
Celecoxib Nanoformulations with Enhanced Solubility, Dissolution Rate, and Oral Bioavailability: Experimental Approaches over In Vitro/In Vivo Evaluation
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Co-Milled Celecoxib Composition and Physical Mixture (PM)
2.3. Preliminary Screening Experiments
2.4. Experimental Design and Optimization
2.4.1. Central Composite Design (CCD)
2.4.2. Bayesian Optimization (BO)
2.5. Characterization of Co-Milled Celecoxib Composition and Physical Mixture
2.5.1. Determination of Solubility
2.5.2. Determination of Intrinsic Dissolution Rate (IDR)
2.5.3. Differential Scanning Calorimetry (DSC)
2.5.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.5.5. Powder X-ray Diffraction Analysis (PXRD)
2.5.6. Determination of Particle Size
2.5.7. Scanning Electron Microscopy (SEM)
2.5.8. Storage Stability Study
2.6. Analytical Methods (High-Performance Liquid Chromatography (HPLC))
2.7. Formulation Studies
2.8. In Vitro Dissolution Study
2.9. Cell Culture Studies
2.9.1. Cell Viability Studies
2.9.2. Permeability Studies
2.10. In Vivo Oral Bioavailability
3. Results and Discussion
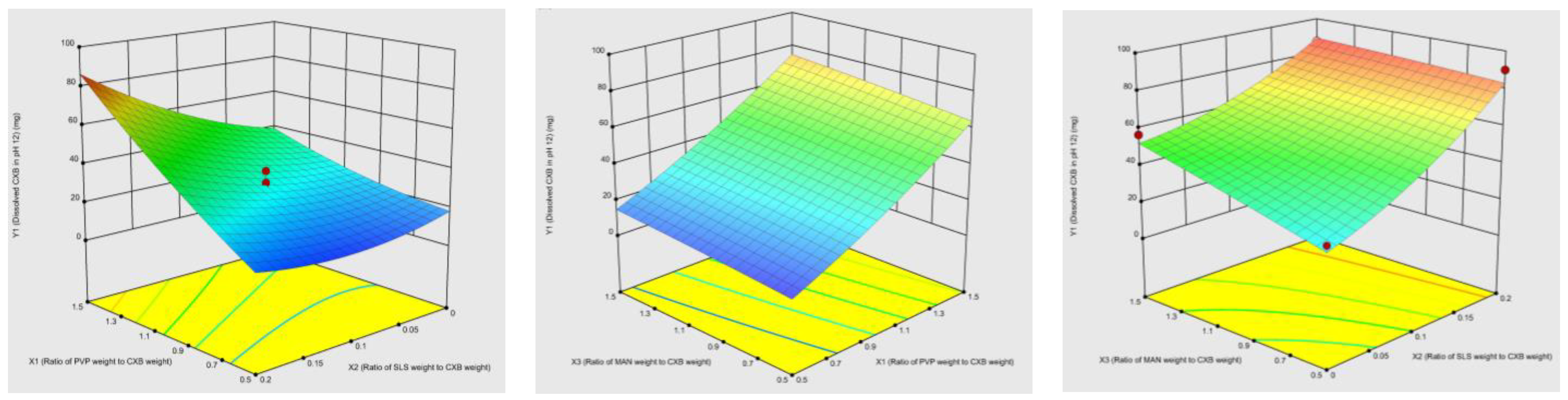
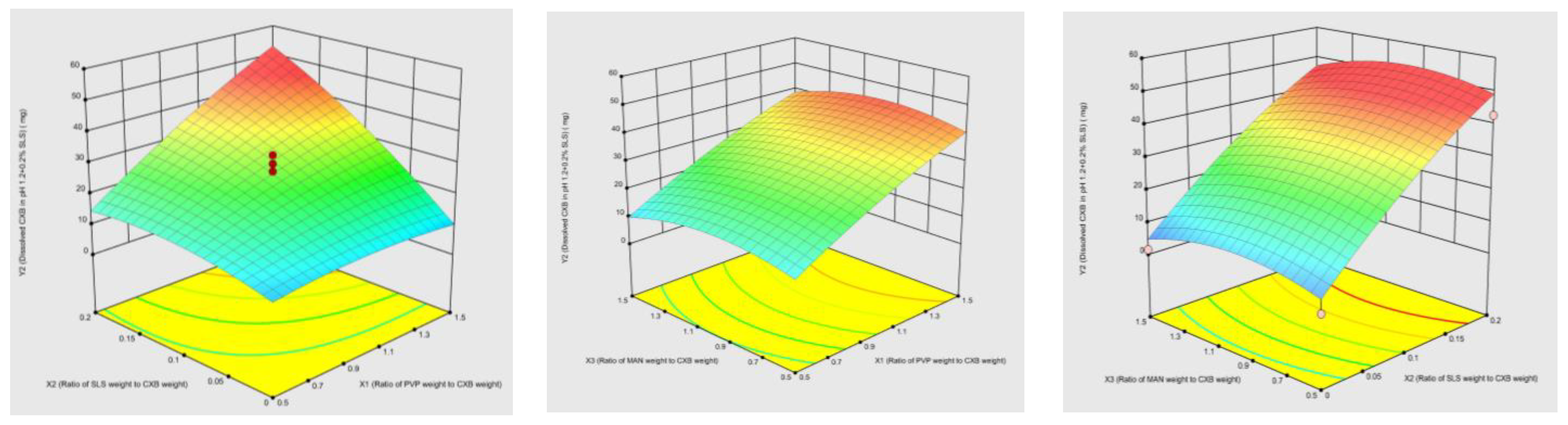
3.1. Optimization of Co-Milled CXB Composition Using CCD
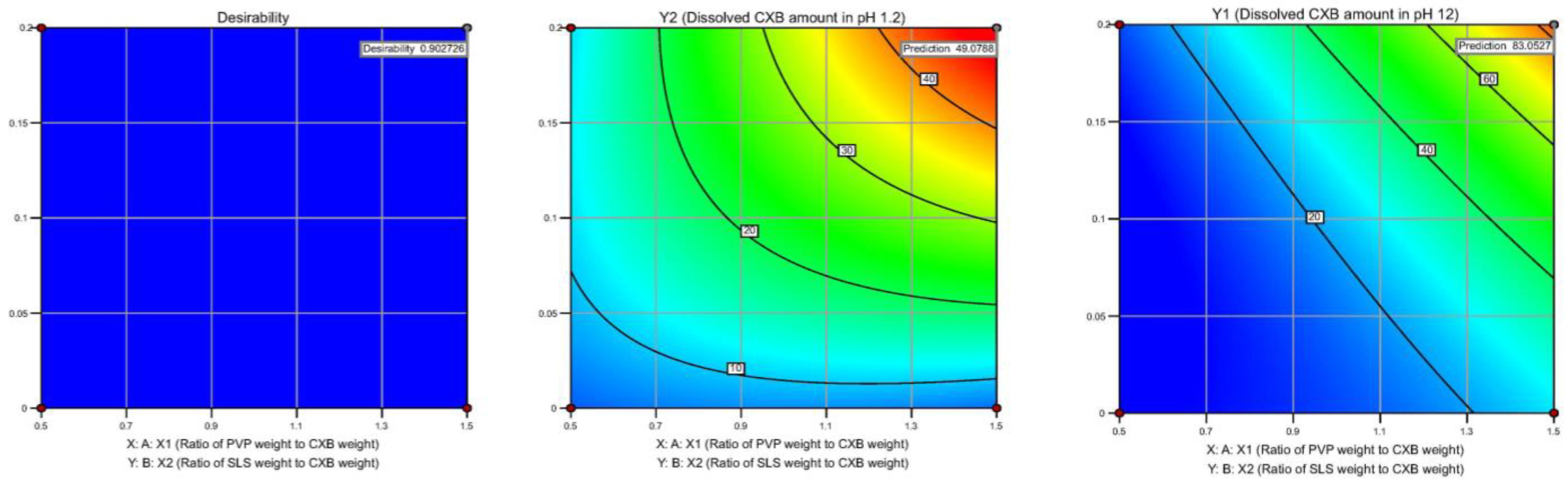
Model Validation and Optimum Composition
3.2. Bayesian Optimization (BO)
3.3. Physicochemical Characterization of Co-Milled Celecoxib Composition
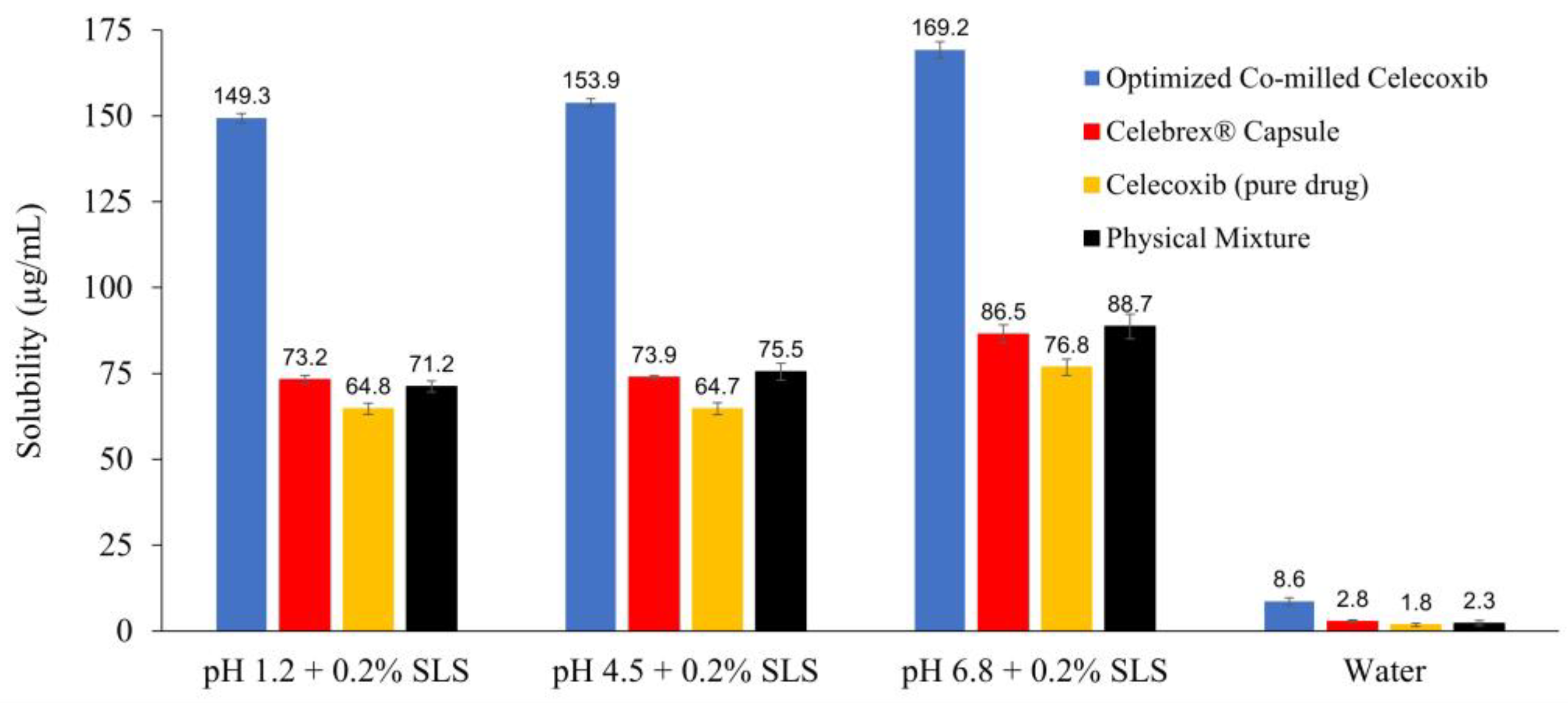
3.3.1. Apparent Solubility
3.3.2. Intrinsic Dissolution Rate (IDR)
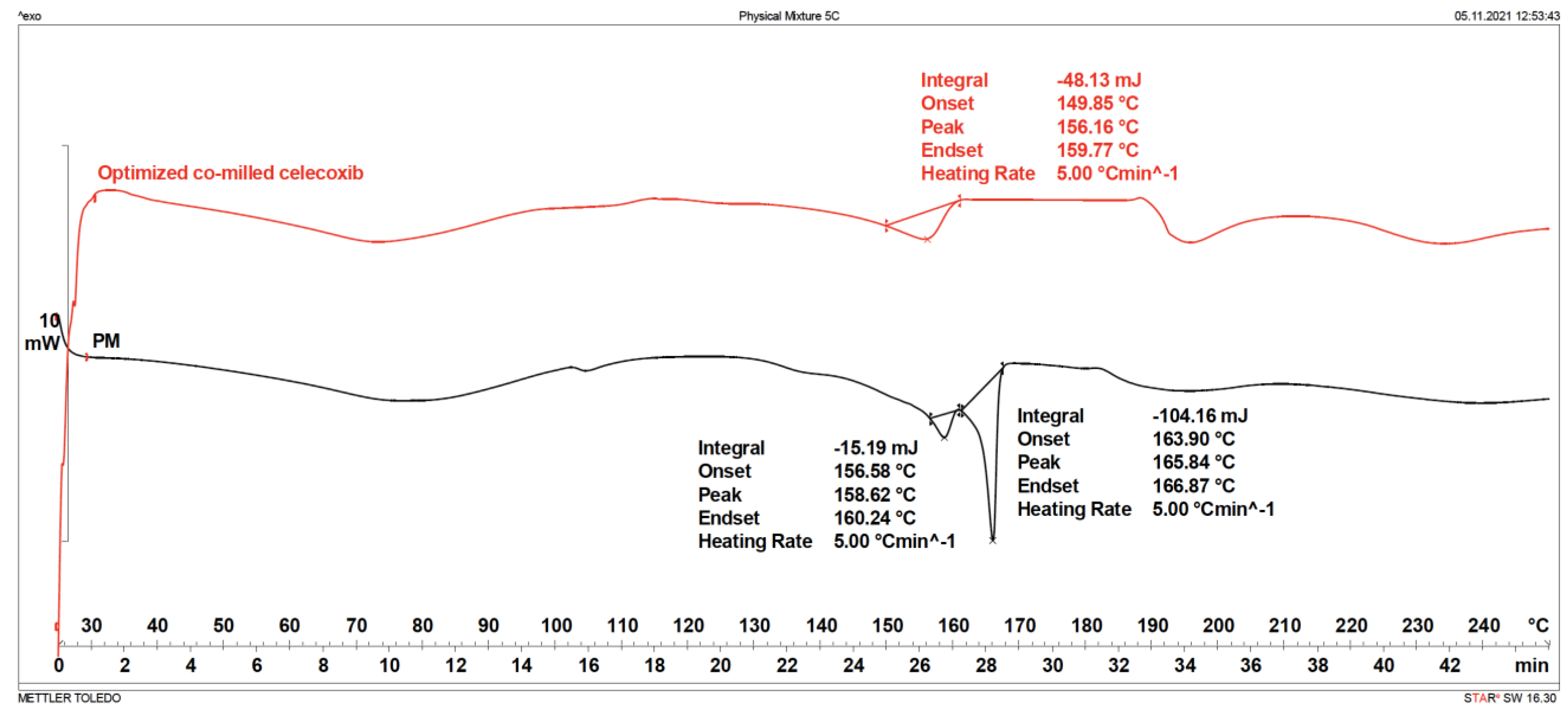
3.3.3. Differential Scanning Calorimetry (DSC)
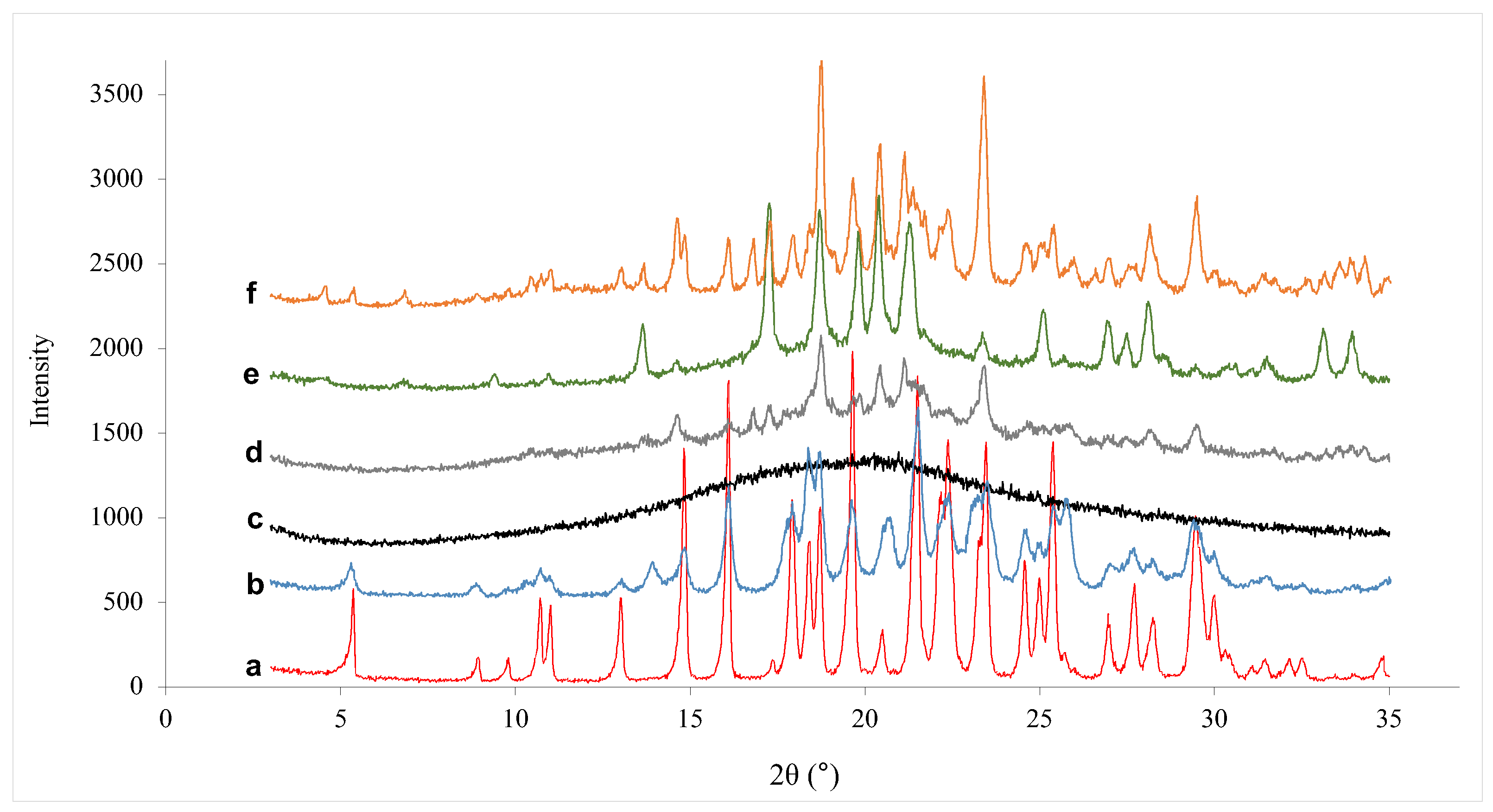
3.3.4. Powder X-ray Diffraction Analysis (PXRD)
3.3.5. Particle Size and Scanning Electron Microscopy (SEM)
