
Current Challenges and Opportunities of Photodynamic Therapy against Cancer
 ,
,
Abstract
:1. Introduction
2. History of Photodynamic Therapy of Cancer
3. The Fundamentals of Photodynamic Therapy: Sensitizers, Light Penetration in Tissues, Light Sources, Photodynamic Effect
3.1. Photosensitizers
3.2. Drug-to-Light Interval
3.3. Attenuation and Propagation of Light in Tissues
3.4. Therapeutic Window of PDT
3.5. Light Sources in PDT
3.6. The Importance of Fluence and Fluence Rate
3.7. Photodynamic Effect
4. The Consequences of Photodynamic Therapy: Cell Death Pathways, DAMPs, ICD, Tissue Disruption, Vascular Disruption, and Immune Activation
4.1. Accidental Necrosis, Regulated Necrosis, Apoptosis, and Autophagy: Direct Damage to Tumor Cells
4.2. Damaging the Tumor Vasculature
4.3. Damage-Associated Molecular Patterns and Immunogenic Cell Death
4.4. Antitumor Immune Responses
5. Current Challenges and Opportunities of Photodynamic Therapy against Cancer
6. Biodistribution of Photosensitizers
6.1. Nanoparticles
6.2. Antibodies for Tumor Targeting
6.3. Peptides
6.4. Extracellular Vesicles
6.5. Sensitizer-Loaded Immune Cells
7. Light Propagation through Tissues
7.1. NIR-Absorbing Sensitizers
7.2. Upconversion Nanoparticles
8. Hypoxia in the Tumor Area
8.1. Sensitizers That Function As Their Own Source of Oxygen
8.2. Hypoxia-Responsive Prodrugs
8.3. Diffusion of Oxygen in the Tumor
9. Vascular Disruption
9.1. Tumor Vasculature Disrupting Agents
9.2. Specific Targeting of the Vasculature
9.3. Using VTP to Enhance Combination Treatments
10. Partial Destruction of the Tumor
10.1. Combinations with Chemotherapeutic Agents
10.2. Combinations with Other Antineoplastic Agents
11. Insufficient PDT-Induced Antitumor Immune Responses Followed by Tumor Progression
11.1. PDT-Generated or Enhanced Tumor Vaccines
11.2. Combination with Immunostimulatory Agents
11.3. Combination with Immune Checkpoint Blockade Antibodies
12. Recent Advances in Clinical Photodynamic Therapy
13. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Raab, O. Uber die Wirkung, fluorescirender Stoffe auf infusorien. Z. Biol. 1900, 39, 524–546. [Google Scholar]
- Tappeiner, H.; Jodlbauer, A. Über die Wirkungen der photodynamischen (fluoreszierenden) Stoffe auf Protozoen und Enzyme. Deutsch. Arch. Klin. Med. 1904, 80, 427. [Google Scholar]
- Figge, F.; Weiland, G.; Manganiello, L. Cancer detection and therapy; affinity of neoplastic, embryonic, and traumatized tissues for porphyrins and metalloporphyrins. Proc. Soc. Exp. Biol. Med. 1948, 68, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Lipson, R.; Baldes, E.; Olsen, A. Hematoporphyrin derivative: A new aid for endoscopic detection of malignant disease. J. Thorac. Cardiovasc. Surg. 1961, 42, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Lipson, R.; Baldes, E.; Olsen, A. The use of a derivative of hematoporhyrin in tumor detection. J. Natl. Cancer Inst. 1961, 26, 1–11. [Google Scholar] [PubMed]
- Gregorie, H.; Horger, E.; Ward, J.; Green, J.; Richards, T.; Robertson, H.; Stevenson, T. Hematoporphyrin-derivative fluorescence in malignant neoplasms. Ann. Surg. 1968, 167, 820–828. [Google Scholar] [CrossRef]
- Diamond, I.; Mcdonagh, A.; Wilson, C.; Granelli, S.; Nielsen, S.; Jaenicke, R. Photodynamic therapy of malignant tumours. Lancet 1972, 300, 1175–1177. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Grindey, G.B.; Fiel, R.; Weishaupt, K.R.; Boyle, D.G. Photoradiation Therapy. II. Cure of Animal Tumors With Hematoporphyrin and Light. JNCI J. Natl. Cancer Inst. 1975, 55, 115–121. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Kaufman, J.E.; Goldfarb, A.; Weishaupt, K.R.; Boyle, D.; Mittleman, A. Photoradiation Therapy for the Treatment of Malignant Tumors. Cancer Res. 1978, 38, 2628–2635. [Google Scholar]
