, stomach, rectum and small intestine cancers d | [110,113,116,117,118,119,120,121] |
| SLC29A1 | ENT1 | Nucleosides, nucleobases, creatinine, guanidine, thiamine | Gemcitabine, cytarabine, 5-fluorouracil, 6-mercaptopurine | Ubiquitous | High expression: ovarian, endometrial and uterine cervix carcinomas c Low expression: pancreatic ductal adenocarcinoma d, prostate cancer d | [35,113,114,115,122,123,124,125] |
| SLC29A2 | ENT2 | Nucleosides, creatinine, thiamine, carnitine | 5-fluorouracil, gemcitabine | Ubiquitous | High expression: mantle-cell lymphoma d, hepatocellular carcinoma d, ovarian, endometrial and uterine cervix carcinomas c | [35,113,114,122,123,124,125,126,127] |
| SLC31A1 | CTR1 | Copper (I) | Cisplatin, carboplatin, oxaliplatin | Liver, lung | High expression: bladder cancer c | [128,129,130,131,132] |
| SLC46A1 | PCFT | Reduced folates, folic acid | Pemetrexed | Small intestine, choroid plexus, kidney, liver, placenta, retinal pigment epithelium | High expression: colorectal cancer d, ER-positive breast cancer d | [51,133,134,135,136,137,138,139,140,141] |
| SLC47A1 | MATE1 | TEA, MPP | Oxaliplatin | Liver, kidney, muscle | Low expression: KRAS-driven colorectal carcinoma c | [142,143,144,145,146] |
| SLC47A2 | MATE2 | TEA, MPP | Oxaliplatin | Kidney | Not found | [143,146,147,148] |
5. SLC Transporters in Cancer
5.1. Drug Transporters
5.1.1. Glucose Transporter 2
5.1.2. Large Neutral Amino Acids Transporter Small Subunit 1
5.1.3. Reduced Folate Transporter
5.1.4. Organic Anion Transporting Polypeptides
5.1.5. Organic Cation Transporters
5.1.6. Organic Cation Transporter Novel Type (OCTNs)
5.1.7. Organic Anion Transporters
5.1.8. Concentrative Nucleoside Transporters
5.1.9. Equilibrative Nucleoside Transporters
5.1.10. Copper Transporter 1
5.1.11. Proton-Coupled Folate Transporter
5.1.12. Multidrug and Toxin Extrusion Proteins 1 and 2
6. Strategies to Overcome Low SLC Transporter Expression-Mediated Drug Resistance
6.1. Modulation of Transporter Expression
6.1.1. Impact of Nuclear Receptors on Transporter Expression
6.1.2. Impact of Epigenetics on Transporter Expression
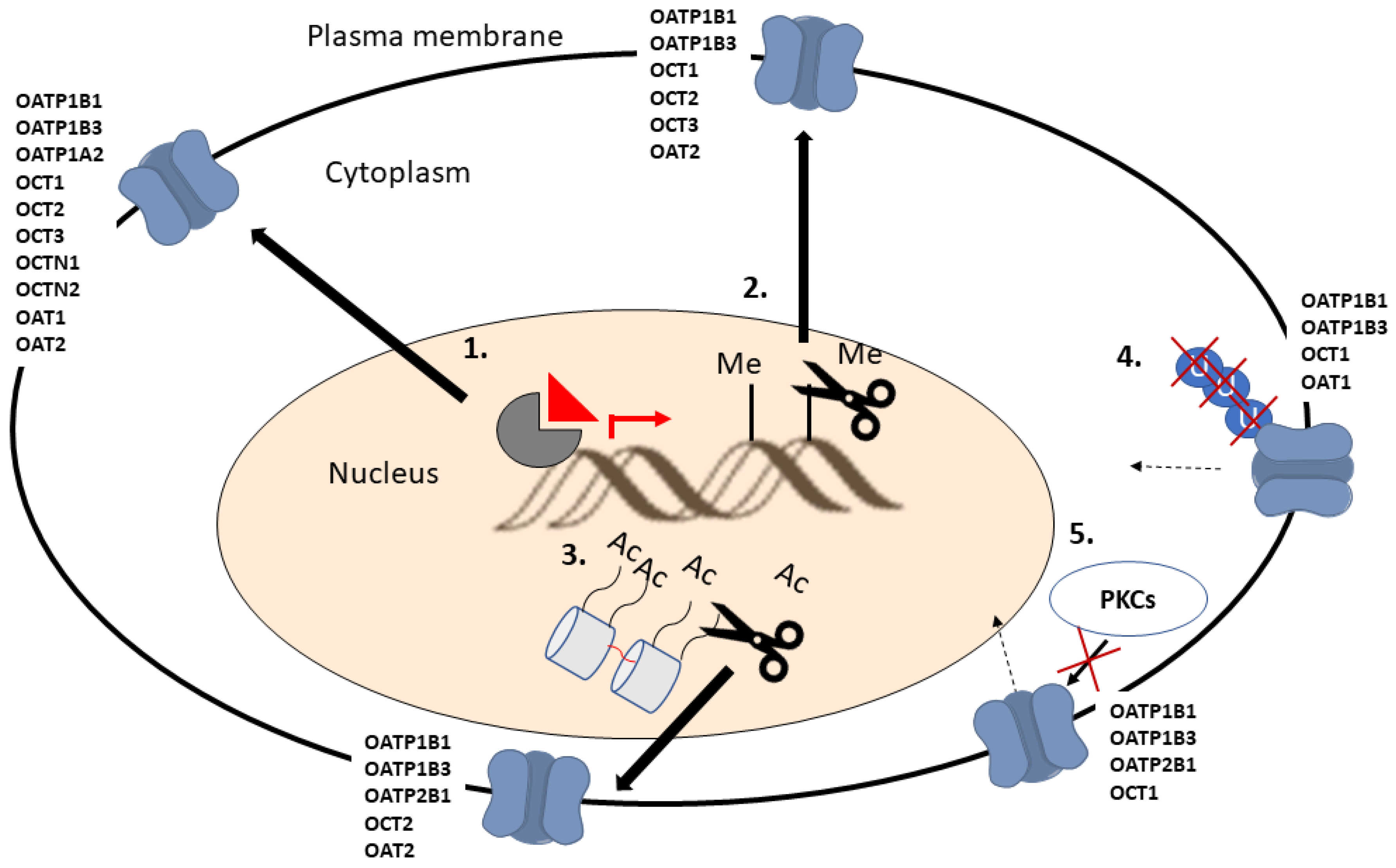
6.1.3. Impact of Post-Translational Modifications on Transporter Expression, Localization and Function
6.1.4. Impact of Anticancer Drugs on Transporter Expression and Function
6.1.5. Tumor Microenvironment Impact on SLC Drug Transporter Expression and Function
6.2. Drug Delivery via Transporters Highly Expressed in Cancer Cells and Exploiting the Cancer Dependence of Transporters
6.2.1. Glucose Transporter 1 (GLUT1 Encoded by SLC2A1)
6.2.2. Monocarboxylate Transporters (MCTs)
6.2.3. Amino Acid Transporters
6.2.4. Proton-Coupled Peptide Transporter 1
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Moller, B. Predicting the future burden of cancer. Nat. Rev. Cancer 2006, 6, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Sutherland, R.; Meeson, A.; Lowes, S. Solute transporters and malignancy: Establishing the role of uptake transporters in breast cancer and breast cancer metastasis. Cancer Metastasis Rev. 2020, 39, 919–932. [Google Scholar] [CrossRef]
- Wu, G.; Wilson, G.; George, J.; Liddle, C.; Hebbard, L.; Qiao, L. Overcoming treatment resistance in cancer: Current understanding and tactics. Cancer Lett. 2017, 387, 69–76. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Aleksakhina, S.N.; Kashyap, A.; Imyanitov, E.N. Mechanisms of acquired tumor drug resistance. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188310. [Google Scholar] [CrossRef]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Konishi, I. Correlation of anti-tumour drug resistance with epigenetic regulation. Br. J. Cancer 2021, 124, 681–682. [Google Scholar] [CrossRef]
- Taylor, S.T.; Hickman, J.A.; Dive, C. Epigenetic determinants of resistance to etoposide regulation of Bcl-X(L) and Bax by tumor microenvironmental factors. J. Natl. Cancer Inst. 2000, 92, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Thwaites, D.T. Hijacking solute carriers for proton-coupled drug transport. Physiology 2010, 25, 364–377. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, L.A.; Caplan, M.J. The cell biology of ion pumps: Sorting and regulation. Eur. J. Cell Biol. 2000, 79, 557–563. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.E.; Kim, J.Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Kim, T.H.; Hyun, D.W.; et al. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef]
- Daskalow, K.; Pfander, D.; Weichert, W.; Rohwer, N.; Thelen, A.; Neuhaus, P.; Jonas, S.; Wiedenmann, B.; Benckert, C.; Cramer, T. Distinct temporospatial expression patterns of glycolysis-related proteins in human hepatocellular carcinoma. Histochem. Cell Biol. 2009, 132, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.; Ulloa, V.; Rodriguez, F.; Reinicke, K.; Yanez, A.J.; Garcia Mde, L.; Medina, R.A.; Carrasco, M.; Barberis, S.; Castro, T.; et al. Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: Ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J. Cell Physiol. 2006, 207, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.N. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev. 1990, 70, 43–77. [Google Scholar] [CrossRef] [PubMed]
- Oxender, D.L.; Christensen, H.N. Evidence for two types of mediation of neutral and amino-acid transport in Ehrlich cells. Nature 1963, 197, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Dus-Szachniewicz, K.; Ostasiewicz, P.; Ziolkowski, P.; Rakus, D.; Mann, M. Absolute Proteome Analysis of Colorectal Mucosa, Adenoma, and Cancer Reveals Drastic Changes in Fatty Acid Metabolism and Plasma Membrane Transporters. J. Proteome Res. 2015, 14, 4005–4018. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Ostasiewicz, P.; Dus, K.; Zielinska, D.F.; Gnad, F.; Mann, M. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol. 2012, 8, 611. [Google Scholar] [CrossRef]
- Nawashiro, H.; Otani, N.; Uozumi, Y.; Ooigawa, H.; Toyooka, T.; Suzuki, T.; Katoh, H.; Tsuzuki, N.; Ohnuki, A.; Shima, K.; et al. High expression of L-type amino acid transporter 1 in infiltrating glioma cells. Brain Tumor Pathol. 2005, 22, 89–91. [Google Scholar] [CrossRef]
- El Ansari, R.; Craze, M.L.; Miligy, I.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. The amino acid transporter SLC7A5 confers a poor prognosis in the highly proliferative breast cancer subtypes and is a key therapeutic target in luminal B tumours. Breast Cancer Res. 2018, 20, 21. [Google Scholar] [CrossRef]
- Maimaiti, M.; Sakamoto, S.; Yamada, Y.; Sugiura, M.; Rii, J.; Takeuchi, N.; Imamura, Y.; Furihata, T.; Ando, K.; Higuchi, K.; et al. Expression of L-type amino acid transporter 1 as a molecular target for prognostic and therapeutic indicators in bladder carcinoma. Sci. Rep. 2020, 10, 1292. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Ichinoe, M.; Mikami, T.; Nakada, N.; Hana, K.; Koizumi, W.; Endou, H.; Okayasu, I. High expression of L-type amino acid transporter 1 (LAT1) predicts poor prognosis in pancreatic ductal adenocarcinomas. J. Clin. Pathol. 2012, 65, 1019–1023. [Google Scholar] [CrossRef]
- Lin, J.; Raoof, D.A.; Thomas, D.G.; Greenson, J.K.; Giordano, T.J.; Robinson, G.S.; Bourner, M.J.; Bauer, C.T.; Orringer, M.B.; Beer, D.G. L-type amino acid transporter-1 overexpression and melphalan sensitivity in Barrett’s adenocarcinoma. Neoplasia 2004, 6, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Greig, N.H.; Sweeney, D.J.; Rapoport, S.I. Melphalan concentration dependent plasma protein binding in healthy humans and rats. Eur. J. Clin. Pharmacol. 1987, 32, 179–185. [Google Scholar] [CrossRef]
- Goldman, I.D. The characteristics of the membrane transport of amethopterin and the naturally occurring folates. Ann. N. Y. Acad. Sci. 1971, 186, 400–422. [Google Scholar] [CrossRef]
- Goldman, I.D.; Lichtenstein, N.S.; Oliverio, V.T. Carrier-mediated transport of the folic acid analogue, methotrexate, in the L1210 leukemia cell. J. Biol. Chem. 1968, 243, 5007–5017. [Google Scholar] [CrossRef]
- Sierra, E.E.; Brigle, K.E.; Spinella, M.J.; Goldman, I.D. pH dependence of methotrexate transport by the reduced folate carrier and the folate receptor in L1210 leukemia cells. Further evidence for a third route mediated at low pH. Biochem. Pharmacol. 1997, 53, 223–231. [Google Scholar] [CrossRef]
- Alam, C.; Hoque, M.T.; Finnell, R.H.; Goldman, I.D.; Bendayan, R. Regulation of Reduced Folate Carrier (RFC) by Vitamin D Receptor at the Blood-Brain Barrier. Mol. Pharm. 2017, 14, 3848–3858. [Google Scholar] [CrossRef]
- Westerhof, G.R.; Schornagel, J.H.; Kathmann, I.; Jackman, A.L.; Rosowsky, A.; Forsch, R.A.; Hynes, J.B.; Boyle, F.T.; Peters, G.J.; Pinedo, H.M.; et al. Carrier- and receptor-mediated transport of folate antagonists targeting folate-dependent enzymes: Correlates of molecular-structure and biological activity. Mol. Pharmacol. 1995, 48, 459–471. [Google Scholar]
- Zhang, X.; Zhang, D.; Huang, L.; Li, G.; Chen, L.; Ma, J.; Li, M.; Wei, M.; Zhou, W.; Zhou, C.; et al. Discovery of Novel Biomarkers of Therapeutic Responses in Han Chinese Pemetrexed-Based Treated Advanced NSCLC Patients. Front. Pharmacol. 2019, 10, 944. [Google Scholar] [CrossRef]
