
Investigating the Effects of Olaparib on the Susceptibility of Glioblastoma Multiforme Tumour Cells to Natural Killer Cell-Mediated Responses
 , ,
, ,  ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Relationship between NKR Ligand Expression and Disease-Free and Overall Survival of GBM Patients
2.2. Cell Culture of T98G Cells
2.3. Identifying the Non-Lethal Dose of Olaparib in T98G Cells Using Cell Counting Kit (CCK) 8 Assay
2.4. Quantifying the Effects of Olaparib on NKR Ligand Expression by T98G Cells
2.5. Assessing the Effects of Olaparib on NK Cell Phenotype
2.6. Quantifying the Immunomodulatory Effects of T98G Cells and the Potential Rescuing Effects of Olaparib on NK Cell Phenotype and Function
2.7. Quantification of Cytokine Secretion and NKR Ligand Shedding on T98G Cells
2.8. Assessing the Effects of Olaparib on NK Cell Chemotaxis towards T98G Cells
2.9. Statistical Analysis
3. Results
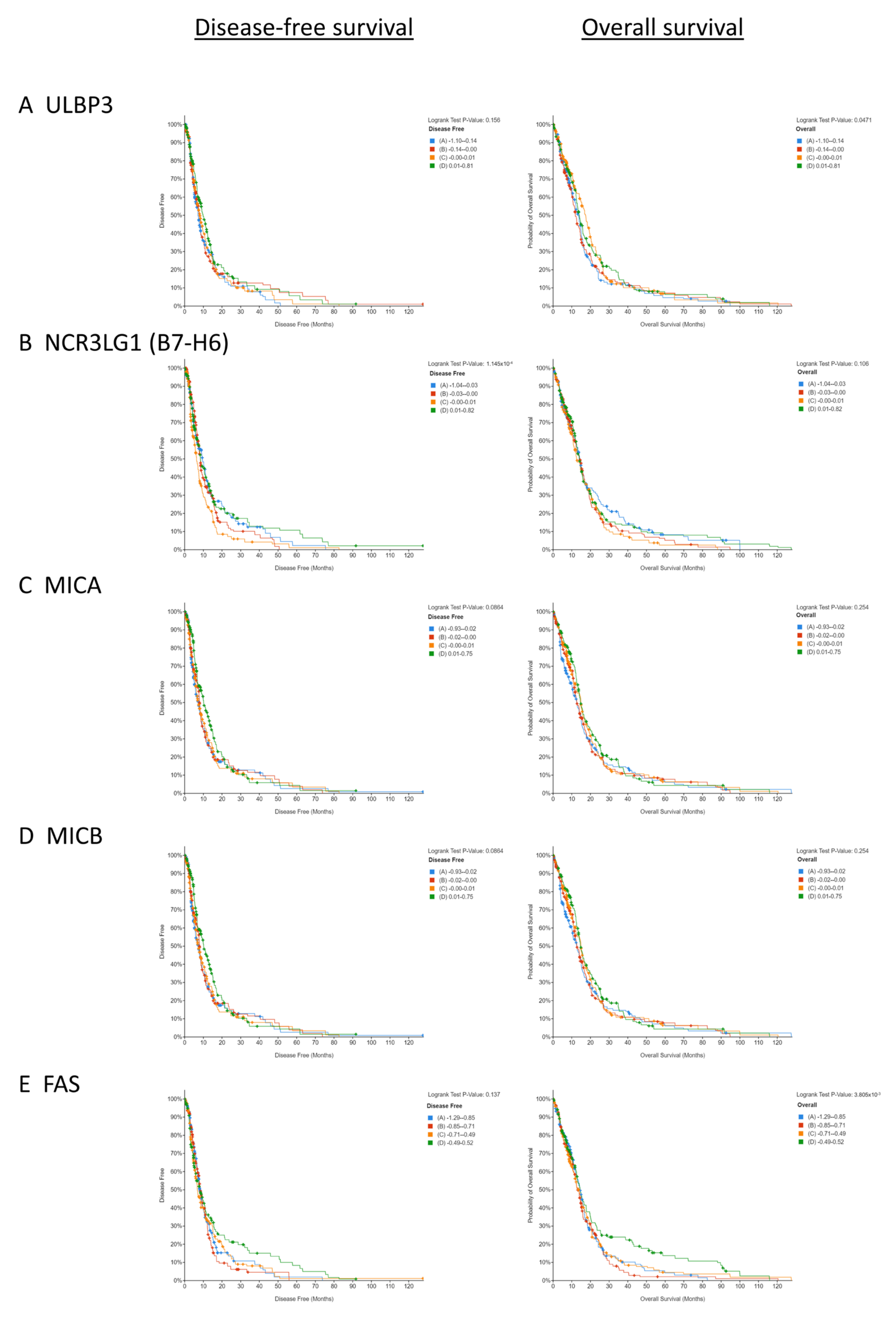
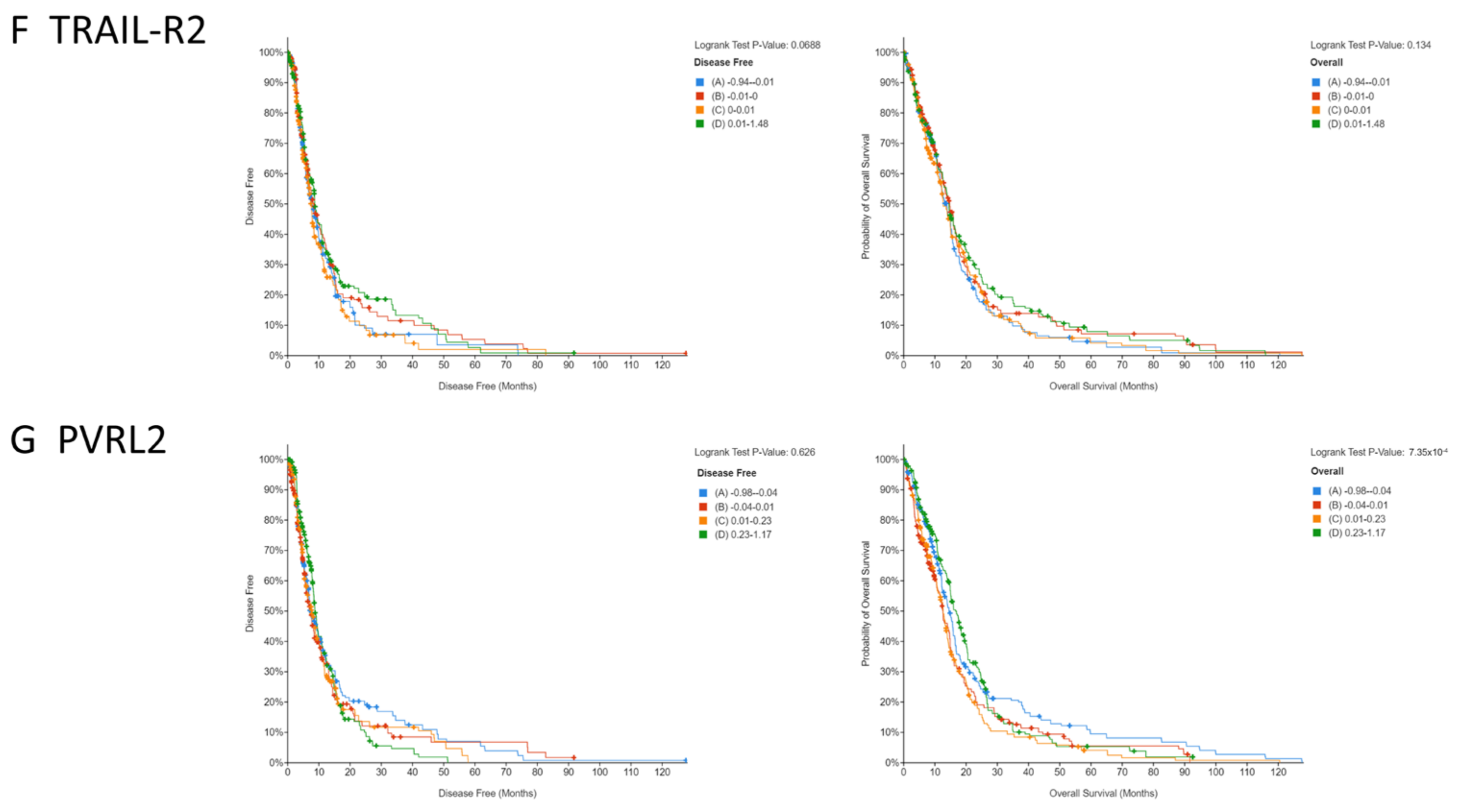
3.1. Low Copy Numbers of Genes Encoding Activating NKR Ligands and Death Receptors Is Associated with Poorer Survival in GBM Patients
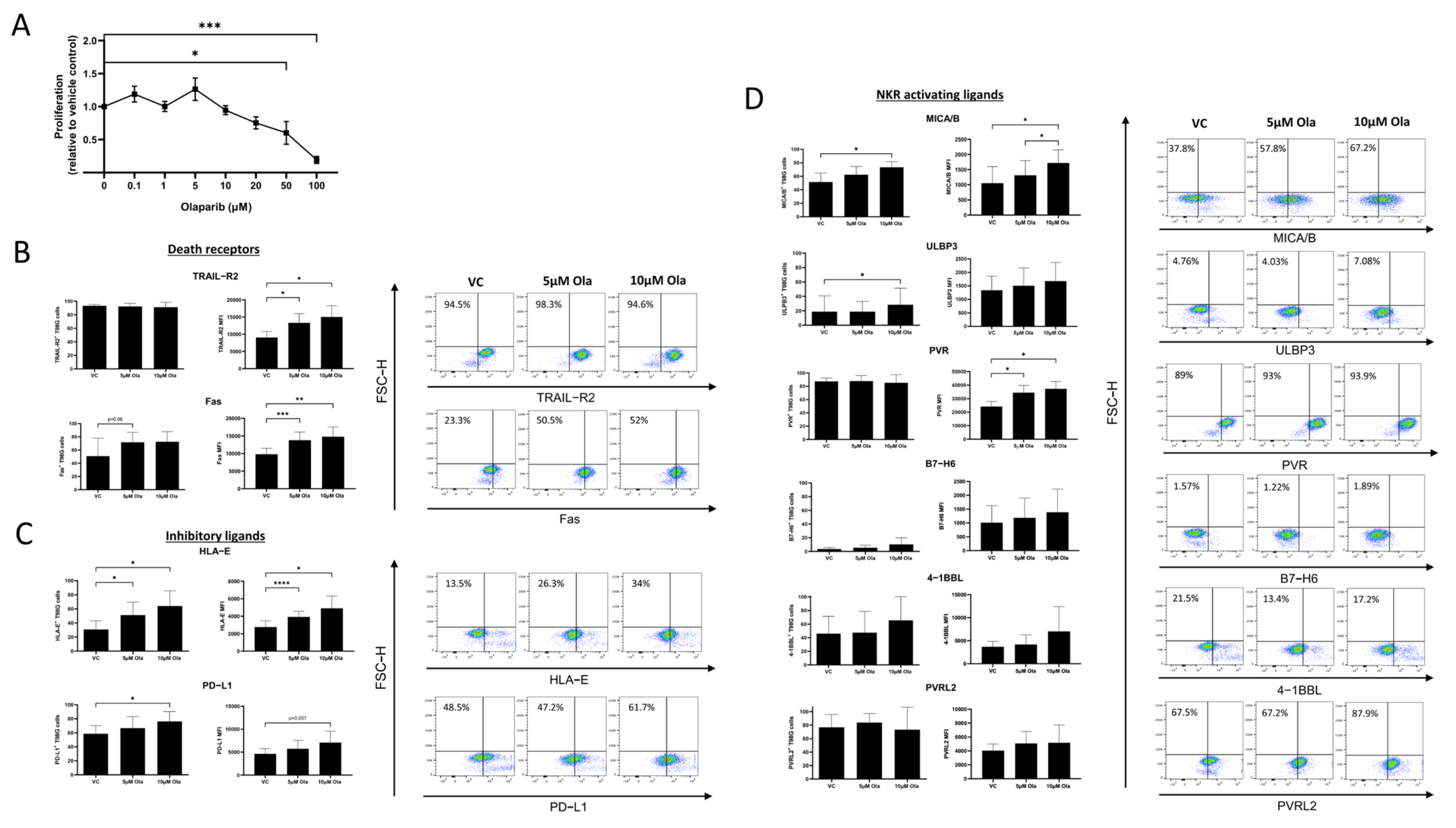
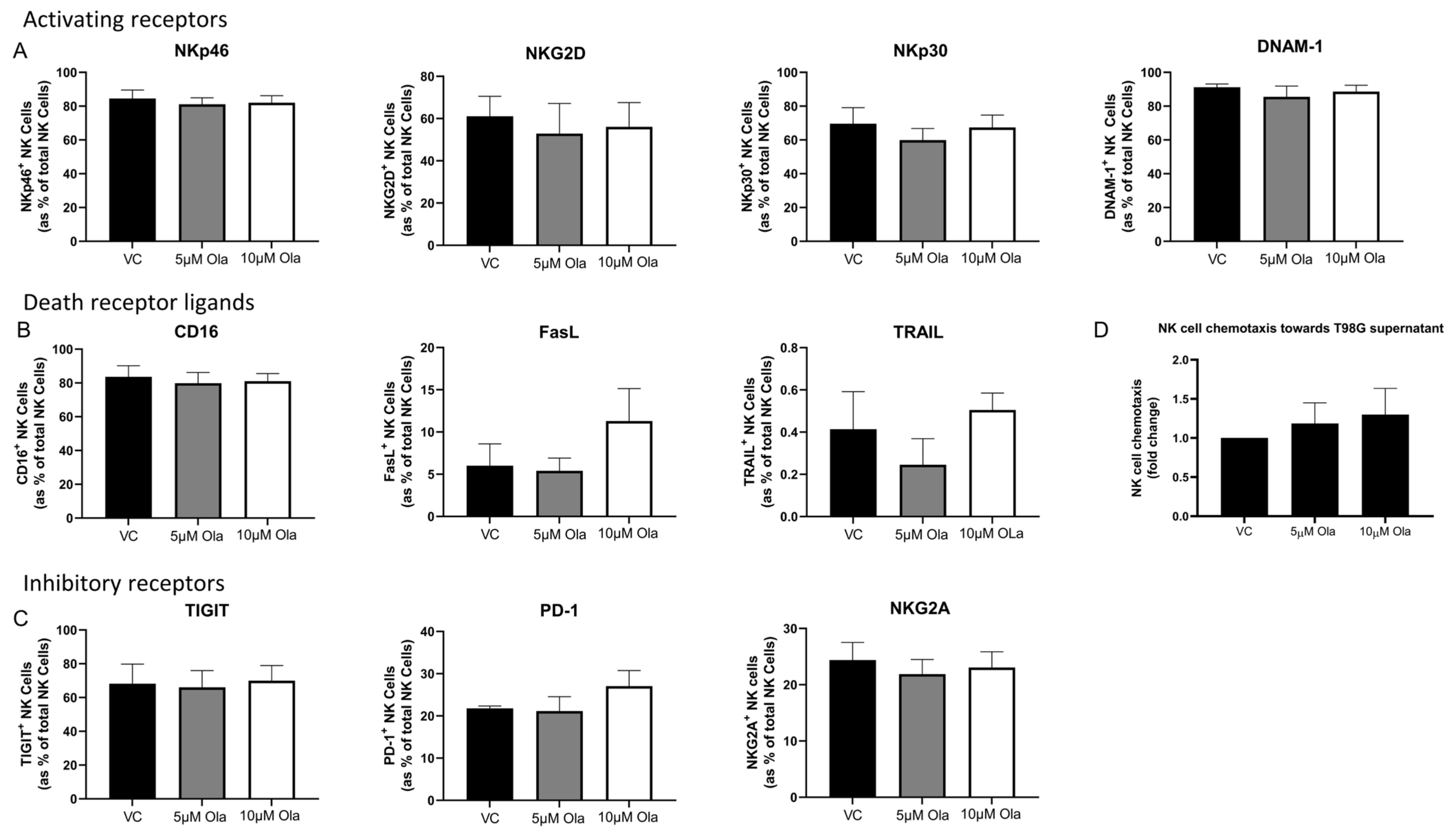
3.2. Olaparib Alters the Surface Expression of Death Receptors and NKR Ligands on T98G Cells
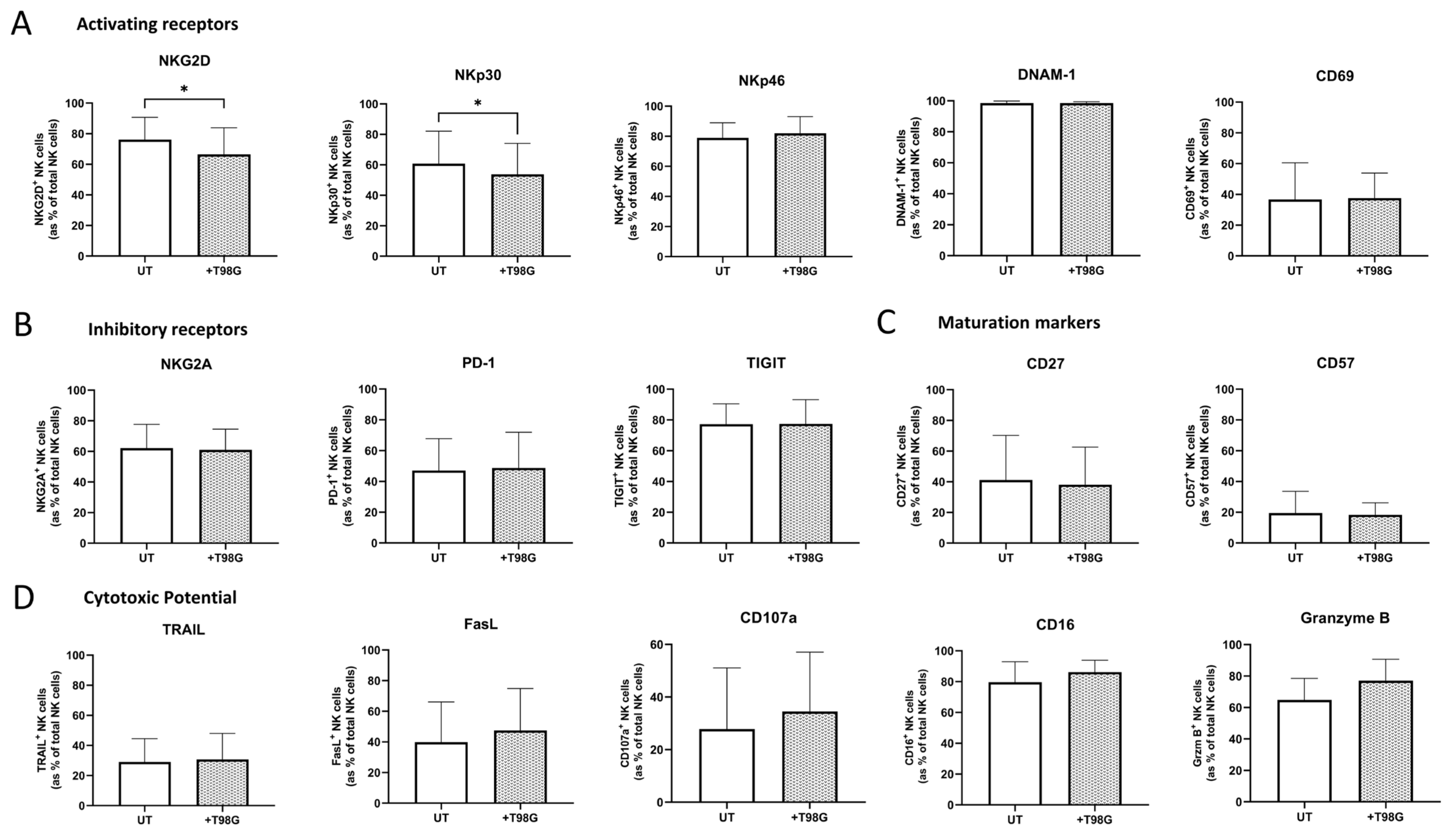
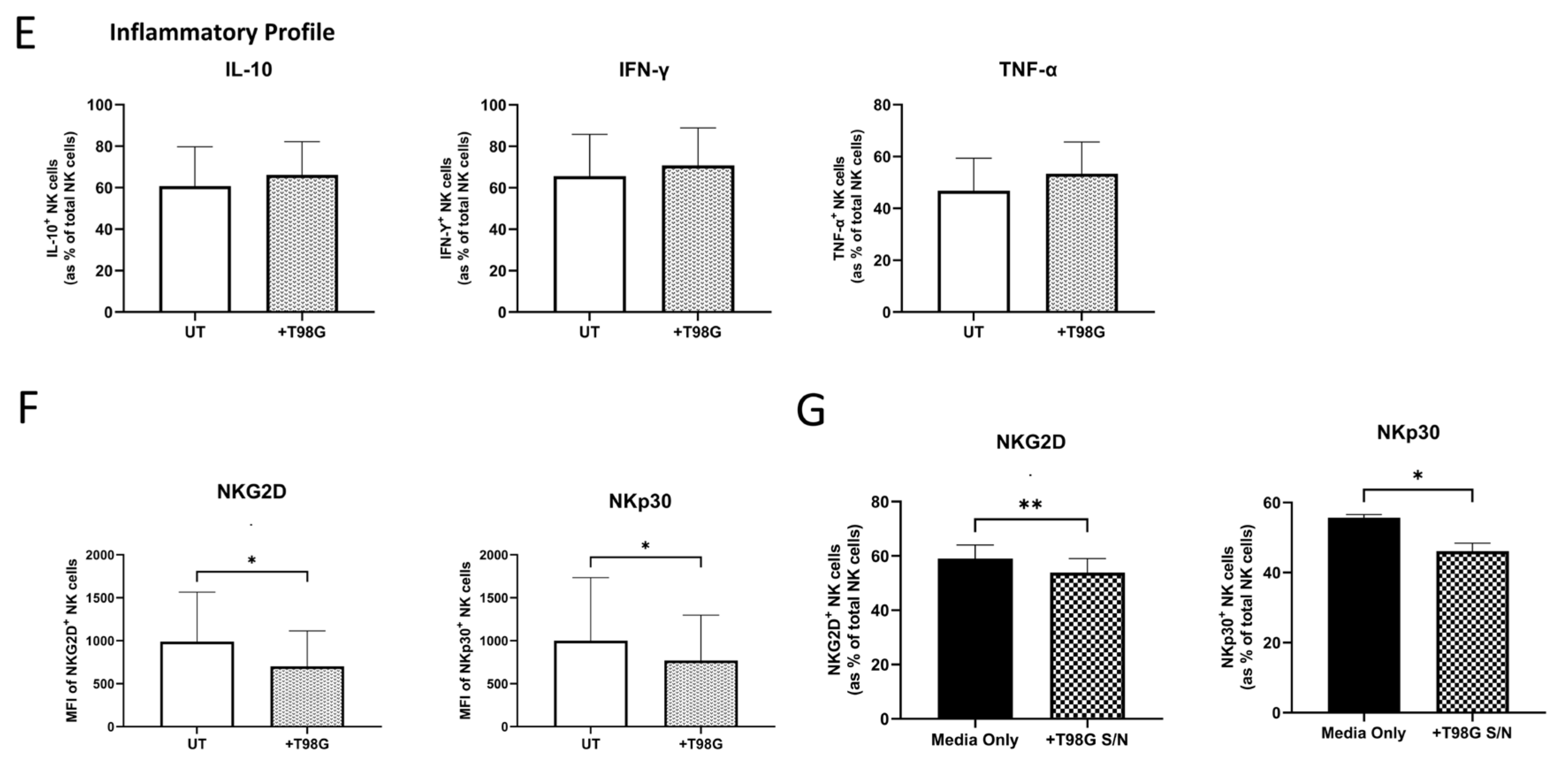