- Dougherty, T.J.; Lawrence, G.; Kaufman, J.H.; Boyle, D.; Weishaupt, K.R.; Goldfarb, A. Photoradiation in the Treatment of Recurrent Breast Carcinoma. JNCI J. Natl. Cancer Inst. 1979, 62, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Gomer, C.J.; Dougherty, T.J. Determination of [3H]- and [14C]Hematoporphyrin Derivative Distribution in Malignant and Normal Tissue. Cancer Res. 1979, 39, 146–151. [Google Scholar] [PubMed]
- Weishaupt, K.R.; Gomer, C.J.; Dougherty, T.J. Identification of Singlet Oxygen as the Cytotoxic Agent in Photo-inactivation of a Murine Tumor. Cancer Res. 1976, 36, 2326–2329. [Google Scholar] [PubMed]
- TJ, W.; TS, M.; VH, F.; TG, H.; MW, R.; TS, C.; VQ, N.; ER, R. Effect of photodynamic therapy on blood flow in normal and tumor vessels. Surgery 1988, 104, 512–517. [Google Scholar]
- Schmidt-Erfurth, U.; Miller, J.; Sickenberg, M.; Bunse, A.; Laqua, H.; Gragoudas, E.; Zografos, L.; Birngruber, R.; van den Bergh, H.; Strong, A.; et al. Photodynamic therapy of subfoveal choroidal neovascularization: Clinical and angiographic examples. Graefe’s Arch. Clin. Exp. Ophthalmol. 1998, 236, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Siegel, M.M.; Tsao, R.; McReynolds, J.H.; Dougherty, T.J. Fast atom bombardment mass spectral analyses of Photofrin II and its synthetic analogs. Biomed. Environ. Mass Spectrom. 1990, 19, 405–414. [Google Scholar] [CrossRef]
- Agarwal, M.L.; Clay, M.E.; Harvey, E.J.; Evans, H.H.; Antunez, A.R.; Oleinick, N.L. Photodynamic Therapy Induces Rapid Cell Death by Apoptosis in L5178Y Mouse Lymphoma Cells. Cancer Res. 1991, 51, 5993–5996. [Google Scholar]
- Kim, H.-R.C.; Luo, Y.; Li, G.; Kessel, D. Enhanced Apoptotic Response to Photodynamic Therapy after bcl-2 Transfection. Cancer Res. 1999, 59, 3429. [Google Scholar]
- Xue, L.Y.; Chiu, S.M.; Oleinick, N.L. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 2001, 20, 3420–3427. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, C.; Senge, M.O.; Arnaut, L.G.; Gomes-da-Silva, L.C. Cell death in photodynamic therapy: From oxidative stress to anti-tumor immunity. Biochim. Biophys. Acta-Rev. Cancer 2019, 1872, 188308. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 306164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding Cell Death Signals in Inflammation and Immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta-Rev. Cancer 2010, 1805, 53–71. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef]
- Nath, S.; Obaid, G.; Hasan, T. The Course of Immune Stimulation by Photodynamic Therapy: Bridging Fundamentals of Photochemically Induced Immunogenic Cell Death to the Enrichment of T-Cell Repertoire. Photochem. Photobiol. 2019, 95, 1288–1305. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Sun, J.; Cecic, I. Photodynamic Therapy-Induced Cell Surface Expression and Release of Heat Shock Proteins: Relevance for Tumor Response. Cancer Res. 2005, 65, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Panzarini, E.; Inguscio, V.; Fimia, G.M.; Dini, L. Rose Bengal Acetate PhotoDynamic Therapy (RBAc-PDT) Induces Exposure and Release of Damage-Associated Molecular Patterns (DAMPs) in Human HeLa Cells. PLoS ONE 2014, 9, e105778. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef] [Green Version]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moan, J. Properties for optimal PDT sensitizers. J. Photochem. Photobiol. B Biol. 1990, 5, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.R.; Downie, G.H.; Cuenca, R.; Hu, X.-H.; Childs, C.J.; Sibata, C.H. Photosensitizers in clinical PDT. Photodiagnosis Photodyn. Ther. 2004, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.N. Introduction to Biophotonics; Wiley-Interscience: New York, NY, USA, 2003. [Google Scholar]