- Lau, D.T.; Flemming, C.L.; Gherardi, S.; Perini, G.; Oberthuer, A.; Fischer, M.; Juraeva, D.; Brors, B.; Xue, C.; Norris, M.D.; et al. MYCN amplification confers enhanced folate dependence and methotrexate sensitivity in neuroblastoma. Oncotarget 2015, 6, 15510–15523. [Google Scholar] [CrossRef] [Green Version]
- Odin, E.; Sonden, A.; Gustavsson, B.; Carlsson, G.; Wettergren, Y. Expression of Folate Pathway Genes in Stage III Colorectal Cancer Correlates with Recurrence Status Following Adjuvant Bolus 5-FU-Based Chemotherapy. Mol. Med. 2015, 21, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.I.; Behrens, C.; Woods, D.M.; Lin, H.; Suraokar, M.; Kadara, H.; Hofstetter, W.; Kalhor, N.; Lee, J.J.; Franklin, W.; et al. High expression of folate receptor alpha in lung cancer correlates with adenocarcinoma histology and EGFR [corrected] mutation. J. Thorac. Oncol. 2012, 7, 833–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, M.K.; Kong, D.S.; Chan, H.Y.; Wong, E.S.; Ip, P.P.; Jiang, L.; Ngan, H.Y.; Le, X.F.; Cheung, A.N. Paradoxical impact of two folate receptors, FRalpha and RFC, in ovarian cancer: Effect on cell proliferation, invasion and clinical outcome. PLoS ONE 2012, 7, e47201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers. Arch. 2004, 447, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilogianni, A.M.; Al-Majdoub, Z.M.; Achour, B.; Annie Peters, S.; Barber, J.; Rostami-Hodjegan, A. Quantitative Proteomics of Hepatic Drug-Metabolizing Enzymes and Transporters in Patients With Colorectal Cancer Metastasis. Clin. Pharmacol. Ther. 2022, 112, 699–710. [Google Scholar] [CrossRef]
- Meyer zu Schwabedissen, H.E.; Tirona, R.G.; Yip, C.S.; Ho, R.H.; Kim, R.B. Interplay between the nuclear receptor pregnane X receptor and the uptake transporter organic anion transporter polypeptide 1A2 selectively enhances estrogen effects in breast cancer. Cancer Res. 2008, 68, 9338–9347. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Wang, G.; Liu, S.; Zhang, Z.; Liu, C.; Li, F.; Liu, X.; Meng, L.; Yang, H.; Li, C.; et al. Highly expressed SLCO1B3 inhibits the occurrence and development of breast cancer and can be used as a clinical indicator of prognosis. Sci. Rep. 2021, 11, 631. [Google Scholar] [CrossRef]
- Ballestero, M.R.; Monte, M.J.; Briz, O.; Jimenez, F.; Gonzalez-San Martin, F.; Marin, J.J. Expression of transporters potentially involved in the targeting of cytostatic bile acid derivatives to colon cancer and polyps. Biochem. Pharmacol. 2006, 72, 729–738. [Google Scholar] [CrossRef]
- Cooper, E.; Woolf, Z.; Swanson, M.E.V.; Correia, J.; Schweder, P.; Mee, E.; Heppner, P.; Turner, C.; Faull, R.L.M.; Scotter, E.L.; et al. Single-cell image analysis reveals over-expression of organic anion transporting polypeptides (OATPs) in human glioblastoma tissue. Neurooncol. Adv. 2022, 4, vdac166. [Google Scholar] [CrossRef]
- Billington, S.; Ray, A.S.; Salphati, L.; Xiao, G.; Chu, X.; Humphreys, W.G.; Liao, M.; Lee, C.A.; Mathias, A.; Hop, C.; et al. Transporter Expression in Noncancerous and Cancerous Liver Tissue from Donors with Hepatocellular Carcinoma and Chronic Hepatitis C Infection Quantified by LC-MS/MS Proteomics. Drug Metab. Dispos. 2018, 46, 189–196. [Google Scholar] [CrossRef]
- Svoboda, M.; Wlcek, K.; Taferner, B.; Hering, S.; Stieger, B.; Tong, D.; Zeillinger, R.; Thalhammer, T.; Jager, W. Expression of organic anion-transporting polypeptides 1B1 and 1B3 in ovarian cancer cells: Relevance for paclitaxel transport. Biomed. Pharmacother. 2011, 65, 417–426. [Google Scholar] [CrossRef]
- Kounnis, V.; Ioachim, E.; Svoboda, M.; Tzakos, A.; Sainis, I.; Thalhammer, T.; Steiner, G.; Briasoulis, E. Expression of organic anion-transporting polypeptides 1B3, 1B1, and 1A2 in human pancreatic cancer reveals a new class of potential therapeutic targets. Onco Targets Ther. 2011, 4, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.L.; Kwon, E.M.; Ostrander, E.A.; Montgomery, R.B.; Lin, D.W.; Vessella, R.; Stanford, J.L.; Mostaghel, E.A. Expression of SLCO transport genes in castration-resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Terakawa, T.; Katsuta, E.; Yan, L.; Turaga, N.; McDonald, K.A.; Fujisawa, M.; Guru, K.A.; Takabe, K. High expression of SLCO2B1 is associated with prostate cancer recurrence after radical prostatectomy. Oncotarget 2018, 9, 14207–14218. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Aspects Med. 2013, 34, 413–435. [Google Scholar] [CrossRef]
- Neul, C.; Schaeffeler, E.; Sparreboom, A.; Laufer, S.; Schwab, M.; Nies, A.T. Impact of Membrane Drug Transporters on Resistance to Small-Molecule Tyrosine Kinase Inhibitors. Trends Pharmacol. Sci. 2016, 37, 904–932. [Google Scholar] [CrossRef]
- Koepsell, H. Polyspecific organic cation transporters: Their functions and interactions with drugs. Trends Pharmacol. Sci. 2004, 25, 375–381. [Google Scholar] [CrossRef]
- Jonker, J.W.; Schinkel, A.H. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J. Pharmacol. Exp. Ther. 2004, 308, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Lautem, A.; Heise, M.; Grasel, A.; Hoppe-Lotichius, M.; Weiler, N.; Foltys, D.; Knapstein, J.; Schattenberg, J.M.; Schad, A.; Zimmermann, A.; et al. Downregulation of organic cation transporter 1 (SLC22A1) is associated with tumor progression and reduced patient survival in human cholangiocellular carcinoma. Int. J. Oncol. 2013, 42, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Schaeffeler, E.; Hellerbrand, C.; Nies, A.T.; Winter, S.; Kruck, S.; Hofmann, U.; van der Kuip, H.; Zanger, U.M.; Koepsell, H.; Schwab, M. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 2011, 3, 82. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Leblanc, A.F.; Uddin, M.E.; Kim, J.Y.; Chen, M.; Eisenmann, E.D.; Gibson, A.A.; Li, Y.; Hong, K.W.; DiGiacomo, D.; et al. Neuronal uptake transporters contribute to oxaliplatin neurotoxicity in mice. J. Clin. Investig. 2020, 130, 4601–4606. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Schmitt, B.M.; Gorboulev, V. Organic cation transporters. Rev. Physiol. Biochem. Pharmacol. 2003, 150, 36–90. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Fisel, P.; Buttner, F.; Rausch, S.; D’Amico, D.; Hennenlotter, J.; Kruck, S.; Nies, A.T.; Stenzl, A.; Junker, K.; et al. Methylomes of renal cell lines and tumors or metastases differ significantly with impact on pharmacogenes. Sci. Rep. 2016, 6, 29930. [Google Scholar] [CrossRef]
- Gu, J.; Wang, L.; Li, T.; Tang, S.; Wang, Y.; Zhang, W.; Jiang, X. Role and mechanism of organic cation transporter 3 in oxaliplatin treatment of colon cancer in vitro and in vivo. Oncol. Rep. 2019, 42, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.; Koepsell, H.; Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860. [Google Scholar] [CrossRef]
- Inazu, M.; Takeda, H.; Matsumiya, T. Expression and functional characterization of the extraneuronal monoamine transporter in normal human astrocytes. J. Neurochem. 2003, 84, 43–52. [Google Scholar] [CrossRef]
- Hayer-Zillgen, M.; Bruss, M.; Bonisch, H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br. J. Pharmacol. 2002, 136, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Sata, R.; Ohtani, H.; Tsujimoto, M.; Murakami, H.; Koyabu, N.; Nakamura, T.; Uchiumi, T.; Kuwano, M.; Nagata, H.; Tsukimori, K.; et al. Functional analysis of organic cation transporter 3 expressed in human placenta. J. Pharmacol. Exp. Ther. 2005, 315, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Kekuda, R.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Chen, J.; Conway, S.J.; Ganapathy, V. Identity of the organic cation transporter OCT3 as the extraneuronal monoamine transporter (uptake2) and evidence for the expression of the transporter in the brain. J. Biol. Chem. 1998, 273, 32776–32786. [Google Scholar] [CrossRef]
- Hsu, C.M.; Lin, P.M.; Chang, J.G.; Lin, H.C.; Li, S.H.; Lin, S.F.; Yang, M.Y. Upregulated SLC22A3 has a potential for improving survival of patients with head and neck squamous cell carcinoma receiving cisplatin treatment. Oncotarget 2017, 8, 74348–74358. [Google Scholar] [CrossRef] [Green Version]
- Yokoo, S.; Masuda, S.; Yonezawa, A.; Terada, T.; Katsura, T.; Inui, K. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab. Dispos. 2008, 36, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Namisaki, T.; Schaeffeler, E.; Fukui, H.; Yoshiji, H.; Nakajima, Y.; Fritz, P.; Schwab, M.; Nies, A.T. Differential expression of drug uptake and efflux transporters in Japanese patients with hepatocellular carcinoma. Drug Metab. Dispos. 2014, 42, 2033–2040. [Google Scholar] [CrossRef] [Green Version]
- Grundemann, D.; Harlfinger, S.; Golz, S.; Geerts, A.; Lazar, A.; Berkels, R.; Jung, N.; Rubbert, A.; Schomig, E. Discovery of the ergothioneine transporter. Proc. Natl. Acad. Sci. USA 2005, 102, 5256–5261. [Google Scholar] [CrossRef] [Green Version]