3.3. Direct Co-Culture or Exposure to the Soluble Environment of T98G Cells Diminished NK Cell-Activating Receptor Expression
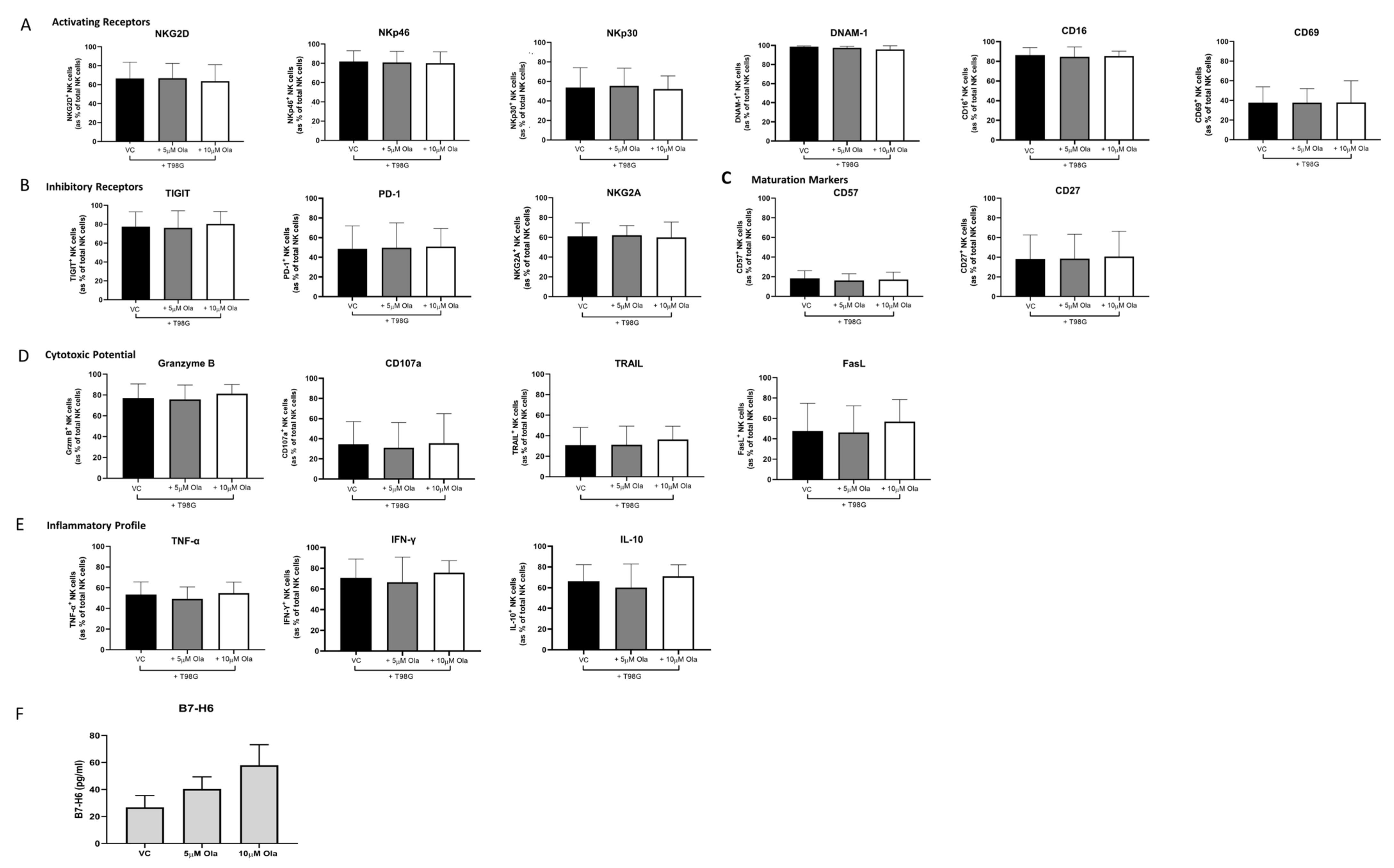
3.4. Concurrent Olaparib Treatment and Direct T98G Co-Culture Did Not Alter NK Cell Phenotype or Function
3.5. Olaparib Treatment Elicits no Significant Effects on the Phenotype, Inflammatory Profile, Cytotoxic Potential or Migratory Capacity of NK Cells