- Niemz, M.H. Laser-Tissue Interactions, 3rd ed.; Springer: New York, NY, USA, 2007. [Google Scholar]
- Anderson, R.R.; Parrish, J.a.; Rox Anderson, B.R.; Parrish, M.D. The Optics of Human Skin. J. Investig. Dermatol. 1981, 77, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juzeniene, A.; Nielsen, K.P.; Moan, J. Biophysical aspects of photodynamic therapy. J. Environ. Pathol. Toxicol. Oncol. 2006, 25, 7–28. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.; Foster, T.; Girotti, A.; Gollnick, S.; Hahn, S.; Hamblin, M.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Van Straten, D.; Mashayekhi, V.; De Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Brancaleon, L.; Moseley, H. Laser and non-laser light sources for photodynamic therapy. Lasers Med. Sci. 2002, 17, 173–186. [Google Scholar] [CrossRef]
- Mang, T.S. Lasers and light sources for PDT: Past, present and future. Photodiagnosis Photodyn. Ther. 2004, 1, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Haedersdal, M.; Togsverd-Bo, K.; Wulf, H.C. Evidence-based review of lasers, light sources and photodynamic therapy in the treatment of acne vulgaris. Jeadv 2008, 22, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Juzeniene, A.; Juzenas, P.; Ma, L.-W.; Iani, V.; Moan, J. Effectiveness of different light sources for 5-aminolevulinic acid photodynamic therapy. Lasers Med. Sci. 2004, 19, 139–149. [Google Scholar] [CrossRef]
- Etcheverry, M.E.; Pasquale, M.A.; Garavaglia, M. Photodynamic therapy of HeLa cell cultures by using LED or laser sources. J. Photochem. Photobiol. B Biol. 2016, 160, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.L.S.; Hovenic, W.; Ball, K.; Zachary, C.B. Daylight photodynamic therapy: The Southern California experience. Lasers Surg. Med. 2015, 47, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-N.; Hsu, R.; Chen, S.; Wong, T.-W. Daylight Photodynamic Therapy: An Update. Molecules 2020, 25, 5195. [Google Scholar] [CrossRef]
- Sorbellini, E.; Rucco, M.; Rinaldi, F. Photodynamic and photobiological effects of light-emitting diode (LED) therapy in dermatological disease: An update. Lasers Med. Sci. 2018, 33, 1431. [Google Scholar] [CrossRef] [Green Version]
- Henderson, B.W.; Busch, T.M.; Snyder, J.W. Fluence rate as a modulator of PDT mechanisms. Lasers Surg. Med. 2006, 38, 489–493. [Google Scholar] [CrossRef]
- Grossman, C.E.; Carter, S.L.; Czupryna, J.; Wang, L.; Putt, M.E.; Busch, T.M. Fluence Rate Differences in Photodynamic Therapy Efficacy and Activation of Epidermal Growth Factor Receptor after Treatment of the Tumor-Involved Murine Thoracic Cavity. Int. J. Mol. Sci. 2016, 17, 101. [Google Scholar] [CrossRef] [Green Version]
- Marian, C.M. Spin-orbit coupling and intersystem crossing in molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- Foote, C.S. Mechanisms of Photosensitized Oxydation. Science 1968, 162, 963–970. [Google Scholar] [CrossRef]
- Henderson, B.W.; Dougherty, T.J. How does photodynamic therapy work? Photochem. Photobiol. 1992, 55, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Photodynamic antimicrobial chemotherapy (PACT). J. Antimicrob. Chemother. 1998, 42, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Jabłoństski, A. Efficiency of Anti-Stokes Fluorescence in Dyes. Nature 1933, 131, 839–840. [Google Scholar] [CrossRef]