- Pochini, L.; Scalise, M.; Galluccio, M.; Pani, G.; Siminovitch, K.A.; Indiveri, C. The human OCTN1 (SLC22A4) reconstituted in liposomes catalyzes acetylcholine transport which is defective in the mutant L503F associated to the Crohn’s disease. Biochim. Biophys. Acta 2012, 1818, 559–565. [Google Scholar] [CrossRef]
- Drenberg, C.D.; Gibson, A.A.; Pounds, S.B.; Shi, L.; Rhinehart, D.P.; Li, L.; Hu, S.; Du, G.; Nies, A.T.; Schwab, M.; et al. OCTN1 Is a High-Affinity Carrier of Nucleoside Analogues. Cancer Res. 2017, 77, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- Garrett, Q.; Xu, S.; Simmons, P.A.; Vehige, J.; Flanagan, J.L.; Willcox, M.D. Expression and localization of carnitine/organic cation transporter OCTN1 and OCTN2 in ocular epithelium. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4844–4849. [Google Scholar] [CrossRef]
- Wada, E.; Koyanagi, S.; Kusunose, N.; Akamine, T.; Masui, H.; Hashimoto, H.; Matsunaga, N.; Ohdo, S. Modulation of peroxisome proliferator-activated receptor-alpha activity by bile acids causes circadian changes in the intestinal expression of Octn1/Slc22a4 in mice. Mol. Pharmacol. 2015, 87, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Meetam, P.; Srimaroeng, C.; Soodvilai, S.; Chatsudthipong, V. Regulatory role of testosterone in organic cation transport: In vivo and in vitro studies. Biol. Pharm. Bull. 2009, 32, 982–987. [Google Scholar] [CrossRef] [Green Version]
- Pochini, L.; Galluccio, M.; Scalise, M.; Console, L.; Indiveri, C. OCTN: A Small Transporter Subfamily with Great Relevance to Human Pathophysiology, Drug Discovery, and Diagnostics. SLAS Discov. 2019, 24, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Lancaster, C.S.; Zuo, Z.; Hu, S.; Chen, Z.; Rubnitz, J.E.; Baker, S.D.; Sparreboom, A. Inhibition of OCTN2-mediated transport of carnitine by etoposide. Mol Cancer Ther. 2012, 11, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H.; Endou, H. The SLC22 drug transporter family. Pflugers. Arch. 2004, 447, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, W.; Prasad, P.D.; Seth, P.; Rajan, D.P.; Leibach, F.H.; Chen, J.; Conway, S.J.; Ganapathy, V. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J. Pharmacol. Exp. Ther. 1999, 290, 1482–1492. [Google Scholar] [PubMed]
- Wang, C.; Uray, I.P.; Mazumdar, A.; Mayer, J.A.; Brown, P.H. SLC22A5/OCTN2 expression in breast cancer is induced by estrogen via a novel intronic estrogen-response element (ERE). Breast Cancer Res. Treat. 2012, 134, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Fink, M.A.; Paland, H.; Herzog, S.; Grube, M.; Vogelgesang, S.; Weitmann, K.; Bialke, A.; Hoffmann, W.; Rauch, B.H.; Schroeder, H.W.S.; et al. L-Carnitine-Mediated Tumor Cell Protection and Poor Patient Survival Associated with OCTN2 Overexpression in Glioblastoma Multiforme. Clin. Cancer Res. 2019, 25, 2874–2886. [Google Scholar] [CrossRef]
- Anzai, N.; Kanai, Y.; Endou, H. Organic anion transporter family: Current knowledge. J. Pharmacol. Sci. 2006, 100, 411–426. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Liu, Z.; Wang, C.; Meng, Q.; Huo, X.; Liu, Q.; Sun, H.; Sun, P.; Yang, X.; Ma, X.; et al. P-gp, MRP2 and OAT1/OAT3 mediate the drug-drug interaction between resveratrol and methotrexate. Toxicol. Appl. Pharmacol. 2016, 306, 27–35. [Google Scholar] [CrossRef]
- Iwaki, M.; Shimada, H.; Irino, Y.; Take, M.; Egashira, S. Inhibition of Methotrexate Uptake via Organic Anion Transporters OAT1 and OAT3 by Glucuronides of Nonsteroidal Anti-inflammatory Drugs. Biol. Pharm. Bull. 2017, 40, 926–931. [Google Scholar] [CrossRef] [Green Version]
- Hosoyamada, M.; Sekine, T.; Kanai, Y.; Endou, H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am. J. Physiol. 1999, 276, F122–F128. [Google Scholar] [CrossRef]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K.I. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar] [CrossRef]
- Whisenant, T.C.; Nigam, S.K. Organic Anion Transporters (OAT) and Other SLC22 Transporters in Progression of Renal Cell Carcinoma. Cancers 2022, 14, 4772. [Google Scholar] [CrossRef]
- Marada, V.V.; Florl, S.; Kuhne, A.; Muller, J.; Burckhardt, G.; Hagos, Y. Interaction of human organic anion transporter 2 (OAT2) and sodium taurocholate cotransporting polypeptide (NTCP) with antineoplastic drugs. Pharmacol. Res. 2015, 91, 78–87. [Google Scholar] [CrossRef]
- Shen, H.; Liu, T.; Morse, B.L.; Zhao, Y.; Zhang, Y.; Qiu, X.; Chen, C.; Lewin, A.C.; Wang, X.T.; Liu, G.; et al. Characterization of Organic Anion Transporter 2 (SLC22A7): A Highly Efficient Transporter for Creatinine and Species-Dependent Renal Tubular Expression. Drug Metab. Dispos. 2015, 43, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Vapurcuyan, A.; Shahidullah, M.; Aleksunes, L.M.; Pelis, R.M. Expression of organic anion transporter 2 in the human kidney and its potential role in the tubular secretion of guanine-containing antiviral drugs. Drug Metab. Dispos. 2012, 40, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Breljak, D.; Ljubojevic, M.; Hagos, Y.; Micek, V.; Balen Eror, D.; Vrhovac Madunic, I.; Brzica, H.; Karaica, D.; Radovic, N.; Kraus, O.; et al. Distribution of organic anion transporters NaDC3 and OAT1-3 along the human nephron. Am. J. Physiol. Renal. Physiol. 2016, 311, F227–F238. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, A.; Geier, E.; Stocker, S.L.; Cropp, C.D.; Grigorenko, E.; Bloomer, M.; Siegenthaler, J.; Xu, L.; Basile, A.S.; Tang-Liu, D.D.; et al. Gene expression profiling of transporters in the solute carrier and ATP-binding cassette superfamilies in human eye substructures. Mol. Pharm. 2013, 10, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.M.; Ng, A.M.; Yao, S.Y.; Labedz, K.A.; Knaus, E.E.; Wiebe, L.I.; Cass, C.E.; Baldwin, S.A.; Chen, X.Z.; Karpinski, E.; et al. Electrophysiological characterization of a recombinant human Na+-coupled nucleoside transporter (hCNT1) produced in Xenopus oocytes. J. Physiol. 2004, 558, 807–823. [Google Scholar] [CrossRef]
- Garcia-Manteiga, J.; Molina-Arcas, M.; Casado, F.J.; Mazo, A.; Pastor-Anglada, M. Nucleoside transporter profiles in human pancreatic cancer cells: Role of hCNT1 in 2’,2’-difluorodeoxycytidine- induced cytotoxicity. Clin. Cancer Res. 2003, 9, 5000–5008. [Google Scholar]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. 5), v7–v12. [Google Scholar] [CrossRef]
- Huang, Q.Q.; Harvey, C.M.; Paterson, A.R.; Cass, C.E.; Young, J.D. Functional expression of Na(+)-dependent nucleoside transport systems of rat intestine in isolated oocytes of Xenopus laevis. Demonstration that rat jejunum expresses the purine-selective system N1 (cif) and a second, novel system N3 having broad specificity for purine and pyrimidine nucleosides. J. Biol. Chem. 1993, 268, 20613–20619. [Google Scholar] [PubMed]
- Anderson, C.M.; Xiong, W.; Young, J.D.; Cass, C.E.; Parkinson, F.E. Demonstration of the existence of mRNAs encoding N1/cif and N2/cit sodium/nucleoside cotransporters in rat brain. Brain Res. Mol. Brain Res. 1996, 42, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Pennycooke, M.; Chaudary, N.; Shuralyova, I.; Zhang, Y.; Coe, I.R. Differential expression of human nucleoside transporters in normal and tumor tissue. Biochem. Biophys. Res. Commun. 2001, 280, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Farre, X.; Guillen-Gomez, E.; Sanchez, L.; Hardisson, D.; Plaza, Y.; Lloberas, J.; Casado, F.J.; Palacios, J.; Pastor-Anglada, M. Expression of the nucleoside-derived drug transporters hCNT1, hENT1 and hENT2 in gynecologic tumors. Int. J. Cancer 2004, 112, 959–966. [Google Scholar] [CrossRef]
- Mohelnikova-Duchonova, B.; Brynychova, V.; Hlavac, V.; Kocik, M.; Oliverius, M.; Hlavsa, J.; Honsova, E.; Mazanec, J.; Kala, Z.; Melichar, B.; et al. The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 669–682. [Google Scholar] [CrossRef]
- Lang, T.T.; Selner, M.; Young, J.D.; Cass, C.E. Acquisition of human concentrative nucleoside transporter 2 (hcnt2) activity by gene transfer confers sensitivity to fluoropyrimidine nucleosides in drug-resistant leukemia cells. Mol. Pharmacol. 2001, 60, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.M.; Slugoski, M.D.; Cass, C.E.; Baldwin, S.A.; Karpinski, E.; Young, J.D. Cation coupling properties of human concentrative nucleoside transporters hCNT1, hCNT2 and hCNT3. Mol. Membr. Biol. 2007, 24, 53–64. [Google Scholar] [CrossRef]
- Che, M.; Ortiz, D.F.; Arias, I.M. Primary structure and functional expression of a cDNA encoding the bile canalicular, purine-specific Na(+)-nucleoside cotransporter. J. Biol. Chem. 1995, 270, 13596–13599. [Google Scholar] [CrossRef] [Green Version]
- Guillen-Gomez, E.; Calbet, M.; Casado, J.; de Lecea, L.; Soriano, E.; Pastor-Anglada, M.; Burgaya, F. Distribution of CNT2 and ENT1 transcripts in rat brain: Selective decrease of CNT2 mRNA in the cerebral cortex of sleep-deprived rats. J. Neurochem. 2004, 90, 883–893. [Google Scholar] [CrossRef]