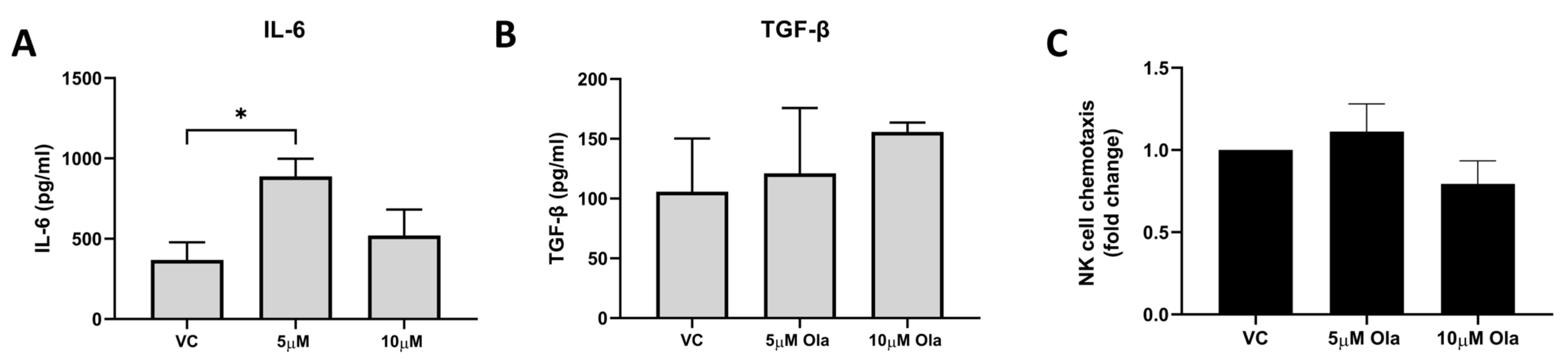
3.6. Olaparib Treatment Enhanced IL-6 Secretion by T98G Cells but Did Not Negatively Affect Chemoattraction of NK Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Brain Tumor Society. About Glioblastoma. 2022. Available online: https://braintumor.org/events/glioblastoma-awareness-day/about-glioblastoma/#:~:text=The%20five%2Dyear%20survival%20rate,been%20virtually%20unchanged%20for%20decades (accessed on 24 November 2022).