- Moan, J. On the diffusion length of singlet oxygen in cells and tissues. J. Photochem. Photobiol. B Biol. 1990, 6, 343–347. [Google Scholar] [CrossRef]
- Dysart, J.S.; Patterson, M.S. Characterization of Photofrin photobleaching for singlet oxygen dose estimation during photodynamic therapy of MLL cells in vitro. Phys. Med. Biol. 2005, 50, 2597–2616. [Google Scholar] [CrossRef]
- Moan, J.; Berg, K.; Kvam, E.; Western, A.; Malik, Z.; Rück, A.; Schneckenburger, H. Intracellular localization of photosensitizers. Ciba Found. Symp. 1989, 146, 95–107. [Google Scholar]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta-Rev. Cancer 2007, 1776, 86–107. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbelik, M. PDT-associated host response and its role in the therapy outcome. Lasers Surg. Med. 2006, 38, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Luo, Y.; Deng, Y.; Chang, C.K. The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem. Photobiol. 1997, 65, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Agostinis, P.; Buytaert, E.; Breyssens, H.; Hendrickx, N. Regulatory pathways in photodynamic therapy induced apoptosis. Photochem. Photobiol. Sci. 2004, 3, 721–729. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Carvalho, A.P.; Duarte, C.B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.J.; Wu, C.C.; Chang, C.J.; Yu, J.S. Subcellular localization of photofrin® determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: When plasma membranes are the main targets. J. Cell. Physiol. 2003, 194, 363–375. [Google Scholar] [CrossRef]
- Fabris, C.; Valduga, G.; Miotto, G.; Borsetto, L.; Jori, G.; Garbisa, S.; Reddi, E. Photosensitization with Zinc (II) Phthalocyanine as a Switch in the Decision between Apoptosis and Necrosis Photosensitization with Zinc (II) Phthalocyanine as a Switch in the Decision between Apoptosis and Necrosis. Cancer Res. 2001, 61, 7495–7500. [Google Scholar]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Vanden Berghe, T.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Akimoto, J.; Moritake, K.; Hironaka, C.; Fujiwara, Y. Photodynamic therapy using talaporfin sodium induces concentration-dependent programmed necroptosis in human glioblastoma T98G cells. Lasers Med. Sci. 2015, 30, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Fettweis, G.; Di Valentin, E.; L’homme, L.; Lassence, C.; Dequiedt, F.; Fillet, M.; Coupienne, I.; Piette, J. RIP3 antagonizes a TSC2-mediated pro-survival pathway in glioblastoma cell death. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Coupienne, I.; Fettweis, G.; Rubio, N.; Agostinis, P.; Piette, J. 5-ALA-PDT induces RIP3-dependent necrosis in glioblastoma. Photochem. Photobiol. Sci. 2011, 10, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329. [Google Scholar] [CrossRef] [PubMed]
- Moor, A.C. Signaling pathways in cell death and survival after photodynamic therapy. J. Photochem. Photobiol. B Biol. 2000, 57, 1–13. [Google Scholar] [CrossRef]
- Mai, N.N.H.; Yamaguchi, Y.; Choijookhuu, N.; Matsumoto, J.; Nanashima, A.; Takagi, H.; Sato, K.; Le, Q.T.; Hishikawa, Y. Photodynamic Therapy Using a Novel Phosphorus Tetraphenylporphyrin Induces an Anticancer. Effect via Bax/Bcl-xL-related Mitochondrial Apoptosis in Biliary Cancer Cells. Acta Histochem. Cytochem. 2020, 53, 61–72. [Google Scholar] [CrossRef]
- Kessel, D.; Castelli, M. Evidence that bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem. Photobiol. 2001, 74, 318–322. [Google Scholar] [CrossRef]
- Usuda, J.; Chiu, S.M.; Murphy, E.S.; Lam, M.; Nieminen, A.L.; Oleinick, N.L. Domain-dependent photodamage to Bcl-2: A membrane anchorage region is needed to form the target of phthalocyanine photosensitization. J. Biol. Chem. 2003, 278, 2021–2029. [Google Scholar] [CrossRef]