- Minuesa, G.; Purcet, S.; Erkizia, I.; Molina-Arcas, M.; Bofill, M.; Izquierdo-Useros, N.; Casado, F.J.; Clotet, B.; Pastor-Anglada, M.; Martinez-Picado, J. Expression and functionality of anti-human immunodeficiency virus and anticancer drug uptake transporters in immune cells. J. Pharmacol. Exp. Ther. 2008, 324, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, D.; Zhang, C.; Zhang, Z.; Chen, X.; Lian, J.; Liu, J.; Wang, G.; Yuan, W.; Sun, Z.; et al. Identification of liver metastasis-associated genes in human colon carcinoma by mRNA profiling. Chin. J. Cancer Res. 2018, 30, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Yao, S.Y.; Ng, A.M.; Cass, C.E.; Baldwin, S.A.; Young, J.D. Equilibrative nucleoside transporters: Mapping regions of interaction for the substrate analogue nitrobenzylthioinosine (NBMPR) using rat chimeric proteins. Biochemistry 2001, 40, 8146–8151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Visser, F.; King, K.M.; Baldwin, S.A.; Young, J.D.; Cass, C.E. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 2007, 26, 85–110. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K. Molecular Basis of Nucleobase Transport Systems in Mammals. Biol. Pharm. Bull. 2017, 40, 1130–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Nakanishi, T.; Tajima, H.; Yamazaki, M.; Yokono, R.; Takabayashi, M.; Shimada, T.; Sawamoto, K.; Miyamoto, K.; Kitagawa, H.; et al. Saturable Hepatic Extraction of Gemcitabine Involves Biphasic Uptake Mediated by Nucleoside Transporters Equilibrative Nucleoside Transporter 1 and 2. J. Pharm. Sci. 2015, 104, 3162–3169. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, E.; Fernandez, V.; Moreno, V.; Valls, J.; Hernandez, L.; Bosch, F.; Abrisqueta, P.; Klapper, W.; Dreyling, M.; Hoster, E.; et al. Five-gene model to predict survival in mantle-cell lymphoma using frozen or formalin-fixed, paraffin-embedded tissue. J. Clin. Oncol. 2008, 26, 4966–4972. [Google Scholar] [CrossRef]
- Chen, C.F.; Hsu, E.C.; Lin, K.T.; Tu, P.H.; Chang, H.W.; Lin, C.H.; Chen, Y.J.; Gu, D.L.; Lin, C.H.; Wu, J.Y.; et al. Overlapping high-resolution copy number alterations in cancer genomes identified putative cancer genes in hepatocellular carcinoma. Hepatology 2010, 52, 1690–1701. [Google Scholar] [CrossRef]
- Kim, H.; Wu, X.; Lee, J. SLC31 (CTR) family of copper transporters in health and disease. Mol. Aspects Med. 2013, 34, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Kilari, D.; Iczkowski, K.A.; Pandya, C.; Robin, A.J.; Messing, E.M.; Guancial, E.; Kim, E.S. Copper Transporter-CTR1 Expression and Pathological Outcomes in Platinum-treated Muscle-invasive Bladder Cancer Patients. Anticancer Res. 2016, 36, 495–501. [Google Scholar]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [Green Version]
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol. Pharmacol. 2010, 77, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef]
- Zhao, R.; Goldman, I.D. Folate and thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. Mol. Aspects Med. 2013, 34, 373–385. [Google Scholar] [CrossRef]
- Desmoulin, S.K.; Hou, Z.; Gangjee, A.; Matherly, L.H. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol. Ther. 2012, 13, 1355–1373. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Min, S.H.; Jansen, M.; Malhotra, U.; Tsai, E.; Cabelof, D.C.; Matherly, L.H.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Rodent intestinal folate transporters (SLC46A1): Secondary structure, functional properties, and response to dietary folate restriction. Am. J. Physiol. Cell Physiol. 2007, 293, C1669–C1678. [Google Scholar] [CrossRef] [Green Version]
- Urquhart, B.L.; Gregor, J.C.; Chande, N.; Knauer, M.J.; Tirona, R.G.; Kim, R.B. The human proton-coupled folate transporter (hPCFT): Modulation of intestinal expression and function by drugs. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G248–G254. [Google Scholar] [CrossRef] [Green Version]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [Green Version]
- Gnana-Prakasam, J.P.; Reddy, S.K.; Veeranan-Karmegam, R.; Smith, S.B.; Martin, P.M.; Ganapathy, V. Polarized distribution of heme transporters in retinal pigment epithelium and their regulation in the iron-overload disease hemochromatosis. Invest Ophthalmol. Vis. Sci. 2011, 52, 9279–9286. [Google Scholar] [CrossRef]
- Zhao, R.; Min, S.H.; Wang, Y.; Campanella, E.; Low, P.S.; Goldman, I.D. A role for the proton-coupled folate transporter (PCFT-SLC46A1) in folate receptor-mediated endocytosis. J. Biol. Chem. 2009, 284, 4267–4274. [Google Scholar] [CrossRef] [Green Version]
- Hlavac, V.; Vaclavikova, R.; Brynychova, V.; Dvorak, P.; Elsnerova, K.; Kozevnikovova, R.; Raus, K.; Kopeckova, K.; Mestakova, S.; Vrana, D.; et al. SLC46A1 Haplotype with Predicted Functional Impact has Prognostic Value in Breast Carcinoma. Mol. Diagn. Ther. 2021, 25, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Inui, K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013, 15, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Terada, T.; Inui, K. Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A). Biochem. Pharmacol. 2008, 75, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresser, M.J.; Leabman, M.K.; Giacomini, K.M. Transporters involved in the elimination of drugs in the kidney: Organic anion transporters and organic cation transporters. J. Pharm. Sci. 2001, 90, 397–421. [Google Scholar] [CrossRef]
- Xie, J.; Xia, L.; Xiang, W.; He, W.; Yin, H.; Wang, F.; Gao, T.; Qi, W.; Yang, Z.; Yang, X.; et al. Metformin selectively inhibits metastatic colorectal cancer with the KRAS mutation by intracellular accumulation through silencing MATE1. Proc. Natl. Acad. Sci. USA 2020, 117, 13012–13022. [Google Scholar] [CrossRef]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Hiasa, M.; Miyaji, T.; Kanamoto, T.; Matsumoto, T.; Otsuka, M.; Moriyama, Y.; Omote, H. Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int. J. Biochem. Cell Biol. 2011, 43, 913–918. [Google Scholar] [CrossRef]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A guide to plasma membrane solute carrier proteins. FEBS J. 2021, 288, 2784–2835. [Google Scholar] [CrossRef]
- Chen, M.; Neul, C.; Schaeffeler, E.; Frisch, F.; Winter, S.; Schwab, M.; Koepsell, H.; Hu, S.; Laufer, S.; Baker, S.D.; et al. Sorafenib Activity and Disposition in Liver Cancer Does Not Depend on Organic Cation Transporter 1. Clin. Pharmacol. Ther. 2020, 107, 227–237. [Google Scholar] [CrossRef]
- Swift, B.; Nebot, N.; Lee, J.K.; Han, T.; Proctor, W.R.; Thakker, D.R.; Lang, D.; Radtke, M.; Gnoth, M.J.; Brouwer, K.L. Sorafenib hepatobiliary disposition: Mechanisms of hepatic uptake and disposition of generated metabolites. Drug Metab. Dispos. 2013, 41, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Belkhiri, A.; Lockhart, A.C.; Merchant, N.; Glaeser, H.; Harris, E.I.; Washington, M.K.; Brunt, E.M.; Zaika, A.; Kim, R.B.; et al. Overexpression of OATP1B3 confers apoptotic resistance in colon cancer. Cancer Res. 2008, 68, 10315–10323. [Google Scholar] [CrossRef] [Green Version]
- Haberkorn, B.; Oswald, S.; Kehl, N.; Gessner, A.; Taudte, R.V.; Dobert, J.P.; Zunke, F.; Fromm, M.F.; Konig, J. Cancer-type organic anion transporting polypeptide 1B3 (Ct-OATP1B3) is localized in lysosomes and mediates resistance against kinase inhibitors. Mol. Pharmacol. 2022, 102, 248–258. [Google Scholar] [CrossRef]
- Czuba, L.C.; Hillgren, K.M.; Swaan, P.W. Post-translational modifications of transporters. Pharmacol. Ther. 2018, 192, 88–99. [Google Scholar] [CrossRef]
- Schnedl, W.J.; Ferber, S.; Johnson, J.H.; Newgard, C.B. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 1994, 43, 1326–1333. [Google Scholar] [CrossRef]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Eddy, R.L.; Fukushima, Y.; Byers, M.G.; Shows, T.B.; Bell, G.I. Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc. Natl. Acad. Sci. USA 1988, 85, 5434–5438. [Google Scholar] [CrossRef] [Green Version]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef]
- Thorens, B.; Wu, Y.J.; Leahy, J.L.; Weir, G.C. The loss of GLUT2 expression by glucose-unresponsive beta cells of db/db mice is reversible and is induced by the diabetic environment. J. Clin. Investig. 1992, 90, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueckler, M. Facilitative glucose transporters. Eur. J. Biochem. 1994, 219, 713–725. [Google Scholar] [CrossRef]
- Yan, R.; Zhao, X.; Lei, J.; Zhou, Q. Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature 2019, 568, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef] [PubMed]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.J.; Sali, A.; Giacomini, K.M. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Aspects Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [Green Version]
- Badagnani, I.; Castro, R.A.; Taylor, T.R.; Brett, C.M.; Huang, C.C.; Stryke, D.; Kawamoto, M.; Johns, S.J.; Ferrin, T.E.; Carlson, E.J.; et al. Interaction of methotrexate with organic-anion transporting polypeptide 1A2 and its genetic variants. J. Pharmacol. Exp. Ther. 2006, 318, 521–529. [Google Scholar] [CrossRef]