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; Van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.S.; Ding, Z.N.; Meng, G.X.; Yan, L.J.; Liu, H.; Li, H.C.; Yao, S.Y.; Tian, B.W.; Dong, Z.R.; Chen, Z.Q.; et al. The Prognostic Value of Natural Killer Cells and Their Receptors/Ligands in Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 872353. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, V.; Srdic, T.; Konjevic, G.; Markovic, O.; Colovic, M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med. Oncol. 2007, 24, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Habif, G.; Crinier, A.; Andre, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Mylod, E.; Lysaght, J.; Conroy, M.J. Natural killer cell therapy: A new frontier for obesity-associated cancer. Cancer Lett. 2022, 535, 215620. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstadt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef]
- Elahi, R.; Heidary, A.H.; Hadiloo, K.; Esmaeilzadeh, A. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev. Rep. 2021, 17, 2081–2106. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Nguyen, B.; Genet, M.; Kim, M.; Chen, P. Tumor Cell IDO Enhances Immune Suppression and Decreases Survival Independent of Tryptophan Metabolism in Glioblastoma. Clin. Cancer Res. 2021, 27, 6514–6528. [Google Scholar] [CrossRef] [PubMed]
- Slattery, K.; Woods, E.; Zaiatz-Bittencourt, V.; Marks, S.; Chew, S.; Conroy, M.; Goggin, C.; MacEochagain, C.; Kennedy, J.; Lucas, S.; et al. TGFbeta drives NK cell metabolic dysfunction in human metastatic breast cancer. J. Immunother. Cancer 2021, 9, e002044. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef] [Green Version]
- Harmon, C.; Robinson, M.W.; Hand, F.; Almuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Atallah Lanman, N.M.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar] [CrossRef]
- Peng, Y.P.; Zhang, J.J.; Liang, W.B.; Tu, M.; Lu, Z.P.; Wei, J.S.; Jiang, K.R.; Gao, W.T.; Wu, J.L.; Xu, Z.K.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, D.; Hippen, K.L.; Lemire, A.; Hying, S.; Luo, X.; Lenvik, T.; Curtsinger, J.; Davis, Z.; Zhang, B.; Cooley, S.; et al. Adaptive NK Cells Resist Regulatory T-cell Suppression Driven by IL37. Cancer Immunol. Res. 2018, 6, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the alphav integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Mylod, E.; McKenna, E.; Davern, M.; Barr, M.P.; Donlon, N.E.; Bibby, B.A.S.; Bhardwaj, A.; Reynolds, J.V.; Lysaght, J. Investigating the susceptibility of treatment-resistant oesophageal tumours to natural killer cell-mediated responses. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chitadze, G.; Bhat, J.; Lettau, M.; Janssen, O.; Kabelitz, D. Generation of soluble NKG2D ligands: Proteolytic cleavage, exosome secretion and functional implications. Scand. J. Immunol. 2013, 78, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.G.; Steinle, A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holl, E.K.; Frazier, V.N.; Landa, K.; Beasley, G.M.; Hwang, E.S.; Nair, S.K. Examining Peripheral and Tumor Cellular Immunome in Patients With Cancer. Front. Immunol. 2019, 10, 1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Li, H.; Chen, Z.; Fan, L.; Feng, S.; Cai, X.; Wang, H. Identification of 3 subpopulations of tumor-infiltrating immune cells for malignant transformation of low-grade glioma. Cancer Cell Int. 2019, 19, 265. [Google Scholar] [CrossRef] [Green Version]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Natural Killer (NK) Cells in Combination with Interleukin-2 (IL-2) and Transforming Growth Factor Beta (TGFbeta) Receptor I Inhibitor Vactosertib in Cancer. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05400122 (accessed on 24 November 2022).