- Kessel, D.; Castelli, M.; Reiners, J.J. Ruthenium red-mediated suppression of Bcl-2 loss and Ca(2+) release initiated by photodamage to the endoplasmic reticulum: Scavenging of reactive oxygen species. Cell Death Differ. 2005, 12, 502–511. [Google Scholar] [CrossRef]
- Movahedi, M.M.; Alamzadeh, Z.; Hosseini-Nami, S.; Shakeri-Zadeh, A.; Taheripak, G.; Ahmadi, A.; Zare-Sadeghi, A.; Ghaznavi, H.; Mehdizadeh, A. Investigating the mechanisms behind extensive death in human cancer cells following nanoparticle assisted photo-thermo-radiotherapy. Photodiagnosis Photodyn. Ther. 2020, 29, 101600. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Y.; Peng, X.; Huang, J.; Yang, M.; Wang, X. A Novel Photosensitizer Znln2S4 Mediated Photodynamic Therapy Induced-HepG2 Cell Apoptosis. Radiat. Res. 2019, 192, 422–430. [Google Scholar] [CrossRef]
- Cho, H.; Zheng, H.; Sun, Q.; Shi, S.; He, Y.; Ahn, K.; Kim, B.; Kim, H.-E.; Kim, O. Development of Novel Photosensitizer Using the Buddleja officinalis Extract for Head and Neck Cancer. Evid. Based. Complement. Alternat. Med. 2018, 2018, 6917590. [Google Scholar] [CrossRef] [Green Version]
- Buytaert, E.; Callewaert, G.; Hendrickx, N.; Scorrano, L.; Hartmann, D.; Missiaen, L.; Vandenheede, J.R.; Heirman, I.; Grooten, J.; Agostinis, P. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J. 2006, 20, 756–758. [Google Scholar] [CrossRef] [Green Version]
- Chiu, S.-M.; Xue, L.-Y.; Usuda, J.; Azizuddin, K.; Oleinick, N.L. Bax is essential for mitochondrion-mediated apoptosis but not for cell death caused by photodynamic therapy. Br. J. Cancer 2003, 89, 1590–1597. [Google Scholar] [CrossRef] [Green Version]
- Granville, D.J.; Shaw, J.R.; Leong, S.; Carthy, C.M.; Margaron, P.; Hunt, D.W.; McManus, B.M. Release of cytochrome c, Bax migration, Bid cleavage, and activation of caspases 2, 3, 6, 7, 8, and 9 during endothelial cell apoptosis. Am. J. Pathol. 1999, 155, 1021–1025. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.; Ahmad, N.; Gupta, S.; Mukhtar, H. Involvement of Bcl-2 and Bax in Photodynamic Therapy-mediated Apoptosis. Antisense Bcl-2 oligonucleotide sensitizes RIF 1 cells to photodynamic therapy apoptosis. J. Biol. Chem. 2001, 276, 15481–15488. [Google Scholar] [CrossRef] [Green Version]
- Belzacq, A.S.; Jacotot, E.; Vieira, H.L.A.; Mistro, D.; Granville, D.J.; Xie, Z.; Reed, J.C.; Kroemer, G.; Brenner, C. Apoptosis induction by the photosensitizer verteporfin: Identification of mitochondrial adenine nucleotide translocator as a critical target. Cancer Res. 2001, 61, 1260–1264. [Google Scholar]
- MacDonald, I.J.; Morgan, J.; Bellnier, D.A.; Paszkiewicz, G.M.; Whitaker, J.E.; Litchfield, D.J.; Dougherty, T.J. Subcellular localization patterns and their relationship to photodynamic activity of pyropheophorbide—A derivatives. Photochem. Photobiol. 1999, 70, 789–797. [Google Scholar] [CrossRef]
- Furre, I.E.; Shahzidi, S.; Luksiene, Z.; Møller, M.T.N.; Borgen, E.; Morgan, J.; Tkacz-Stachowska, K.; Nesland, J.M.; Peng, Q. Targeting PBR by hexaminolevulinate-mediated photodynamic therapy induces apoptosis through translocation of apoptosis-inducing factor in human leukemia cells. Cancer Res. 2005, 65, 11051–11060. [Google Scholar] [CrossRef]
- Lam, M.; Oleinick, N.L.; Nieminen, A.L. Photodynamic therapy-induced apoptosis in epidermoid carcinoma cells. Reactive oxygen species and mitochondrial inner membrane permeabilization. J. Biol. Chem. 2001, 276, 47379–47386. [Google Scholar] [CrossRef] [Green Version]