- Bauer, M.; Matsuda, A.; Wulkersdorfer, B.; Philippe, C.; Traxl, A.; Ozvegy-Laczka, C.; Stanek, J.; Nics, L.; Klebermass, E.M.; Poschner, S.; et al. Influence of OATPs on Hepatic Disposition of Erlotinib Measured With Positron Emission Tomography. Clin. Pharmacol. Ther. 2018, 104, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Windt, T.; Toth, S.; Patik, I.; Sessler, J.; Kucsma, N.; Szepesi, A.; Zdrazil, B.; Ozvegy-Laczka, C.; Szakacs, G. Identification of anticancer OATP2B1 substrates by an in vitro triple-fluorescence-based cytotoxicity screen. Arch. Toxicol. 2019, 93, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Schulte, R.R.; Ho, R.H. Organic Anion Transporting Polypeptides: Emerging Roles in Cancer Pharmacology. Mol. Pharmacol. 2019, 95, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.M.; Pu, Z.; He, C.; Liang, D.; Jia, Y.; Yuan, X.; Wang, G.; Xie, H. Effect of OATP1B1 genetic polymorphism on the uptake of tamoxifen and its metabolite, endoxifen. Oncol. Rep. 2017, 38, 1124–1132. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib-glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, C.S.; Sprowl, J.A.; Walker, A.L.; Hu, S.; Gibson, A.A.; Sparreboom, A. Modulation of OATP1B-type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol. Cancer Ther. 2013, 12, 1537–1544. [Google Scholar] [CrossRef]
- Koepsell, H. Substrate recognition and translocation by polyspecific organic cation transporters. Biol. Chem. 2011, 392, 95–101. [Google Scholar] [CrossRef]
- Ciarimboli, G. Role of organic cation transporters in drug-induced toxicity. Expert Opin. Drug Metab. Toxicol. 2011, 7, 159–174. [Google Scholar] [CrossRef]
- Okabe, M.; Szakacs, G.; Reimers, M.A.; Suzuki, T.; Hall, M.D.; Abe, T.; Weinstein, J.N.; Gottesman, M.M. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008, 7, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Zhang, T.; Guo, L.; Wu, B. Liver Receptor Homolog-1 Regulates Organic Anion Transporter 2 and Docetaxel Pharmacokinetics. Drug Metab. Dispos. 2018, 46, 980–988. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Ohshiro, N.; Sakai, R.; Ohbayashi, M.; Kohyama, N.; Yamamoto, T. Transport mechanism and substrate specificity of human organic anion transporter 2 (hOat2 [SLC22A7]). J. Pharm. Pharmacol. 2005, 57, 573–578. [Google Scholar] [CrossRef]
- Choudhuri, S.; Cherrington, N.J.; Li, N.; Klaassen, C.D. Constitutive expression of various xenobiotic and endobiotic transporter mRNAs in the choroid plexus of rats. Drug Metab. Dispos. 2003, 31, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Buist, S.C.; Cherrington, N.J.; Choudhuri, S.; Hartley, D.P.; Klaassen, C.D. Gender-specific and developmental influences on the expression of rat organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 301, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Sweet, D.H.; Miller, D.S.; Pritchard, J.B.; Fujiwara, Y.; Beier, D.R.; Nigam, S.K. Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (Oat3 (Slc22a8)) knockout mice. J. Biol. Chem. 2002, 277, 26934–26943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonson, G.D.; Vincent, A.C.; Roberg, K.J.; Huang, Y.; Iwanij, V. Molecular cloning and characterization of a novel liver-specific transport protein. J. Cell Sci. 1994, 107((Pt. 4)), 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.H.; Owen, R.P.; Giacomini, K.M. The concentrative nucleoside transporter family, SLC28. Pflugers. Arch. 2004, 447, 728–734. [Google Scholar] [CrossRef]
- Podgorska, M.; Kocbuch, K.; Pawelczyk, T. Recent advances in studies on biochemical and structural properties of equilibrative and concentrative nucleoside transporters. Acta Biochim. Pol. 2005, 52, 749–758. [Google Scholar] [CrossRef]
- Kang, N.; Jun, A.H.; Bhutia, Y.D.; Kannan, N.; Unadkat, J.D.; Govindarajan, R. Human equilibrative nucleoside transporter-3 (hENT3) spectrum disorder mutations impair nucleoside transport, protein localization, and stability. J. Biol. Chem. 2010, 285, 28343–28352. [Google Scholar] [CrossRef] [Green Version]
- Greenhalf, W.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Lamb, R.F.; Garner, E.; Campbell, F.; Mackey, J.R.; Costello, E.; et al. Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. J. Natl. Cancer Inst. 2014, 106, djt347. [Google Scholar] [CrossRef]
- Tsujie, M.; Nakamori, S.; Nakahira, S.; Takahashi, Y.; Hayashi, N.; Okami, J.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; et al. Human equilibrative nucleoside transporter 1, as a predictor of 5-fluorouracil resistance in human pancreatic cancer. Anticancer Res. 2007, 27, 2241–2249. [Google Scholar]
- Hubeek, I.; Stam, R.W.; Peters, G.J.; Broekhuizen, R.; Meijerink, J.P.; van Wering, E.R.; Gibson, B.E.; Creutzig, U.; Zwaan, C.M.; Cloos, J.; et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br. J. Cancer 2005, 93, 1388–1394. [Google Scholar] [CrossRef] [Green Version]
- Aller, S.G.; Eng, E.T.; De Feo, C.J.; Unger, V.M. Eukaryotic CTR copper uptake transporters require two faces of the third transmembrane domain for helix packing, oligomerization, and function. J. Biol. Chem. 2004, 279, 53435–53441. [Google Scholar] [CrossRef] [Green Version]
- De Feo, C.J.; Aller, S.G.; Siluvai, G.S.; Blackburn, N.J.; Unger, V.M. Three-dimensional structure of the human copper transporter hCTR1. Proc. Natl. Acad. Sci. USA 2009, 106, 4237–4242. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Pena, M.M.; Nose, Y.; Thiele, D.J. Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 2002, 277, 4380–4387. [Google Scholar] [CrossRef] [Green Version]
- Sinani, D.; Adle, D.J.; Kim, H.; Lee, J. Distinct mechanisms for Ctr1-mediated copper and cisplatin transport. J. Biol. Chem. 2007, 282, 26775–26785. [Google Scholar] [CrossRef] [Green Version]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef]
- Zhao, R.; Qiu, A.; Tsai, E.; Jansen, M.; Akabas, M.H.; Goldman, I.D. The proton-coupled folate transporter: Impact on pemetrexed transport and on antifolates activities compared with the reduced folate carrier. Mol. Pharmacol. 2008, 74, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Omote, H.; Hiasa, M.; Matsumoto, T.; Otsuka, M.; Moriyama, Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 2006, 27, 587–593. [Google Scholar] [CrossRef]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Fujita, S.; Hirota, T.; Sakiyama, R.; Baba, M.; Ieiri, I. Identification of drug transporters contributing to oxaliplatin-induced peripheral neuropathy. J. Neurochem. 2019, 148, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, K.L.R.; Evers, R.; Hayden, E.; Hu, S.; Li, C.Y.; Meyer Zu Schwabedissen, H.E.; Neuhoff, S.; Oswald, S.; Piquette-Miller, M.; Saran, C.; et al. Regulation of Drug Transport Proteins-From Mechanisms to Clinical Impact: A White Paper on Behalf of the International Transporter Consortium. Clin. Pharmacol. Ther. 2022, 112, 461–484. [Google Scholar] [CrossRef]
- Zhou, S.; Shu, Y. Transcriptional Regulation of Solute Carrier (SLC) Drug Transporters. Drug Metab. Dispos. 2022, 50, 1238–1250. [Google Scholar] [CrossRef]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Honkakoski, P.; Sueyoshi, T.; Negishi, M. Drug-activated nuclear receptors CAR and PXR. Ann. Med. 2003, 35, 172–182. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, M. OATP1B1 Plays an Important Role in the Transport and Treatment Efficacy of Sorafenib in Hepatocellular Carcinoma. Dis. Markers 2021, 2021, 9711179. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, D.; Li, X.; Cao, Y.; Yi, C.; Wiredu Ocansey, D.K.; Zhou, Y.; Mao, F. Farnesoid-X receptor as a therapeutic target for inflammatory bowel disease and colorectal cancer. Front. Pharmacol. 2022, 13, 1016836. [Google Scholar] [CrossRef]
- Deuschle, U.; Schuler, J.; Schulz, A.; Schluter, T.; Kinzel, O.; Abel, U.; Kremoser, C. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS ONE 2012, 7, e43044. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wang, B.; Chen, R.; Zhong, S.; Gao, F.; Zhang, Y.; Niu, Y.; Li, C.; Shi, G. The Nuclear Farnesoid X Receptor Reduces p53 Ubiquitination and Inhibits Cervical Cancer Cell Proliferation. Front Cell Dev. Biol. 2021, 9, 583146. [Google Scholar] [CrossRef]
- Girisa, S.; Henamayee, S.; Parama, D.; Rana, V.; Dutta, U.; Kunnumakkara, A.B. Targeting Farnesoid X receptor (FXR) for developing novel therapeutics against cancer. Mol. Biomed. 2021, 2, 21. [Google Scholar] [CrossRef]
- Murray, M.; Zhou, F. Trafficking and other regulatory mechanisms for organic anion transporting polypeptides and organic anion transporters that modulate cellular drug and xenobiotic influx and that are dysregulated in disease. Br. J. Pharmacol. 2017, 174, 1908–1924. [Google Scholar] [CrossRef] [Green Version]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Interactions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Li, K.; Jiang, J.; Wang, X.; Chai, Y.; Zhang, C.; Deng, Q.; Shuai, L.; Feng, K.; Ma, K.; et al. Low expression of organic anion-transporting polypeptide 1B3 predicts a poor prognosis in hepatocellular carcinoma. World J. Surg. Oncol. 2020, 18, 127. [Google Scholar] [CrossRef]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef]
- Laurencikiene, J.; Ryden, M. Liver X receptors and fat cell metabolism. Int. J. Obes. 2012, 36, 1494–1502. [Google Scholar] [CrossRef] [Green Version]
- Meyer Zu Schwabedissen, H.E.; Bottcher, K.; Chaudhry, A.; Kroemer, H.K.; Schuetz, E.G.; Kim, R.B. Liver X receptor alpha and farnesoid X receptor are major transcriptional regulators of OATP1B1. Hepatology 2010, 52, 1797–1807. [Google Scholar] [CrossRef]