3.3.6. Storage Stability
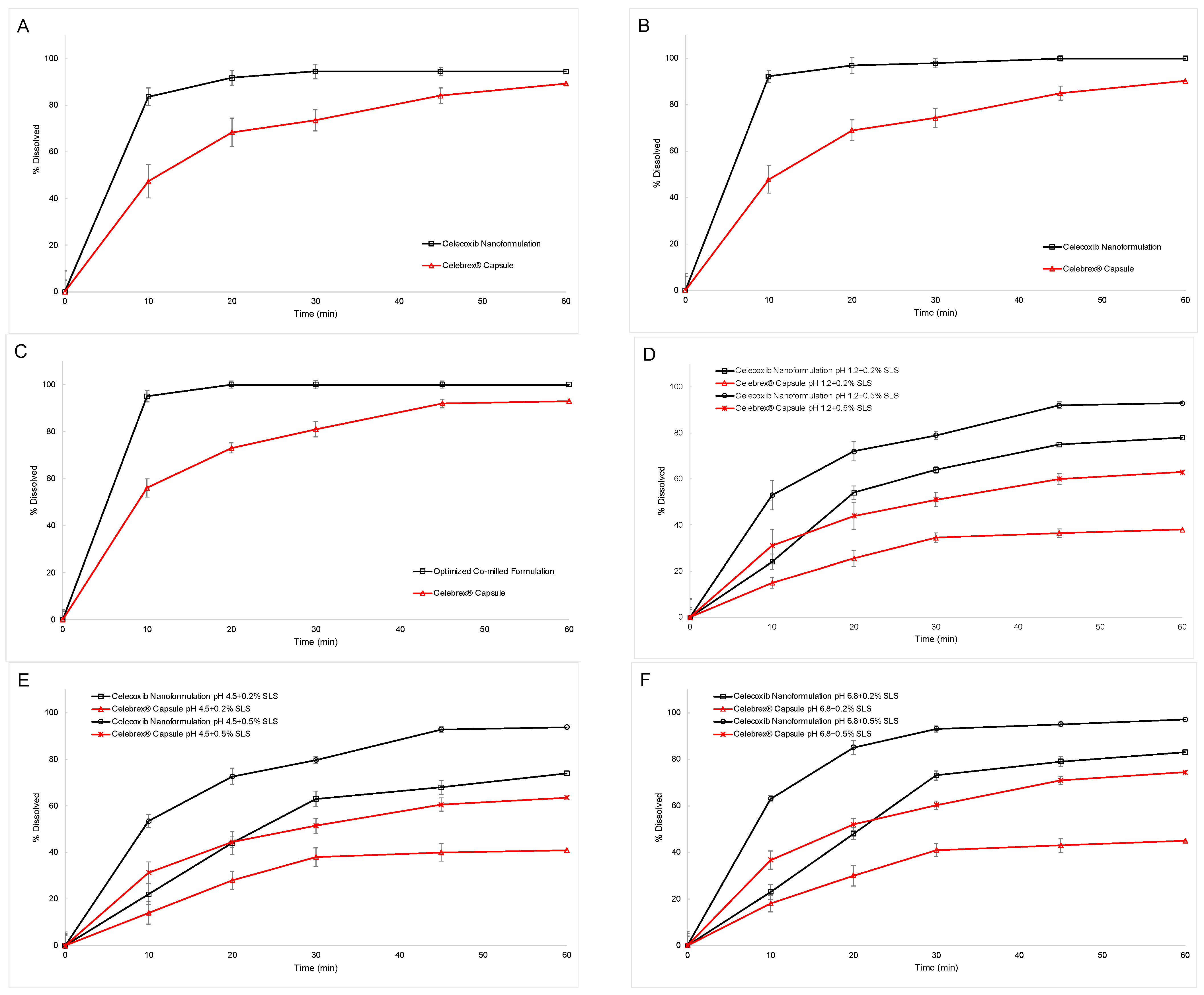
3.4. In Vitro Dissolution Study
3.5. Cell Culture
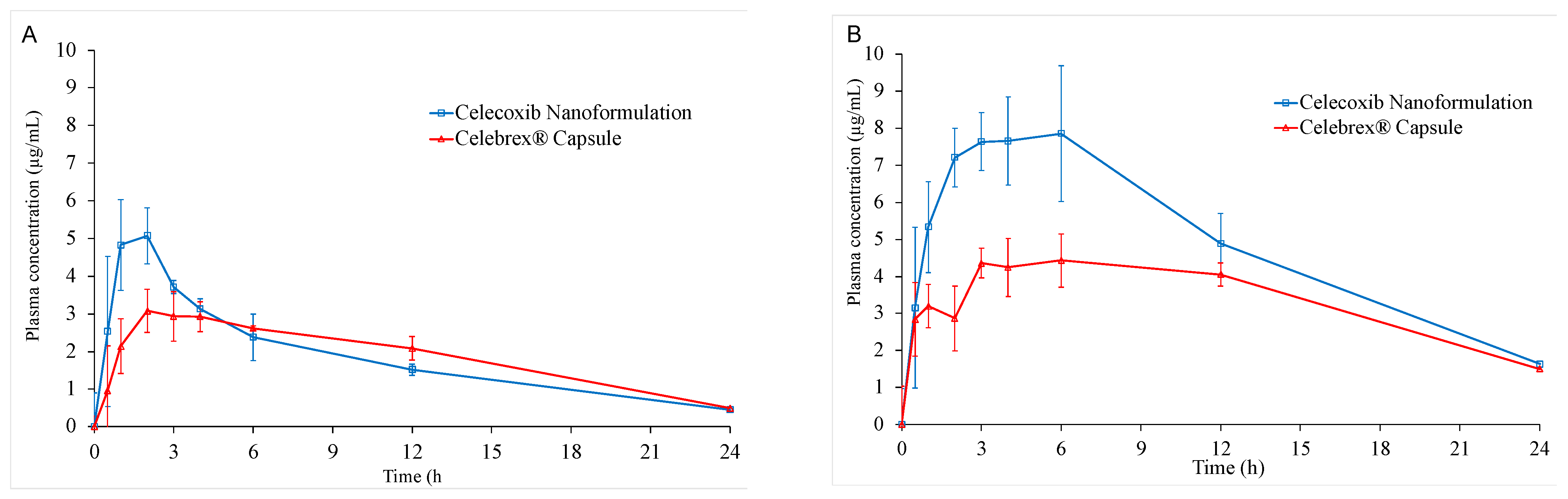
3.6. In Vivo Oral Bioavailability
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malamatari, M.; Taylor, K.M.G.; Malamataris, S.; Douroumis, D.; Kachrimanis, K. Pharmaceutical nanocrystals: Production by wet milling and applications. Drug. Discov. Today 2018, 23, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Merisko-Liversidge, E.M.; Liversidge, G.G. Drug Nanoparticles: Formulating Poorly Water-Soluble Compounds. Toxicol. Pathol. 2008, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.K.; Jackson, J.D.; Bell, R.G.; Martin, G. Poorly Soluble Drugs: Dissolution and Drug Release. Dissolution Technol. 2017, 24, 42. [Google Scholar]
- Pergolizzi, J.V., Jr.; Raffa, R.B.; Nalamachu, S.; Taylor, R., Jr. Evolution to low-dose NSAID therapy. Pain. Manag. 2016, 6, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Terada, K. Rate- and Extent-Limiting Factors of Oral Drug Absorption: Theory and Applications. J. Pharm. Sci. 2015, 104, 2777–2788. [Google Scholar] [CrossRef]
- Sugano, K.; Okazaki, A.; Sugimoto, S.; Tavornvipas, S.; Omura, A.; Mano, T. Solubility and dissolution profile assessment in drug discovery. Drug. Metab. Pharm. 2007, 22, 225–254. [Google Scholar] [CrossRef]
- Alshora, D.H.; Ibrahim, M.A.; Alanazi, F.K. Chapter 6—Nanotechnology from particle size reduction to enhancing aqueous solubility. In Surface Chemistry of Nanobiomaterials; Grumezescu, A.M., Ed.; William Andrew Publishing: New York, NY, USA, 2016; pp. 163–191. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharm. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]