- Faivre, S.; Santoro, A.; Kelley, R.K.; Gane, E.; Costentin, C.E.; Gueorguieva, I.; Smith, C.; Cleverly, A.; Lahn, M.M. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019, 39, 1468–1477. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhu, X.; Kong, L.; Wang, M.; Spanoudis, C.; Chaturvedi, P.; George, V.; Jiao, J.; You, L.; Egan, J.O.; et al. Bifunctional TGF-beta trap/IL-15 protein complex elicits potent NK cell and CD8(+) T cell immunity against solid tumors. Mol. Ther. 2021, 29, 2949–2962. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Fenerty, K.E.; Padget, M.; Wolfson, B.; Gameiro, S.R.; Su, Z.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Hodge, J.W. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J. Immunother. Cancer 2018, 6, 133. [Google Scholar] [CrossRef]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 2018, 8, 670. [Google Scholar] [CrossRef] [Green Version]
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.P.; Chneiweiss, H.; Castera, L.; Müller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 3664. [Google Scholar] [CrossRef]
- Fulton, B.; Short, S.C.; James, A.; Nowicki, S.; McBain, C.; Jefferies, S.; Kelly, C.; Stobo, J.; Morris, A.; Williamson, A.; et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Transl. Radiat. Oncol. 2018, 8, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpel-Massler, G.; Pareja, F.; Aimé, P.; Shu, C.; Chau, L.; Westhoff, M.A.; Halatsch, M.E.; Crary, J.F.; Canoll, P.; Siegelin, M.D. PARP inhibition restores extrinsic apoptotic sensitivity in glioblastoma. PLoS ONE 2014, 9, e114583. [Google Scholar] [CrossRef]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.B.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020, 22, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kane, L.E.; Mellotte, G.S.; Mylod, E.O.; Brien, R.M.; O’Connell, F.; Buckley, C.E.; Arlow, J.; Nguyen, K.; Mockler, D.; Meade, A.; et al. Diagnostic Accuracy of Blood-based Biomarkers for Pancreatic Cancer: A Systematic Review and Meta-analysis. Cancer Res. Commun. 2022, 2, 1229–1243. [Google Scholar] [CrossRef]
- Milani, R.; Brognara, E.; Fabbri, E.; Finotti, A.; Borgatti, M.; Lampronti, I.; Marzaro, G.; Chilin, A.; Lee, K.K.H.; Kok, S.H.L.; et al. Corilagin Induces High Levels of Apoptosis in the Temozolomide-Resistant T98G Glioma Cell Line. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 1307–1315. [Google Scholar] [CrossRef]
- Ghorai, A.; Mahaddalkar, T.; Thorat, R.; Dutt, S. Sustained inhibition of PARP-1 activity delays glioblastoma recurrence by enhancing radiation-induced senescence. Cancer Lett. 2020, 490, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.; Lee, J.H.; Kim, S.H.; Go, K.O.; Ji, S.Y.; Han, J.H.; Kim, C.Y. The Combination PARP Inhibitor Olaparib With Temozolomide in an Experimental Glioblastoma Model. In Vivo 2021, 35, 2015–2023. [Google Scholar] [CrossRef]
- Cruz-Munoz, M.E.; Valenzuela-Vazquez, L.; Sanchez-Herrera, J.; Santa-Olalla Tapia, J. From the “missing self” hypothesis to adaptive NK cells: Insights of NK cell-mediated effector functions in immune surveillance. J. Leukoc. Biol. 2019, 105, 955–971. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Malmberg, K.J.; Carlsten, M.; Björklund, A.; Sohlberg, E.; Bryceson, Y.T.; Ljunggren, H.G. Natural killer cell-mediated immunosurveillance of human cancer. Semin. Immunol. 2017, 31, 20–29. [Google Scholar] [CrossRef]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.A.; Yagita, H.; Takeda, K.; van Dommelen, S.L.H.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef]
- Miller, J.S.; Lanier, L.L. Natural Killer Cells in Cancer Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 77–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhou, Z.; Wang, X.; Zhang, C.; Jiang, X. Natural killer cell awakening: Unleash cancer-immunity cycle against glioblastoma. Cell Death Dis. 2022, 13, 588. [Google Scholar] [CrossRef]
- Golan, I.; Rodriguez de la Fuente, L.; Costoya, J.A. NK Cell-Based Glioblastoma Immunotherapy. Cancers 2018, 10, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, E.; Tsuboi, K.; Saijo, K.; Harada, H.; Takano, S.; Nose, T.; Ohno, T. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer. Res. 2004, 24, 1861–1871. [Google Scholar] [PubMed]
- Celularity. Celularity Announces Expansion of Human Placental Hematopoietic Stem Cell Derived Natural Killer Cells (CYNK-001) Phase 1 Trial in Patients with Acute Myeloid Leukemia. 2022. Available online: https://celularity.com/celularity-announces-expansion-of-human-placental-hematopoietic-stem-cell-derived-natural-killer-cells-cynk-001-phase-1-trial-in-patients-with-acute-myeloid-leukemia/ (accessed on 4 May 2022).
- ClinicalTrials.gov. A Phase I Study of Human Placental Hematopoietic Stem Cell Derived Natural Killer Cells (CYNK-001) in Adults With Recurrent Glioblastoma Multiforme (GBM). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04489420 (accessed on 14 January 2022).