- Lin, Z.; Xia, S.; Liang, Y.; Ji, L.; Pan, Y.; Jiang, S.; Wan, Z.; Tao, L.; Chen, J.; Lin, C.; et al. LXR activation potentiates sorafenib sensitivity in HCC by activating microRNA-378a transcription. Theranostics 2020, 10, 8834–8850. [Google Scholar] [CrossRef]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Austin, G.; Holcroft, A.; Rinne, N.; Wang, L.; Clark, R.E. Evidence that the pregnane X and retinoid receptors PXR, RAR and RXR may regulate transcription of the transporter hOCT1 in chronic myeloid leukaemia cells. Eur. J. Haematol. 2015, 94, 74–78. [Google Scholar] [CrossRef]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Giannoudis, A.; Austin, G.; Clark, R.E. Peroxisome proliferator-activated receptor activation increases imatinib uptake and killing of chronic myeloid leukemia cells. Exp. Hematol. 2012, 40, 811–819.e812. [Google Scholar] [CrossRef]
- Allenby, G.; Janocha, R.; Kazmer, S.; Speck, J.; Grippo, J.F.; Levin, A.A. Binding of 9-cis-retinoic acid and all-trans-retinoic acid to retinoic acid receptors alpha, beta, and gamma. Retinoic acid receptor gamma binds all-trans-retinoic acid preferentially over 9-cis-retinoic acid. J. Biol. Chem. 1994, 269, 16689–16695. [Google Scholar] [CrossRef]
- le Maire, A.; Teyssier, C.; Balaguer, P.; Bourguet, W.; Germain, P. Regulation of RXR-RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells 2019, 8, 1392. [Google Scholar] [CrossRef] [Green Version]
- Le Vee, M.; Jouan, E.; Stieger, B.; Fardel, O. Differential regulation of drug transporter expression by all-trans retinoic acid in hepatoma HepaRG cells and human hepatocytes. Eur. J. Pharm. Sci. 2013, 48, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.S.; Chen, H.W.; Lin, T.Y.; Lin, A.H.; Lii, C.K. Shikonin upregulates the expression of drug-metabolizing enzymes and drug transporters in primary rat hepatocytes. J. Ethnopharmacol. 2018, 216, 18–25. [Google Scholar] [CrossRef]
- Le Vee, M.; Jouan, E.; Stieger, B.; Lecureur, V.; Fardel, O. Regulation of human hepatic drug transporter activity and expression by diesel exhaust particle extract. PLoS ONE 2015, 10, e0121232. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Zhang, L. The Role of the Aryl Hydrocarbon Receptor (AhR) and Its Ligands in Breast Cancer. Cancers 2022, 14, 5574. [Google Scholar] [CrossRef]
- Zhu, P.; Zhou, K.; Lu, S.; Bai, Y.; Qi, R.; Zhang, S. Modulation of aryl hydrocarbon receptor inhibits esophageal squamous cell carcinoma progression by repressing COX2/PGE2/STAT3 axis. J. Cell Commun. Signal. 2020, 14, 175–192. [Google Scholar] [CrossRef]
- Wu, X.G.; Peng, S.B.; Huang, Q. Transcriptional regulation of breast cancer resistance protein. Yi Chuan 2012, 34, 1529–1536. [Google Scholar] [CrossRef]
- Mahringer, A.; Bernd, A.; Miller, D.S.; Fricker, G. Aryl hydrocarbon receptor ligands increase ABC transporter activity and protein expression in killifish (Fundulus heteroclitus) renal proximal tubules. Biol. Chem. 2019, 400, 1335–1345. [Google Scholar] [CrossRef]
- Honkakoski, P. Searching for Constitutive Androstane Receptor Modulators. Drug Metab. Dispos. 2022, 50, 1002–1009. [Google Scholar] [CrossRef]
- Bae, S.D.W.; Nguyen, R.; Qiao, L.; George, J. Role of the constitutive androstane receptor (CAR) in human liver cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188516. [Google Scholar] [CrossRef]
- Jigorel, E.; Le Vee, M.; Boursier-Neyret, C.; Parmentier, Y.; Fardel, O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab. Dispos. 2006, 34, 1756–1763. [Google Scholar] [CrossRef] [Green Version]
- Ichihara, S.; Kikuchi, R.; Kusuhara, H.; Imai, S.; Maeda, K.; Sugiyama, Y. DNA methylation profiles of organic anion transporting polypeptide 1B3 in cancer cell lines. Pharm. Res. 2010, 27, 510–516. [Google Scholar] [CrossRef]
- Imai, S.; Kikuchi, R.; Tsuruya, Y.; Naoi, S.; Nishida, S.; Kusuhara, H.; Sugiyama, Y. Epigenetic regulation of organic anion transporting polypeptide 1B3 in cancer cell lines. Pharm. Res. 2013, 30, 2880–2890. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Marmorstein, R.; Trievel, R.C. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim. Biophys. Acta 2009, 1789, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, Q.; Hu, H.; Zhu, H.; Yang, B.; He, Q.; Yu, L.; Zeng, S. Upregulation of histone acetylation reverses organic anion transporter 2 repression and enhances 5-fluorouracil sensitivity in hepatocellular carcinoma. Biochem. Pharmacol. 2021, 188, 114546. [Google Scholar] [CrossRef]
- Zhu, Q.; Yu, L.; Qin, Z.; Chen, L.; Hu, H.; Zheng, X.; Zeng, S. Regulation of OCT2 transcriptional repression by histone acetylation in renal cell carcinoma. Epigenetics 2019, 14, 791–803. [Google Scholar] [CrossRef]
- Pelis, R.M.; Suhre, W.M.; Wright, S.H. Functional influence of N-glycosylation in OCT2-mediated tetraethylammonium transport. Am. J. Physiol. Renal Physiol. 2006, 290, F1118–F1126. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G.; Struwe, K.; Arndt, P.; Gorboulev, V.; Koepsell, H.; Schlatter, E.; Hirsch, J.R. Regulation of the human organic cation transporter hOCT1. J. Cell Physiol. 2004, 201, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, I.; Ciarimboli, G.; Yalcinkaya, G.; Mehrens, T.; Velic, A.; Hirsch, J.R.; Gorboulev, V.; Koepsell, H.; Schlatter, E. Regulation of human organic cation transporter hOCT2 by PKA, PI3K, and calmodulin-dependent kinases. Am. J. Physiol. Renal Physiol. 2003, 284, F293–F302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Ha, J.M.; Sugiyama, Y. Post-translational regulation of the major drug transporters in the families of organic anion transporters and organic anion-transporting polypeptides. J. Biol. Chem. 2020, 295, 17349–17364. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Korkuc, P.; Walther, D. Towards understanding the crosstalk between protein post-translational modifications: Homo- and heterotypic PTM pair distances on protein surfaces are not random. Proteins 2017, 85, 78–92. [Google Scholar] [CrossRef]
- Alam, K.; Farasyn, T.; Crowe, A.; Ding, K.; Yue, W. Treatment with proteasome inhibitor bortezomib decreases organic anion transporting polypeptide (OATP) 1B3-mediated transport in a substrate-dependent manner. PLoS ONE 2017, 12, e0186924. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; You, G. Proteasome Inhibitors Bortezomib and Carfilzomib Stimulate the Transport Activity of Human Organic Anion Transporter 1. Mol. Pharmacol. 2020, 97, 384–391. [Google Scholar] [CrossRef]
- Mayati, A.; Moreau, A.; Le Vee, M.; Stieger, B.; Denizot, C.; Parmentier, Y.; Fardel, O. Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression. Int. J. Mol. Sci. 2017, 18, 764. [Google Scholar] [CrossRef] [Green Version]
- Hayden, E.R.; Chen, M.; Pasquariello, K.Z.; Gibson, A.A.; Petti, J.J.; Shen, S.; Qu, J.; Ong, S.S.; Chen, T.; Jin, Y.; et al. Regulation of OATP1B1 Function by Tyrosine Kinase-mediated Phosphorylation. Clin. Cancer Res. 2021, 27, 4301–4310. [Google Scholar] [CrossRef]
- Sun, J.; Damaraju, V.L.; Cass, C.E.; Sawyer, M. Inhibition of nucleoside transporters by tyrosine kinase inhibitors and its effects on chemotherapy efficacy. Cancer Cell Microenviron. 2014, 1, e389. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.T. Role of OCTN1 (SLC22A4) in the Disposition of Nucleoside Analogs in AML; The Ohio State University, Graduate Program in Pharmaceutical Sciences: Columbus, OH, USA, 2019. [Google Scholar]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [Green Version]
- Quintero, M.; Mackenzie, N.; Brennan, P.A. Hypoxia-inducible factor 1 (HIF-1) in cancer. Eur. J. Surg Oncol. 2004, 30, 465–468. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [Green Version]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Casanello, P.; Torres, A.; Sanhueza, F.; Gonzalez, M.; Farias, M.; Gallardo, V.; Pastor-Anglada, M.; San Martin, R.; Sobrevia, L. Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ. Res. 2005, 97, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Abdulla, P.; Hoffman, E.; Hamilton, K.E.; Daniels, D.; Schonfeld, C.; Loffler, M.; Reyes, G.; Duszenko, M.; Karhausen, J.; et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 2005, 202, 1493–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, R.; Paul, A.; Zastre, J. Hypoxia induced upregulation and function of the thiamine transporter, SLC19A3 in a breast cancer cell line. Cancer Biol. Ther. 2010, 10, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Wu, J.B.; Chu, G.C.; Li, Q.; Wang, R.; Zhang, C.; Zhang, Y.; Kim, H.L.; Wang, J.; Zhau, H.E.; et al. Heptamethine carbocyanine dye-mediated near-infrared imaging of canine and human cancers through the HIF-1alpha/OATPs signaling axis. Oncotarget 2014, 5, 10114–10126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Tashiro, K.; Dixit, A.; Soni, A.; Vogel, K.; Hall, B.; Shafqat, I.; Slaughter, J.; Param, N.; Le, A.; et al. Loss of HIF1A From Pancreatic Cancer Cells Increases Expression of PPP1R1B and Degradation of p53 to Promote Invasion and Metastasis. Gastroenterology 2020, 159, 1882–1897.e1885. [Google Scholar] [CrossRef] [PubMed]