- Narvekar, M.; Xue, H.Y.; Eoh, J.Y.; Wong, H.L. Nanocarrier for poorly water-soluble anticancer drugs--barriers of translation and solutions. AAPS PharmSciTech 2014, 15, 822–833. [Google Scholar] [CrossRef]
- Gigliobianco, M.R.; Casadidio, C.; Censi, R.; Di Martino, P. Nanocrystals of Poorly Soluble Drugs: Drug Bioavailability and Physicochemical Stability. Pharmaceutics 2018, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Placha, D.; Jampilek, J. Chronic Inflammatory Diseases, Anti-Inflammatory Agents and Their Delivery Nanosystems. Pharmaceutics 2021, 13, 64. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Johnston, K.P.; Williams III, R.O. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug. Dev. Ind. Pharm. 2004, 30, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Dixit, M.; Kini, A.G.; Kulkarni, P. Enhancing solubility and dissolution of celecoxib by spray drying using Pluronic® F 127. Indian J. Pharm. Educ. 2011, 45, 346–352. [Google Scholar]
- Manca, M.L.; Lai, F.; Pireddu, R.; Valenti, D.; Schlich, M.; Pini, E.; Ailuno, G.; Fadda, A.M.; Sinico, C. Impact of nanosizing on dermal delivery and antioxidant activity of quercetin nanocrystals. J. Drug. Deliv. Sci. Technol. 2020, 55, 101482. [Google Scholar] [CrossRef]
- Rasenack, N.; Müller, B.W. Micron—Size Drug Particles: Common and Novel Micronization Techniques. Pharm. Dev. Technol. 2004, 9, 1–13. [Google Scholar] [CrossRef]
- Albetawi, S.; Abdalhafez, A.; Abu-Zaid, A.; Matrouk, A.; Alhourani, N. Recent solubility and dissolution enhancement techniques for repaglinide a BCS class II drug: A review. Pharmacia 2021, 68, 573–583. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug. Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef]