- Faraoni, I.; Aloisio, F.; De Gabrieli, A.; Consalvo, M.I.; Lavorgna, S.; Voso, M.T.; Lo-Coco, F.; Graziani, G. The poly(ADP-ribose) polymerase inhibitor olaparib induces up-regulation of death receptors in primary acute myeloid leukemia blasts by NF-kappaB activation. Cancer Lett. 2018, 423, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- McGilvray, R.W.; Eagle, R.A.; Rolland, P.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int. J. Cancer 2010, 127, 1412–1420. [Google Scholar] [CrossRef]
- Watson, N.F.; Spendlove, I.; Madjd, Z.; McGilvray, R.; Green, A.R.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Expression of the stress-related MHC class I chain-related protein MICA is an indicator of good prognosis in colorectal cancer patients. Int. J. Cancer 2006, 118, 1445–1452. [Google Scholar] [CrossRef]
- Zhu, Z.; Teng, K.Y.; Zhou, J.; Xu, Y.; Zhang, L.; Zhao, H.; Zhang, X.; Tian, L.; Li, Z.; Lu, T.; et al. B7H6 Serves as a Negative Prognostic Marker and an Immune Modulator in Human Pancreatic Cancer. Front. Oncol. 2022, 12, 814312. [Google Scholar] [CrossRef]
- Bisheshar, S.K.; De Ruiter, E.J.; Devriese, L.A.; Willems, S.M. The prognostic role of NK cells and their ligands in squamous cell carcinoma of the head and neck: A systematic review and meta-analysis. Oncoimmunology 2020, 9, 1747345. [Google Scholar] [CrossRef]
- De Kruijf, E.M.; Sajet, A.; Van Nes, J.G.H.; Putter, H.; Smit, V.T.H.B.M.; Eagle, R.; Jafferji, I.; Trowsdale, J.; Liefers, G.J.; Van de Helde, C.J.H.; et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: An observational study. BMC Cancer 2012, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.C.; Bai, Y.; Lu, X.Z.; Qi, C.J. Effects and Mechanism of PARP Inhibitor Olaparib on the Expression of NKG2D Ligands in HL-60 Cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zou, C.; Guan, G.; Guo, Q.; Yan, Z.; Liu, T.; Shen, S.; Xu, X.; Chen, C.; Lin, Z.; et al. Development and validation of an interferon signature predicting prognosis and treatment response for glioblastoma. Oncoimmunology 2019, 8, e1621677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Cretney, E.; Hayakawa, Y.; Ota, T.; Akiba, H.; Ogasawara, K.; Yagita, H.; Kinoshita, K.; Okumura, K.; Smyth, M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 2005, 105, 2082–2089. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Parihar, R.; Dierksheide, J.; Hu, Y.; Carson, W.E. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J. Clin. Investig. 2002, 110, 983–992. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; Van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef]
- Zhu, V.F.; Yang, J.; Lebrun, D.G.; Li, M. Understanding the role of cytokines in Glioblastoma Multiforme pathogenesis. Cancer Lett. 2012, 316, 139–150. [Google Scholar] [CrossRef]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.G.; Kiessling, R.; Malmberg, K.J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-beta signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar] [PubMed]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; Von Sttrandmann, E.P.; Textor, S.; et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, S.; Oliviero, B.; Lombardi, A.; Varchetta, S.; Mele, D.; Sangiovanni, A.; Rossi, G.; Donadon, M.; Torzilli, G.; Soldani, C.; et al. Deficient Natural Killer Cell NKp30-Mediated Function and Altered NCR3 Splice Variants in Hepatocellular Carcinoma. Hepatology 2019, 69, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology 2015, 4, e1001224. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, M.; Rusakiewicz, S.; Zitvogel, L.; Kroemer, G. Natural killer cell mediated immunosurveillance of pediatric neuroblastoma. Oncoimmunology 2015, 4, e1042202. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Messina, L.; Ashiru, O.; Boutet, P.; Agüera-González, S.; Skepper, J.N.; Reyburn, H.T.; Valés-Gómez, M. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J. Biol. Chem. 2010, 285, 8543–8551. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Vulpis, E.; Loconte, L.; Santoni, A. NKG2D Ligand Shedding in Response to Stress: Role of ADAM10. Front. Immunol. 2020, 11, 447. [Google Scholar] [CrossRef]
- Ponath, V.; Hoffmann, N.; Bergmann, L.; Mäder, C.; Alhamwe, B.A.; Preuber, C.; Von Strandmann, E.P. Secreted Ligands of the NK Cell Receptor NKp30: B7-H6 Is in Contrast to BAG6 Only Marginally Released via Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 2189. [Google Scholar] [CrossRef]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Gao, F.X.; Wang, C.; Qin, M.; Han, F.; Xu, T.; Hu, Z.; Long, Y.; He, X.M.; Deng, X.; et al. IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 321. [Google Scholar] [CrossRef] [PubMed]
- Shou, Q.; Fu, H.; Huang, X.; Yang, Y. PARP-1 controls NK cell recruitment to the site of viral infection. J. Clin. Investig. 2019, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Pietra, G.; Romagnani, C.; Manzini, C.; Moretta, L.; Mingari, M.C. The emerging role of HLA-E-restricted CD8+ T lymphocytes in the adaptive immune response to pathogens and tumors. J. Biomed. Biotechnol. 2010, 2010, 907092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterbach, N.; Wieten, L.; Popeijus, H.E.; Voorter, C.E.; Tilanus, M.G. HLA-E regulates NKG2C+ natural killer cell function through presentation of a restricted peptide repertoire. Hum. Immunol. 2015, 76, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Urbani, E.; André, P.; Mancusi, A.; Tosti, A.; Topini, F.; Bléry, M.; Animobono, L.; Romagné, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2016, 101, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, P.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Chen, S.; Huang, Z.; et al. The Synergistic Effect of PARP Inhibitors and Immune Checkpoint Inhibitors. Clin. Med. Insights Oncol. 2021, 15, 1179554921996288. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moran, J.; Mylod, E.; Kane, L.E.; Marion, C.; Keenan, E.; Mekhaeil, M.; Lysaght, J.; Dev, K.K.; O’Sullivan, J.; Conroy, M.J. Investigating the Effects of Olaparib on the Susceptibility of Glioblastoma Multiforme Tumour Cells to Natural Killer Cell-Mediated Responses. Pharmaceutics 2023, 15, 360. https://doi.org/10.3390/pharmaceutics15020360
Moran J, Mylod E, Kane LE, Marion C, Keenan E, Mekhaeil M, Lysaght J, Dev KK, O’Sullivan J, Conroy MJ. Investigating the Effects of Olaparib on the Susceptibility of Glioblastoma Multiforme Tumour Cells to Natural Killer Cell-Mediated Responses. Pharmaceutics. 2023; 15(2):360. https://doi.org/10.3390/pharmaceutics15020360
Chicago/Turabian StyleMoran, Jennifer, Eimear Mylod, Laura E. Kane, Caroline Marion, Emily Keenan, Marianna Mekhaeil, Joanne Lysaght, Kumlesh K. Dev, Jacintha O’Sullivan, and Melissa J. Conroy. 2023. "Investigating the Effects of Olaparib on the Susceptibility of Glioblastoma Multiforme Tumour Cells to Natural Killer Cell-Mediated Responses" Pharmaceutics 15, no. 2: 360. https://doi.org/10.3390/pharmaceutics15020360