- Hays, A.; Apte, U.; Hagenbuch, B. Organic anion transporting polypeptides expressed in pancreatic cancer may serve as potential diagnostic markers and therapeutic targets for early stage adenocarcinomas. Pharm. Res. 2013, 30, 2260–2269. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal. Transduct. Target Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226, discussion 230–212. [Google Scholar]
- Le Vee, M.; Lecureur, V.; Stieger, B.; Fardel, O. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab. Dispos. 2009, 37, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Le Vee, M.; Gripon, P.; Stieger, B.; Fardel, O. Down-regulation of organic anion transporter expression in human hepatocytes exposed to the proinflammatory cytokine interleukin 1beta. Drug Metab. Dispos. 2008, 36, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Swietach, P. What is pH regulation, and why do cancer cells need it? Cancer Metastasis Rev. 2019, 38, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, T.; Imai, K.; Nezu, J.; Tsuji, A.; Tamai, I. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J. Pharmacol. Exp. Ther. 2004, 308, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Hu, Q.; Qin, Y.; Xu, J.; Zhang, B.; Yu, X.; Wang, W. The Relationship of Redox With Hallmarks of Cancer: The Importance of Homeostasis and Context. Front. Oncol. 2022, 12, 862743. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef]
- Jiang, X.; Xin, H.; Ren, Q.; Gu, J.; Zhu, L.; Du, F.; Feng, C.; Xie, Y.; Sha, X.; Fang, X. Nanoparticles of 2-deoxy-D-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials 2014, 35, 518–529. [Google Scholar] [CrossRef]
- Jiang, X.; Xin, H.; Gu, J.; Du, F.; Feng, C.; Xie, Y.; Fang, X. Enhanced antitumor efficacy by d-glucosamine-functionalized and paclitaxel-loaded poly(ethylene glycol)-co-poly(trimethylene carbonate) polymer nanoparticles. J. Pharm. Sci. 2014, 103, 1487–1496. [Google Scholar] [CrossRef]
- Shao, K.; Ding, N.; Huang, S.; Ren, S.; Zhang, Y.; Kuang, Y.; Guo, Y.; Ma, H.; An, S.; Li, Y.; et al. Smart nanodevice combined tumor-specific vector with cellular microenvironment-triggered property for highly effective antiglioma therapy. ACS Nano 2014, 8, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Awuah, S.G.; Lippard, S.J. Chemical Approach to Positional Isomers of Glucose-Platinum Conjugates Reveals Specific Cancer Targeting through Glucose-Transporter-Mediated Uptake in Vitro and in Vivo. J. Am. Chem. Soc. 2016, 138, 12541–12551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Aspects Med. 2013, 34, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcaino, J.R.; Miranda-Goncalves, V.; Pinheiro, C.; Silva, J.; Pereira, H.; Monteiro, P.; Henrique, R.M.; Reis, R.M.; Lopes, C.; et al. Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer 2011, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes-Sousa, S.; Granja, S.; Pinheiro, C.; Fernandes, D.; Longatto-Filho, A.; Laus, A.C.; Alves, C.D.; Suarez-Penaranda, J.M.; Perez-Sayans, M.; Lopes Carvalho, A.; et al. Prognostic significance of monocarboxylate transporter expression in oral cavity tumors. Cell Cycle 2016, 15, 1865–1873. [Google Scholar] [CrossRef] [Green Version]
- Afonso, J.; Pinto, T.; Simoes-Sousa, S.; Schmitt, F.; Longatto-Filho, A.; Pinheiro, C.; Marques, H.; Baltazar, F. Clinical significance of metabolism-related biomarkers in non-Hodgkin lymphoma—MCT1 as potential target in diffuse large B cell lymphoma. Cell Oncol. 2019, 42, 303–318. [Google Scholar] [CrossRef]
- Venishetty, V.K.; Samala, R.; Komuravelli, R.; Kuncha, M.; Sistla, R.; Diwan, P.V. beta-Hydroxybutyric acid grafted solid lipid nanoparticles: A novel strategy to improve drug delivery to brain. Nanomedicine 2013, 9, 388–397. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Shennan, D.B.; Thomson, J.; Gow, I.F.; Travers, M.T.; Barber, M.C. L-leucine transport in human breast cancer cells (MCF-7 and MDA-MB-231): Kinetics, regulation by estrogen and molecular identity of the transporter. Biochim. Biophys. Acta 2004, 1664, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Puris, E.; Fricker, G.; Gynther, M. Targeting Transporters for Drug Delivery to the Brain: Can We Do Better? Pharm. Res. 2022. [Google Scholar] [CrossRef]
- Ohshima, Y.; Suzuki, H.; Hanaoka, H.; Sasaki, I.; Watanabe, S.; Haba, H.; Arano, Y.; Tsushima, Y.; Ishioka, N.S. Preclinical evaluation of new alpha-radionuclide therapy targeting LAT1: 2-[(211)At]astato-alpha-methyl-L-phenylalanine in tumor-bearing model. Nucl. Med. Biol. 2020, 90–91, 15–22. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, X.; Chi, D.; Xu, Z.; Lin, G.; Liu, H.; Sun, J.; He, Z.; Wang, Y. Single-ligand dual-targeting irinotecan liposomes: Control of targeting ligand display by pH-responsive PEG-shedding strategy to enhance tumor-specific therapy and attenuate toxicity. Int. J. Pharm. 2020, 587, 119680. [Google Scholar] [CrossRef]
- Pocasap, P.; Weerapreeyakul, N.; Timonen, J.; Jarvinen, J.; Leppanen, J.; Karkkainen, J.; Rautio, J. Tyrosine-Chlorambucil Conjugates Facilitate Cellular Uptake through L-Type Amino Acid Transporter 1 (LAT1) in Human Breast Cancer Cell Line MCF-7. Int. J. Mol. Sci. 2020, 21, 2132. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, K.; Kyoko, H.; Toyooka, N.; Kato, A.; Orihashi, M.; Tomi, M.; Tachikawa, M. Evaluation of amino acid-mustard transport as L-type amino acid transporter 1 (LAT1)-mediated alkylating agents. Biol. Pharm. Bull. 2008, 31, 2126–2130. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Fang, Z.; Jung, H.Y.; Yoon, J.H.; Hong, S.S.; Maeng, H.J. Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties. Molecules 2018, 23, 2608. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Subudhi, B.B. Development and characterization of lysine-methotrexate conjugate for enhanced brain delivery. Drug Deliv. 2016, 23, 2327–2337. [Google Scholar] [CrossRef]
- Bhunia, S.; Vangala, V.; Bhattacharya, D.; Ravuri, H.G.; Kuncha, M.; Chakravarty, S.; Sistla, R.; Chaudhuri, A. Large Amino Acid Transporter 1 Selective Liposomes of l-DOPA Functionalized Amphiphile for Combating Glioblastoma. Mol. Pharm. 2017, 14, 3834–3847. [Google Scholar] [CrossRef]
- Ong, Z.Y.; Chen, S.; Nabavi, E.; Regoutz, A.; Payne, D.J.; Elson, D.S.; Dexter, D.T.; Dunlop, I.E.; Porter, A.E. Multibranched Gold Nanoparticles with Intrinsic LAT-1 Targeting Capabilities for Selective Photothermal Therapy of Breast Cancer. ACS Appl. Mater. Interfaces 2017, 9, 39259–39270. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef]
- Zhou, P.; Liang, X.; Zhou, C.; Qin, J.; Hou, C.; Zhu, Z.; Zhang, W.; Wang, S.; Zhong, D. Glutamine-beta-cyclodextrin for targeted doxorubicin delivery to triple-negative breast cancer tumors via the transporter ASCT2. J. Mater. Chem. B 2019, 7, 5363–5375. [Google Scholar] [CrossRef]
- Wang, C.; Wu, J.; Wang, Z.; Yang, Z.; Li, Z.; Deng, H.; Li, L.; Peng, X.; Feng, M. Glutamine addiction activates polyglutamine-based nanocarriers delivering therapeutic siRNAs to orthotopic lung tumor mediated by glutamine transporter SLC1A5. Biomaterials 2018, 183, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.L.; Mager, S. Cloning and functional expression of a human Na(+) and Cl(-)-dependent neutral and cationic amino acid transporter B(0+). J. Biol. Chem. 1999, 274, 23740–23745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Miyauchi, S.; Martindale, R.G.; Herdman, A.V.; Podolsky, R.; Miyake, K.; Mager, S.; Prasad, P.D.; Ganapathy, M.E.; Ganapathy, V. Upregulation of the amino acid transporter ATB0,+ (SLC6A14) in colorectal cancer and metastasis in humans. Biochim. Biophys. Acta 2005, 1741, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Prasad, P.D.; Ghamande, S.; Moore-Martin, P.; Herdman, A.V.; Martindale, R.G.; Podolsky, R.; Mager, S.; Ganapathy, M.E.; Ganapathy, V. Up-regulation of the amino acid transporter ATB(0,+) (SLC6A14) in carcinoma of the cervix. Gynecol. Oncol. 2006, 100, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Gong, P.; Sun, M.; Kou, L.; Ganapathy, V.; Jing, Y.; He, Z.; Sun, J. Transporter occluded-state conformation-induced endocytosis: Amino acid transporter ATB(0,+)-mediated tumor targeting of liposomes for docetaxel delivery for hepatocarcinoma therapy. J. Control Release 2016, 243, 370–380. [Google Scholar] [CrossRef]
- Luo, Q.; Yang, B.; Tao, W.; Li, J.; Kou, L.; Lian, H.; Che, X.; He, Z.; Sun, J. ATB(0,+) transporter-mediated targeting delivery to human lung cancer cells via aspartate-modified docetaxel-loading stealth liposomes. Biomater. Sci. 2017, 5, 295–304. [Google Scholar] [CrossRef]
- Kou, L.; Huang, H.; Lin, X.; Jiang, X.; Wang, Y.; Luo, Q.; Sun, J.; Yao, Q.; Ganapathy, V.; Chen, R. Endocytosis of ATB(0,+)(SLC6A14)-targeted liposomes for drug delivery and its therapeutic application for pancreatic cancer. Expert Opin. Drug Deliv. 2020, 17, 395–405. [Google Scholar] [CrossRef]
- Spanier, B.; Rohm, F. Proton Coupled Oligopeptide Transporter 1 (PepT1) Function, Regulation, and Influence on the Intestinal Homeostasis. Compr. Physiol. 2018, 8, 843–869. [Google Scholar] [CrossRef]