- Loh, Z.H.; Samanta, A.K.; Sia Heng, P.W. Overview of milling techniques for improving the solubility of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 255–274. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, P.J.; Olugemo, K.; Solorio, D.; Young, C.L. Pharmacokinetic properties and tolerability of low-dose SoluMatrix diclofenac. Clin. Ther. 2015, 37, 448–461. [Google Scholar] [CrossRef]
- iCeutica; Dodd, A.; Meiser, F.; Norret, M.; Russell, A.; Bosch, H.W. Diclofenac Formulation. EP2421525, 7 June 2017. [Google Scholar]
- iCeutica; Bosch, H.W. Formulation of Meloxicam. US Patent US9526734B2, 27 December 2017. [Google Scholar]
- iCeutica; Dodd, A.; Meiser, F.; Norret, M.; Russell, A.; Bosch, H.W. Formulation of Indomethacin. US Patent US9095496B2, 4 August 2015. [Google Scholar]
- Politis, S.N.; Colombo, P.; Colombo, G.; Rekkas, D.M. Design of experiments (DoE) in pharmaceutical development. Drug. Dev. Ind. Pharm. 2017, 43, 889–901. [Google Scholar] [CrossRef]
- Sreeharsha, N.; Naveen, N.R.; Anitha, P.; Goudanavar, P.S.; Ramkanth, S.; Fattepur, S.; Telsang, M.; Habeebuddin, M.; Answer, M.K. Development of Nanocrystal Compressed Minitablets for Chronotherapeutic Drug Delivery. Pharmaceuticals 2022, 15, 311. [Google Scholar] [CrossRef]
- Vining, G.; Kowalski, S. An Overview of Composite Designs Run as Split-Plots. In Frontiers in Statistical Quality Control 8; Lenz, H.-J., Wilrich, P.-T., Eds.; Physica-Verlag HD: Berlin/Heidelberg, Germany, 2006; pp. 342–351. [Google Scholar]
- Beg, S.; Rahman, Z. Central Composite Designs and Their Applications in Pharmaceutical Product Development. In Design of Experiments for Pharmaceutical Product Development: Volume I: Basics and Fundamental Principles; Beg, S., Ed.; Springer: Singapore, 2021; pp. 63–76. [Google Scholar] [CrossRef]