- Chaloupka, R.; Petit, P.X.; Israël, N.; Sureau, F. Over-expression of Bcl-2 does not protect cells from hypericin photo-induced mitochondrial membrane depolarization, but delays subsequent events in the apoptotic pathway. FEBS Lett. 1999, 462, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Minamikawa, T.; Sriratana, A.; Williams, D.A.; Bowser, D.N.; Hill, J.S.; Nagley, P. Chloromethyl-X-rosamine (MitoTracker Red) photosensitises mitochondria and induces apoptosis in intact human cells. J. Cell Sci. 1999, 112 Pt 1, 2419–2430. [Google Scholar] [CrossRef]
- Chiu, S.M.; Oleinick, N.L. Dissociation of mitochondrial depolarization from cytochrome c release during apoptosis induced by photodynamic therapy. Br. J. Cancer 2001, 84, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Ikeda, H.; Inokuchi, T.; Sano, K.; Koji, T. Enhanced cell death in NR-S1 tumor by photodynamic therapy: Possible involvement of Fas and Fas ligand system. Lasers Surg. Med. 2000, 26, 449–460. [Google Scholar] [CrossRef]
- Chen, B.; Roskams, T.; Xu, Y.; Agostinis, P.; De Witte, P.A.M. Photodynamic therapy with hypericin induces vascular damage and apoptosis in the RIF-1 mouse tumor model. Int. J. Cancer 2002, 98, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Granville, D.J.; Cassidy, B.A.; Ruehlmann, D.O.; Choy, J.C.; Brenner, C.; Kroemer, G.; van Breemen, C.; Margaron, P.; Hunt, D.W.; McManus, B.M. Mitochondrial release of apoptosis-inducing factor and cytochrome c during smooth muscle cell apoptosis. Am. J. Pathol. 2001, 159, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Furre, I.E.; Møller, M.T.N.; Shahzidi, S.; Nesland, J.M.; Peng, Q. Involvement of both caspase-dependent and -independent pathways in apoptotic induction by hexaminolevulinate-mediated photodynamic therapy in human lymphoma cells. Apoptosis 2006, 11, 2031–2042. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy 2021, 17, 1797280. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.Y.; Chiu, S.M.; Azizuddin, K.; Joseph, S.; Oleinick, N.L. The death of human cancer cells following photodynamic therapy: Apoptosis competence is necessary for Bcl-2 protection but not for induction of autophagy. Photochem. Photobiol. 2007, 83, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Vicente, M.G.H.; Reiners, J.J. Initiation of apoptosis and autophagy by photodynamic therapy. Lasers Surg. Med. 2006, 38, 482–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasnauskiene, A.; Kadziauskas, J.; Vezelyte, N.; Jonusiene, V.; Kirveliene, V. Apoptosis, autophagy and cell cycle arrest following photodamage to mitochondrial interior. Apoptosis 2009, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Jr, J.J.R. Apoptosis and Autophagy After Mitochondrial or Endoplasmic Reticulum Photodamage. Photochem. Photobiol. 2007, 83, 1024–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejak, M.; Price, M.; Kessel, D.H. Apoptotic and autophagic responses to photodynamic therapy in 1c1c7 murine hepatoma cells. Autophagy 2011, 7, 979. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.-Y.; Chiu, S.-M.; Oleinick, N.L. Atg7 deficiency increases resistance of MCF-7 human breast cancer cells to photodynamic therapy. Autophagy 2010, 6, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, D.; Arroyo, A.S. Apoptotic and autophagic responses to Bcl-2 inhibition and photodamage. Photochem. Photobiol. Sci. 2007, 6, 1290–1295. [Google Scholar] [CrossRef] [Green Version]
- Castellani, A.; Pace, G.P.; Concioli, M. Photodynamic effect of haematoporphyrin on blood microcirculation. J. Pathol. Bacteriol. 1963, 86, 99–102. [Google Scholar] [CrossRef]