- Tai, W.; Chen, Z.; Cheng, K. Expression profile and functional activity of peptide transporters in prostate cancer cells. Mol. Pharm. 2013, 10, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Schniers, B.K.; Rajasekaran, D.; Korac, K.; Sniegowski, T.; Ganapathy, V.; Bhutia, Y.D. PEPT1 is essential for the growth of pancreatic cancer cells: A viable drug target. Biochem. J. 2021, 478, 3757–3774. [Google Scholar] [CrossRef]
- Tao, W.; Zhao, D.; Sun, M.; Wang, Z.; Lin, B.; Bao, Y.; Li, Y.; He, Z.; Sun, Y.; Sun, J. Intestinal absorption and activation of decitabine amino acid ester prodrugs mediated by peptide transporter PEPT1 and enterocyte enzymes. Int. J. Pharm. 2018, 541, 64–71. [Google Scholar] [CrossRef]
- Thompson, B.R.; Shi, J.; Zhu, H.J.; Smith, D.E. Pharmacokinetics of gemcitabine and its amino acid ester prodrug following intravenous and oral administrations in mice. Biochem. Pharmacol. 2020, 180, 114127. [Google Scholar] [CrossRef]
- Du, Y.; Tian, C.; Wang, M.; Huang, D.; Wei, W.; Liu, Y.; Li, L.; Sun, B.; Kou, L.; Kan, Q.; et al. Dipeptide-modified nanoparticles to facilitate oral docetaxel delivery: New insights into PepT1-mediated targeting strategy. Drug Deliv. 2018, 25, 1403–1413. [Google Scholar] [CrossRef]
- Landowski, C.P.; Vig, B.S.; Song, X.; Amidon, G.L. Targeted delivery to PEPT1-overexpressing cells: Acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol. Cancer Ther. 2005, 4, 659–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Wu, X.; Wang, T.; Zhao, J.; Liu, X.; Yao, Z.; Zhang, Q.; Jian, X. Targeting PEPT1: A novel strategy to improve the antitumor efficacy of doxorubicin in human hepatocellular carcinoma therapy. Oncotarget 2017, 8, 40454–40468. [Google Scholar] [CrossRef] [Green Version]
- Ancey, P.B.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Chai, Y.J.; Yi, J.W.; Oh, S.W.; Kim, Y.A.; Yi, K.H.; Kim, J.H.; Lee, K.E. Upregulation of SLC2 (GLUT) family genes is related to poor survival outcomes in papillary thyroid carcinoma: Analysis of data from The Cancer Genome Atlas. Surgery 2017, 161, 188–194. [Google Scholar] [CrossRef]
- Younes, M.; Lechago, L.V.; Lechago, J. Overexpression of the human erythrocyte glucose transporter occurs as a late event in human colorectal carcinogenesis and is associated with an increased incidence of lymph node metastases. Clin. Cancer Res. 1996, 2, 1151–1154. [Google Scholar]
- Airley, R.; Loncaster, J.; Davidson, S.; Bromley, M.; Roberts, S.; Patterson, A.; Hunter, R.; Stratford, I.; West, C. Glucose transporter glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin. Cancer Res. 2001, 7, 928–934. [Google Scholar]
- Shan, X.H.; Hu, H.; Xiong, F.; Gu, N.; Geng, X.D.; Zhu, W.; Lin, J.; Wang, Y.F. Targeting Glut1-overexpressing MDA-MB-231 cells with 2-deoxy-D-g1ucose modified SPIOs. Eur. J. Radiol. 2012, 81, 95–99. [Google Scholar] [CrossRef]
- Xiong, F.; Zhu, Z.Y.; Xiong, C.; Hua, X.Q.; Shan, X.H.; Zhang, Y.; Gu, N. Preparation, characterization of 2-deoxy-D-glucose functionalized dimercaptosuccinic acid-coated maghemite nanoparticles for targeting tumor cells. Pharm. Res. 2012, 29, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; Feron, O. Lactate shuttles at a glance: From physiological paradigms to anti-cancer treatments. Dis. Model Mech. 2011, 4, 727–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Broer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350((Pt. 1)), 219–227. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers. Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, I.; Bang, H.; Kim, H.C.; Lee, W.Y.; Yun, S.H.; Lee, J.; Lee, S.J.; Park, Y.S.; Kim, K.M.; et al. MCT4 Expression Is a Potential Therapeutic Target in Colorectal Cancer with Peritoneal Carcinomatosis. Mol. Cancer Ther. 2018, 17, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Coelho, F.; Nunes, C.; Gouveia-Fernandes, S.; Rosas, R.; Silva, F.; Gameiro, P.; Carvalho, T.; Gomes da Silva, M.; Cabecadas, J.; Dias, S.; et al. Monocarboxylate transporter 1 (MCT1), a tool to stratify acute myeloid leukemia (AML) patients and a vehicle to kill cancer cells. Oncotarget 2017, 8, 82803–82823. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Pereira, C.; Medeiros, R. ASCT2 and LAT1 Contribution to the Hallmarks of Cancer: From a Molecular Perspective to Clinical Translation. Cancers 2021, 13, 203. [Google Scholar] [CrossRef]
- Rodriguez, C.F.; Escudero-Bravo, P.; Diaz, L.; Bartoccioni, P.; Garcia-Martin, C.; Gilabert, J.G.; Boskovic, J.; Guallar, V.; Errasti-Murugarren, E.; Llorca, O.; et al. Structural basis for substrate specificity of heteromeric transporters of neutral amino acids. Proc. Natl. Acad. Sci. USA 2021, 118, e2113573118. [Google Scholar] [CrossRef]
- Kaneda-Nakashima, K.; Zhang, Z.; Manabe, Y.; Shimoyama, A.; Kabayama, K.; Watabe, T.; Kanai, Y.; Ooe, K.; Toyoshima, A.; Shirakami, Y.; et al. alpha-Emitting cancer therapy using (211) At-AAMT targeting LAT1. Cancer Sci. 2021, 112, 1132–1140. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Cysteine is not a substrate but a specific modulator of human ASCT2 (SLC1A5) transporter. FEBS Lett. 2015, 589, 3617–3623. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef]
- Broer, S. Amino Acid Transporters as Disease Modifiers and Drug Targets. SLAS Discov. 2018, 23, 303–320. [Google Scholar] [CrossRef] [Green Version]
- Sikder, M.O.F.; Yang, S.; Ganapathy, V.; Bhutia, Y.D. The Na(+)/Cl(-)-Coupled, Broad-Specific, Amino Acid Transporter SLC6A14 (ATB(0,+)): Emerging Roles in Multiple Diseases and Therapeutic Potential for Treatment and Diagnosis. AAPS J. 2017, 20, 12. [Google Scholar] [CrossRef]



| Gene Name | Protein Name | Natural Substrates | Tissue Expression | High Expression in Cancer | Utilization in Anticancer Drug Delivery | References |
|---|---|---|---|---|---|---|
| SLC2A1 | GLUT1 | Glucose, galactose, mannose, glucosamine | Erythrocytes, brain, (endothelial cells), blood-tissue barrier, several fetal tissues | Liver, brain, renal, pancreatic, lung, breast, esophageal, endometrial, ovarian, colorectal and cervical cancers | dGlu-conjugated nanoparticles, D-glucosamine-conjugated nanoparticles, DHA-conjugated micelles, polymeric micelles. glucose-platinum conjugates | [35,278,279,280,281,282,283] |
| SLC16A1 | MCT1 | lactate, pyruvate, ketone bodies | Ubiquitous | Prostate cancer, lymphoma, peritoneal carcinomatosis and oral cavity cancer | β-hydroxybutyric acid -conjugated nanoparticles, 3-bromopyruvate | [35,284,285,286,287,288,289,290] |
| SLC7A5 | LAT1 | Phenylalanine, leucine, tryptophan | Brain (endothelial cells), testis, retina, esophagus, testis, placenta and bone marrow | Colorectal cancer, gliomablastoma, triple-negative breast cancer and HER2-positive breast cancers and MYC driver ER-positive breast cancer | L-phenylalanine prodrug of melphalan, threonine-derivative of gemcitabine, aspartate derivative of doxorubicin, liposomes of L-Dopa functionalized amphiphile, lysine-conjugated methotrexate, L- and D-Dopa conjugated anisotropic gold nanoparticles, lysine- and aromatic amino acid-mustards, α-methyl-L-tyrosine conjugate of Astatine-211, tyrosine-conjugated liposomes, L-tyrosine ester- and amide-conjugates of chlorambucil | [35,164,290,291,292,293,294,295,296,297,298,299] |
| SLC1A5 | ASCT2 | L-alanine, L-serine, L-threonine, L-glutamine, L- asparagine | Lung, skeletal muscle, large intestine, kidney, testis, adipose tissue | Colon, kidney, liver, lung, ovarian, pancreatic, stomach and cutaneous cancers | Glutamine-conjugated β-cyclodextrin inclusion complexes of doxorubicin, polyglutamine for siRNA delivery | [35,300,301,302] |
| SLC6A14 | ATB0,+ | Neutral, cationic amino acids | Lung, trachea, salivary gland, mammary gland, stomach, pituitary gland, intestine, uterus, prostate, testis | Colorectal, pancreatic and cervical cancer | Lysine-conjugated liposomes, lysine and polyoxyethylene stearate -conjugated liposomes, aspartate-polyoxyethylene stearate -conjugated liposomes, | [35,303,304,305,306,307,308] |
| SLC15A1 | PEPT1 | Di- and tripeptides, protons, beta-lactam antibiotics | Small intestine, kidney, pancreas, bile duct, liver | Prostate cancer, hepatocellular carcinoma, pancreatic adenocarcinoma | Prolyl and lysyl floxuridine prodrugs, Gly-Gly-Gly conjugate of doxorubicin | [35,309,310,311,312,313,314,315,316] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puris, E.; Fricker, G.; Gynther, M. The Role of Solute Carrier Transporters in Efficient Anticancer Drug Delivery and Therapy. Pharmaceutics 2023, 15, 364. https://doi.org/10.3390/pharmaceutics15020364
Puris E, Fricker G, Gynther M. The Role of Solute Carrier Transporters in Efficient Anticancer Drug Delivery and Therapy. Pharmaceutics. 2023; 15(2):364. https://doi.org/10.3390/pharmaceutics15020364
Chicago/Turabian StylePuris, Elena, Gert Fricker, and Mikko Gynther. 2023. "The Role of Solute Carrier Transporters in Efficient Anticancer Drug Delivery and Therapy" Pharmaceutics 15, no. 2: 364. https://doi.org/10.3390/pharmaceutics15020364