- Wagner, J.R.; Mount, E.M.; Giles, H.F. 25—Design of Experiments. In Extrusion, 2nd ed.; Wagner, J.R., Mount, E.M., Giles, H.F., Eds.; William Andrew Publishing: Oxford, UK, 2014; pp. 291–308. [Google Scholar]
- Witek-Krowiak, A.; Chojnacka, K.; Podstawczyk, D.; Dawiec, A.; Pokomeda, K. Application of response surface methodology and artificial neural network methods in modelling and optimization of biosorption process. Bioresour. Technol. 2014, 160, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Sarrai, A.E.; Hanini, S.; Merzouk, N.K.; Tassalit, D.; Szabó, T.; Hernádi, K.; Nagy, L. Using Central Composite Experimental Design to Optimize the Degradation of Tylosin from Aqueous Solution by Photo-Fenton Reaction. Materials 2016, 9, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mockus, J. Bayesian Approach to Global Optimization, 1st ed.; Springer: Dordrecht, The Netherlands, 1989; p. 270. [Google Scholar]
- Terayama, K.; Sumita, M.; Tamura, R.; Tsuda, K. Black-Box Optimization for Automated Discovery. Acc. Chem. Res. 2021, 54, 1334–1346. [Google Scholar] [CrossRef]
- Sano, S.; Kadowaki, T.; Tsuda, K.; Kimura, S. Application of Bayesian Optimization for Pharmaceutical Product Development. J. Pharm. Innov. 2020, 15, 333–343. [Google Scholar] [CrossRef]
- Pfizer. Celebrex Product SPC. Available online: https://labeling.pfizer.com/ShowLabeling.aspx?id=793 (accessed on 16 January 2023).
- FDA. Biopharmaceutical Review Celebrex. Clinical Pharmacology and Biopharmaceutics Review For Celebrex (Celecoxib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21156-S007_Celebrex_biopharmr.pdf (accessed on 16 January 2023).
- Sciences, B. Elyxyb SPC. 2019. Highlights of Prescribing Information. Available online: https://www.elyxyb.com/images/global/elyxyb-prescribing-info.pdf (accessed on 16 January 2023).