- Star, W.M.; Marijnissen, H.P.A.; van den Berg-Blok, A.E.; Versteeg, J.A.; Franken, K.A.; Reinhold, H.S. Destruction of rat mammary tumor and normal tissue microcirculation by hematoporphyrin derivative photoradiation observed in vivo in sandwich observation chambers. Cancer Res. 1986, 46, 2532–2540. [Google Scholar]
- Tseng, M.T.; Reed, M.W.; Ackermann, D.M.; Schuschke, D.A.; Wieman, T.J.; Miller, F.N. Photodynamic therapy induced ultrastructural alterations in microvasculature of the rat cremaster muscle. Photochem. Photobiol. 1988, 48, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Huis in ’t Veld, R.V.; Ritsma, L.; Kleinovink, J.W.; Que, I.; Ossendorp, F.; Cruz, L.J. Photodynamic cancer therapy enhances accumulation of nanoparticles in tumor-associated myeloid cells. J. Control. Release 2020, 320, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, V.; Hoog, C.O.; Oliveira, S. Vascular targeted photodynamic therapy: A review of the efforts towards molecular targeting of tumor vasculature. J. Porphyr. Phthalocyanines 2019, 23, 1229–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomer, C.J.; Rucker, N.; Murphree, A.L. Differential cell photosensitivity following porphyrin photodynamic therapy. Cancer Res. 1988, 48, 4539–4542. [Google Scholar]
- West, C.M.; West, D.C.; Kumar, S.; Moore, J. V A comparison of the sensitivity to photodynamic treatment of endothelial and tumour cells in different proliferative states. Int. J. Radiat. Biol. 1990, 58, 145–156. [Google Scholar] [CrossRef]
- Lepor, H. Vascular Targeted Photodynamic Therapy for Localized Prostate Cancer. Rev. Urol. 2008, 10, 254. [Google Scholar]
- Preise, D.; Scherz, A.; Salomon, Y. Antitumor immunity promoted by vascular occluding therapy: Lessons from vascular-targeted photodynamic therapy (VTP). Photochem. Photobiol. Sci. 2011, 10, 681–688. [Google Scholar] [CrossRef]
- Coleman, J.; Sjoberg, D.D.; Demac, Q.; ODea, C.; McGill, M.; Tracey, A.; Nogueira, L.; Vickers, A.; Estes, C.; Fine, S.; et al. Phase 2b trial results of padeliporfin (WST11 or Tookad) vascular-targeted photodynamic therapy for partial gland ablation in men with intermediate-risk prostate cancer. J. Clin. Oncol. 2021, 39, e17006. [Google Scholar] [CrossRef]
- Azzouzi, A.-R.; Vincendeau, S.; Barret, E.; Cicco, A.; Kleinclauss, F.; van der Poel, H.G.; Stief, C.G.; Rassweiler, J.; Salomon, G.; Solsona, E.; et al. Padeliporfin vascular-targeted photodynamic therapy versus active surveillance in men with low-risk prostate cancer (CLIN1001 PCM301): An open-label, phase 3, randomised controlled trial. Lancet. Oncol. 2017, 18, 181–191. [Google Scholar] [CrossRef]
- Fingar, V.H.; Wieman, T.J.; Wiehle, S.A.; Cerrito, P.B. The role of microvascular damage in photodynamic therapy: The effect of treatment on vessel constriction, permeability, and leukocyte adhesion. Cancer Res. 1992, 52, 4914–4921. [Google Scholar]
- Chen, B.; Pogue, B.W.; Luna, J.M.; Hardman, R.L.; Hoopes, P.J.; Hasan, T. Tumor Vascular Permeabilization by Vascular-Targeting Photosensitization: Effects, Mechanism, and Therapeutic Implications. Clin. Cancer Res. 2006, 12, 917–923. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Foster, T.H. In vivo confocal fluorescence imaging of the intratumor distribution of the photosensitizer mono-L-aspartylchlorin-e6. Neoplasia 2008, 10, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Z.; Tang, W.; Chuang, Y.J.; Todd, T.; Zhang, W.; Lin, X.; Niu, G.; Liu, G.; Wang, L.; Pan, Z.; et al. Tumor vasculature targeted photodynamic therapy for enhanced delivery of nanoparticles. ACS Nano 2014, 8, 6004–6013. [Google Scholar] [CrossRef]