- Garry, G.; Grahama, A.R.O.D. Coxibs. Compend. Inflamm. Dis. 2016. [Google Scholar] [CrossRef]
- Ha, E.S.; Choo, G.H.; Baek, I.H.; Kim, M.S. Formulation, characterization, and in vivo evaluation of celecoxib-PVP solid dispersion nanoparticles using supercritical antisolvent process. Molecules 2014, 19, 20325–20339. [Google Scholar] [CrossRef] [Green Version]
- Homayouni, A.; Sadeghi, F.; Nokhodchi, A.; Varshosaz, J.; Afrasiabi Garekani, H. Preparation and characterization of celecoxib dispersions in soluplus(®): Comparison of spray drying and conventional methods. Iran. J. Pharm. Res. 2015, 14, 35–50. [Google Scholar]
- Homayouni, A.; Sadeghi, F.; Nokhodchi, A.; Varshosaz, J.; Garekani, H.A. Preparation and characterization of celecoxib solid dispersions; comparison of poloxamer-188 and PVP-K30 as carriers. Iran. J. Basic. Med. Sci. 2014, 17, 322–331. [Google Scholar]
- Knopp, M.M.; Nguyen, J.H.; Becker, C.; Francke, N.M.; Jørgensen, E.B.; Holm, P.; Holm, R.; Mu, H.; Rades, T.; Langguth, P. Influence of polymer molecular weight on in vitro dissolution behavior and in vivo performance of celecoxib:PVP amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2016, 101, 145–151. [Google Scholar] [CrossRef]
- Kwon, H.J.; Heo, E.J.; Kim, Y.H.; Kim, S.; Hwang, Y.H.; Byun, J.M.; Cheon, S.H.; Park, S.Y.; Kim, D.Y.; Cho, K.H.; et al. Development and Evaluation of Poorly Water-Soluble Celecoxib as Solid Dispersions Containing Nonionic Surfactants Using Fluidized-Bed Granulation. Pharmaceutics 2019, 11, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, S.Y.; Martins, S.M.; Brandl, M.; Bauer-Brandl, A. Solid Phospholipid Dispersions for Oral Delivery of Poorly Soluble Drugs: Investigation Into Celecoxib Incorporation and Solubility-In Vitro Permeability Enhancement. J. Pharm. Sci. 2016, 105, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Han, Y.; Xu, G.; Yin, L.; Ngandeu Neubi, M.; Zhou, J.; Ding, Y. Preparation and evaluation of celecoxib nanosuspensions for bioavailability enhancement. RSC Adv. 2017, 7, 13053–13064. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.C.; Kim, D.S.; Jin, S.G.; Youn, Y.S.; Oh, K.T.; Li, D.X.; Yong, C.S.; Oh Kim, J.; Kim, K.S.; Choi, H.G. Development of a novel celecoxib-loaded nanosuspension using a wet media milling process. Pharmazie 2018, 73, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Moghimipour, E. Preparation and evaluation of celecoxib nanoemulsion for ocular drug delivery. Asian J. Pharm. 2017, 11, 543–550. [Google Scholar]
- Nguyen, T.H.; Tan, A.; Santos, L.; Ngo, D.; Edwards, G.A.; Porter, C.J.; Prestidge, C.A.; Boyd, B.J. Silica-lipid hybrid (SLH) formulations enhance the oral bioavailability and efficacy of celecoxib: An in vivo evaluation. J. Control Release 2013, 167, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Jain, S.K. Solubility enhancement of celecoxib using β-cyclodextrin inclusion complexes. Eur. J. Pharm. Biopharm. 2004, 57, 263–267. [Google Scholar] [CrossRef]
- Mandracchia, D.; Trapani, A.; Perteghella, S.; Sorrenti, M.; Catenacci, L.; Torre, M.L.; Trapani, G.; Tripodo, G. pH-sensitive inulin-based nanomicelles for intestinal site-specific and controlled release of celecoxib. Carbohydr. Polym. 2018, 181, 570–578. [Google Scholar] [CrossRef]
- Matbou Riahi, M.; Sahebkar, A.; Sadri, K.; Nikoofal-Sahlabadi, S.; Jaafari, M.R. Stable and sustained release liposomal formulations of celecoxib: In vitro and in vivo anti-tumor evaluation. Int. J. Pharm. 2018, 540, 89–97. [Google Scholar] [CrossRef]
- Hoyle, P.C.; Waymack, P. Ameliorating Drug-Induced Elevations in Blood Pressure by Adjunctive use of Antihypertensive Drugs. U.S. Patent 9,408,837, 9 July 2017. [Google Scholar]
- Burmeister, C.F.; Kwade, A. Process engineering with planetary ball mills. Chem. Soc. Rev. 2013, 42, 7660–7667. [Google Scholar] [CrossRef]
- Hutter, F.; Kotthoff, L.; Vanschoren, J. (Eds.) Automated Machine Learning, 1st ed.; Springer: Cham, Switerland, 2019. [Google Scholar]
- Williams, C.K.I.; Rasmussen, C.E. Gaussian Processes for Machine Learning; The MIT Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Wada, T.; Hino, H. Bayesian Optimization for Multi-Objective Optimization and Multi-Point Search. arXiv 2019, arXiv:1905.02370. [Google Scholar]
- Dezani, A.; Dezani, T.; Caffaro, A.; Reis, J.; Serra, C. Equilibrium solubility versus intrinsic dissolution: Characterization of lamivudine, stavudine and zidovudine for BCS classification. Braz. J. Pharm. Sci. 2013, 49, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-I.; Park, S.Y.; Park, S.J.; Lee, J.; Cho, K.H.; Jee, J.-P.; Kim, H.-C.; Maeng, H.-J.; Jang, D.-J. Development and evaluation of a reconstitutable dry suspension to improve the dissolution and oral absorption of poorly water-soluble celecoxib. Pharmaceutics 2018, 10, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksoy, D.O.; Sagol, E. Application of central composite design method to coal flotation: Modelling, optimization and verification. Fuel 2016, 183, 609–616. [Google Scholar] [CrossRef]
- Ghaedi, M.; Mazaheri, H.; Khodadoust, S.; Hajati, S.; Purkait, M.K. Application of central composite design for simultaneous removal of methylene blue and Pb2+ ions by walnut wood activated carbon. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 135, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Bejaoui, M.; Oueslati, H.; Galai, H. Ternary Solid Dispersion Strategy for Solubility Enhancement of Poorly Soluble Drugs by Co-Milling Technique. In Chitin and Chitosan—Physicochemical Properties and Industrial Applications; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Chen, X.; Partheniadis, I.; Nikolakakis, I.; Al-Obaidi, H. Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling. Polymers 2020, 12, 854. [Google Scholar] [CrossRef] [Green Version]
- Slámová, M.; Prausová, K.; Epikaridisová, J.; Brokešová, J.; Kuentz, M.; Patera, J.; Zámostný, P. Effect of co-milling on dissolution rate of poorly soluble drugs. Int. J. Pharm. 2021, 597, 120312. [Google Scholar] [CrossRef]
- Bhatt, V.; Shete, G.; Bansal, A.K. Mechanism of generation of drug nanocrystals in celecoxib: Mannitol nanocrystalline solid dispersion. Int. J. Pharm. 2015, 495, 132–139. [Google Scholar] [CrossRef]
- Kataoka, M.; Yano, K.; Hamatsu, Y.; Masaoka, Y.; Sakuma, S.; Yamashita, S. Assessment of absorption potential of poorly water-soluble drugs by using the dissolution/permeation system. Eur. J. Pharm. Biopharm. 2013, 85, 1317–1324. [Google Scholar] [CrossRef]
- Bozdağ-Pehlivan, S.; Subaşi, B.; Vural, I.; Unlü, N.; Capan, Y. Evaluation of drug-excipient interaction in the formulation of celecoxib tablets. Acta Pol. Pharm. 2011, 68, 423–433. [Google Scholar]
- Ventura, C.A.; Giannone, I.; Paolino, D.; Pistarà, V.; Corsaro, A.; Puglisi, G. Preparation of celecoxib-dimethyl-beta-cyclodextrin inclusion complex: Characterization and in vitro permeation study. Eur. J. Med. Chem. 2005, 40, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Yazdanian, M.; Briggs, K.; Jankovsky, C.; Hawi, A. The “high solubility” definition of the current FDA Guidance on Biopharmaceutical Classification System may be too strict for acidic drugs. Pharm. Res. 2004, 21, 293–299. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Factors | Design Level | ||
|---|---|---|---|
| Coded | Actual Parameters | Coded Value | Actual Value |
| X1 | PVP/CXB weight ratio | −1.633 | 0.1835 |
| −1 | 0.5 | ||
| 0 | 1 | ||
| +1 | 1.5 | ||
| +1.633 | 1.8165 | ||
| X2 | SLS/CXB weight ratio | −1.633 | −0.0633 |
| −1 | 0 | ||
| 0 | 0.1 | ||
| +1 | 0.2 | ||
| +1.633 | 0.2633 | ||
| X3 | MAN/CXB weight ratio | −1.633 | 0.1835 |
| −1 | 0.5 | ||
| 0 | 1 | ||
| +1 | 1.5 | ||
| +1.633 | 1.8165 | ||
| Independent Factors | Response Variables | ||
|---|---|---|---|
| X1 | X2 | X3 | Y2 |
| 1 | 0.1 | 1 | 20.00 |
| 1 | 0.1 | 1 | 27.50 |
| 1.5 | 0.2 | 0.5 | 42.70 |
| 0.5 | 0.2 | 0.5 | 5.00 |
| 1.5 | 0 | 0.5 | 1.70 |
| 1 | 0.1 | 1 | 30.00 |
| 0.5 | 0 | 1.5 | 1.20 |
| 1.5 | 0.2 | 1.5 | 42.30 |
| Run Order | Formulation Code | Independent Factors | ||
|---|---|---|---|---|
| X1 | X2 | X3 | ||
| 1 | BO01 | 1.23 | 0.14 | 0.79 |
| 2 | BO02 | 1.92 | 0.02 | 0.64 |
| 3 | BO03 | 0.81 | 0.19 | 1.13 |
| 4 | BO04 | 1.63 | 0.15 | 1.04 |
| 5 | BO05 | 1.44 | 0.16 | 0.9 |
| 6 | BO06 | 1.55 | 0.05 | 1.28 |
| 7 | BO07 | 1.99 | 0.175 | 0.81 |
| 8 | BO08 | 1.56 | 0.156 | 0.85 |
| 9 | BO09 | 1.85 | 0.16 | 1.95 |
| 10 | BO10 | 1.7 | 0.16 | 0.9 |
| 11 | BO11 | 1.9 | 0.16 | 1.2 |
| 12 | BO12 | 2.0 | 0.14 | 0.5 |
| Run Order | Formulation Code | Independent Factors | Response Variables | |||
|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 | ||
| 1 | CC01 | 1 | 0.1 | 1 | 90 | 42.7 |
| 2 | CC02 | 1 | −0.0633 | 1 | 56.8 | 1.5 |
| 3 | CC03 | 1 | 0.1 | 0.1835 | 13.9 | 3.7 |
| 4 | CC04 | 1 | 0.1 | 1 | 26 | 20 |
| 5 | CC05 | 0.1835 | 0.1 | 1 | 8.7 | 1 |
| 6 | CC06 | 1 | 0.1 | 1.8165 | 29 | 1.7 |
| 7 | CC07 | 1 | 0.2633 | 1 | 24 | 1.2 |
| 8 | CC08 | 1.8165 | 0.1 | 1 | 28 | 27.5 |
| 9 | CC09 | 1 | 0.1 | 1 | 30 | 30 |
| 10 | CC10 | 1 | 0.1 | 1 | 37.8 | 32.8 |
| 11 | CC11 | 1.5 | 0.2 | 0.5 | 87 | 42.3 |
| 12 | CC12 | 0,5 | 0.2 | 0.5 | 9.8 | 5 |
| 13 | CC13 | 1.5 | 0 | 0.5 | 65.4 | 46 |
| 14 | CC14 | 1 | 0.1 | 1 | 26.3 | 1.8 |
| 15 | CC15 | 0.5 | 0 | 1.5 | 45.1 | 16.7 |
| 16 | CC16 | 1.5 | 0.2 | 1.5 | 31.6 | 23.1 |
| 17 | CC17 | 1.5 | 0 | 1.5 | 6.8 | 10.6 |
| 18 | CC18 | 0.5 | 0 | 0.5 | 11.8 | 22 |
| 19 | CC19 | 1 | 0.1 | 1 | 30 | 19.8 |
| 20 | CC20 | 0.5 | 0.2 | 1.5 | 69.5 | 43.3 |
| Y1: Dissolved CXB Amount in pH 12 after 30 min | Y2: Dissolved CXB Amount in pH 1.2 + 0.2% SLS | |||||||
|---|---|---|---|---|---|---|---|---|
| Sum of Squares | Degree of Freedom | F-Value | p Value | Sum of Squares | Degree of Freedom | F-Value | p Value | |
| Model | 11,565.2 | 9 | 32.96 | <0.0001 | 143.23 | 1 | 11.76 | 0.0006 |
| X1 | 7151.9 | 1 | 183.42 | <0.0001 | 4361.28 | 9 | 31.096 | 0.0003 |
| X2 | 1599.8 | 1 | 41.03 | 0.0001 | 1281.16 | 1 | 46.88 | <0.0001 |
| X3 | 728.8 | 1 | 18.69 | 0.0019 | 1931.49 | 1 | 0.19 | 0.6691 |
| X1X2 | 1255.0 | 1 | 32.19 | 0.0003 | 8.04 | 1 | 17.20 | 0.0025 |
| X1X3 | 3.6 | 1 | 0.09 | 0.7667 | 708.76 | 1 | 0.00 | 0.9786 |
| X2X3 | 220.5 | 1 | 5.66 | 0.0414 | 0.03 | 1 | 0.01 | 0.9274 |
| X12 | 135.4 | 1 | 3.47 | 0.0953 | 0.36 | 1 | 1.21 | 0.2986 |
| X22 | 489.7 | 1 | 12.56 | 0.0063 | 50.14 | 1 | 3.07 | 0.1139 |
| X32 | 2.5 | 1 | 0.06 | 0.8056 | 126.28 | 1 | 7.38 | 0.02376 |
| Residual | 350.9 | 9 | 304.09 | 1 | ||||
| Lack of Fit | 269.6 | 5 | 2.65 | 0.1829 | 370.79 | 9 | 2.29 | 0.2212 |
| Pure Error | 81.3 | 4 | 274.79 | 5 | ||||
| Cor Total | 11,920.4 | 19 | 96.01 | 4 | ||||
| R2 = 0.9706 | R2 = 0.9216 | |||||||
| Formulation Trial | Predicted and Experimental Results | |||||
|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 | ||
| 1 | 1.6 | 0.15 | 1.0 | Predicted | 77.3 | 46.6 |
| Experimental | 83.2 | 49.1 | ||||
| Error | 7.63 | 5.3 | ||||
| Desirability | 0.92 | |||||
| 2 | 1.5 | 0.2 | 1.5 | Predicted | 88.6 | 47.2 |
| Experimental | 87.0 | 42.3 | ||||
| Error | 1.8 | 10.3 | ||||
| Desirability | 0.99 | |||||
| 3 | 1.5 | 0.2 | 0.5 | Predicted | 83.1 | 49.1 |
| Experimental | 90.0 | 47.2 | ||||
| Error | 8.3 | 3.8 | ||||
| Desirability | 0.90 | |||||
| Formulation Code | Independent Factors | Response Variable | ||
|---|---|---|---|---|
| X1 | X2 | X3 | Y2 | |
| BO01 | 1.23 | 0.14 | 0.79 | 45.0 |
| BO02 | 1.92 | 0.02 | 0.64 | 1.7 |
| BO03 | 0.81 | 0.19 | 1.13 | 16.4 |
| BO04 | 1.63 | 0.15 | 1.04 | 49.1 |
| BO05 | 1.44 | 0.16 | 0.9 | 47.8 |
| BO06 | 1.55 | 0.05 | 1.28 | 9.6 |
| BO07 | 1.99 | 0.175 | 0.81 | 46.2 |
| BO08 | 1.56 | 0.156 | 0.85 | 46.1 |
| BO09 | 1.85 | 0.16 | 1.95 | 36 |
| BO10 | 1.7 | 0.16 | 0.9 | 48.5 |
| BO11 | 1.9 | 0.16 | 1.2 | 48 |
| BO12 | 2.0 | 0.14 | 0.5 | 48.4 |
| Optimized Co-Milled CXB | Time (Month) | Assay (%) | Dissolved CXB (mg/cm2) | Z-Average (nm) | PDI |
|---|---|---|---|---|---|
| 40 °C, 75% BN | 0 | 100.5 | 92.2 | 183.50 | 0.24 |
| 1 | 101 | 85.8 | |||
| 2 | 98.7 | 83.2 | |||
| 3 | 98.2 | 77.2 | 219.47 | 0.30 | |
| 25 °C, 60% BN | 3 | 99.8 | 84.0 | 217.33 | 0.33 |
| PM | Time (month) | Assay (%) | Dissolved CXB (mg/cm2) | Z-Average (nm) | PDI |
| 40 °C, 75% BN | 0 | 103 | 22.6 | 1438.33 | 0.21 |
| 1 | 101 | 22.0 | |||
| 2 | 99.5 | 17.2 | |||
| 3 | 99.8 | 13.8 | 1406.67 | 0.30 | |
| 25 °C, 60% BN | 3 | 102 | 24.8 | 1493.67 | 0.25 |
| PK Parameters | 10 mg/kg | 40 mg/kg | ||
|---|---|---|---|---|
| Celecoxib Nanoformulation | Celebrex® Capsule | Celecoxib Nanoformulation | Celebrex® Capsule | |
| Cmaks (µg/mL) | 5.51 ± 1.06 | 3.42 ± 0.35 | 8.23 ± 0.82 | 5.08 ± 0.38 |
| tmaks (h) | 1.60 ± 0.55 | 2.60 ± 1.34 | 3.80 ± 2.28 | 6.00 ± 3.67 |
| AUC0–24 (h·µg/mL) | 41.75 ± 5.27 | 41.97 ± 4.27 | 110.94 ± 25.22 | 76.42 ± 9.14 |
| AUC0–∞ (h·µg/mL) | 46.81 ± 4.16 | 45.30 ± 6.82 | 142.45 ± 41.83 | 99.27 ± 6.04 |
| t1/2 (h) | 7.35 ± 1.45 | 6.45 ± 1.46 | 9.91 ± 4.47 | 10.40 * |
| MRT (h) | 7.93 ± 0.39 | 9.21 ± 0.98 | 9.33 ± 0.89 | 10.44 ± 0.42 |
| ka (h−1) | 2.38 ± 1.28 | 1.76 ± 1.52 | 2.00 ± 1.60 | 0.96 * |
| ke (h−1) | 0.10 ± 0.02 | 0.11 ± 0.02 | 0.08 ± 0.03 | 0.07 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arslan, A.; Yet, B.; Nemutlu, E.; Akdağ Çaylı, Y.; Eroğlu, H.; Öner, L. Celecoxib Nanoformulations with Enhanced Solubility, Dissolution Rate, and Oral Bioavailability: Experimental Approaches over In Vitro/In Vivo Evaluation. Pharmaceutics 2023, 15, 363. https://doi.org/10.3390/pharmaceutics15020363
Arslan A, Yet B, Nemutlu E, Akdağ Çaylı Y, Eroğlu H, Öner L. Celecoxib Nanoformulations with Enhanced Solubility, Dissolution Rate, and Oral Bioavailability: Experimental Approaches over In Vitro/In Vivo Evaluation. Pharmaceutics. 2023; 15(2):363. https://doi.org/10.3390/pharmaceutics15020363
Chicago/Turabian StyleArslan, Aslıhan, Barbaros Yet, Emirhan Nemutlu, Yağmur Akdağ Çaylı, Hakan Eroğlu, and Levent Öner. 2023. "Celecoxib Nanoformulations with Enhanced Solubility, Dissolution Rate, and Oral Bioavailability: Experimental Approaches over In Vitro/In Vivo Evaluation" Pharmaceutics 15, no. 2: 363. https://doi.org/10.3390/pharmaceutics15020363