- Snyder, J.W.; Greco, W.R.; Bellnier, D.A.; Vaughan, L.; Henderson, B.W. Photodynamic Therapy: A Means to Enhanced Drug Delivery to Tumors. Cancer Res. 2003, 63, 8126–8131. [Google Scholar]
- Gao, W.; Wang, Z.; Lv, L.; Yin, D.; Chen, D.; Han, Z.; Ma, Y.; Zhang, M.; Yang, M.; Gu, Y. Photodynamic Therapy Induced Enhancement of Tumor Vasculature Permeability Using an Upconversion Nanoconstruct for Improved Intratumoral Nanoparticle Delivery in Deep Tissues. Theranostics 2016, 6, 1131–1144. [Google Scholar] [CrossRef] [Green Version]
- Firczuk, M.; Nowis, D.; Gołąb, J. PDT-induced inflammatory and host responses. Photochem. Photobiol. Sci. 2011, 10, 653–663. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Huis In’t Veld, R.V.; Da Silva, C.G.; Jager, M.J.; Cruz, L.J.; Ossendorp, F. Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models. Pharmaceutics 2021, 13, 1470. [Google Scholar] [CrossRef]
- Li, W.; Yang, J.; Luo, L.; Jiang, M.; Qin, B.; Yin, H.; Zhu, C.; Yuan, X.; Zhang, J.; Luo, Z.; et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Vabulas, R.M.; Wagner, H.; Schild, H. Heat shock proteins as ligands of Toll-like receptors. Curr. Top. Microbiol. Immunol. 2002, 270, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Flechtner, J.B.; Cohane, K.P.; Mehta, S.; Slusarewicz, P.; Leonard, A.K.; Barber, B.H.; Levey, D.L.; Andjelic, S. High-Affinity Interactions between Peptides and Heat Shock Protein 70 Augment CD8+ T Lymphocyte Immune Responses. J. Immunol. 2006, 177, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimu, J.; Spary, L.K.; Al-Taei, S.; Clayton, A.; Mason, M.D.; Staffurth, J.; Tabi, Z. Cross-Presentation of the Oncofetal Tumor Antigen 5T4 from Irradiated Prostate Cancer Cells--A Key Role for Heat-Shock Protein 70 and Receptor CD91. Cancer Immunol. Res. 2015, 3, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Mitra, S.; Goren, E.M.; Frelinger, J.G.; Foster, T.H. Activation of heat shock protein 70 promoter with meso-tetrahydroxyphenyl chlorin photodynamic therapy reported by green fluorescent protein in vitro and in vivo. Photochem. Photobiol. 2003, 78, 615–622. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. DAMPs and PDT-mediated photo-oxidative stress: Exploring the unknown. Photochem. Photobiol. Sci. 2011, 10, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [Green Version]
- Riteau, N.; Baron, L.; Villeret, B.; Guillou, N.; Savigny, F.; Ryffel, B.; Rassendren, F.; Le Bert, M.; Gombault, A.; Couillin, I. ATP release and purinergic signaling: A common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012, 3, e403. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Cecic, I.; Parkins, C.S.; Korbelik, M. Neutrophils as inflammatory and immune effectors in photodynamic therapy-treated mouse SCCVII tumours. Photochem. Photobiol. Sci. 2002, 1, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.-C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, A.C.S.; Gomes-da-Silva, L.C.; Rodrigues-Santos, P.; Cabrita, A.; Santos-Rosa, M.; Arnaut, L.G. Immune Responses after Vascular Photodynamic Therapy with Redaporfin. J. Clin. Med. 2020, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- De Vree, W.J.A.; Essers, M.C.; Koster, J.F.; Koster, F. Role of Interleukin 1 and Granulocyte Colony-Stimulating Factor in Photofrin-based Photodynamic Therapy of Rat Rhabdomyosarcoma Tumors Advances in Brief Role of Interleukin 1 and Granulocyte Colony-Stimulating Factor in Photofrin based Photodynamic. Cancer Res. 1997, 53, 2555–2558. [Google Scholar]
- Krosl, G.; Korbelik, M.; Dougherty, G.J. Induction of immune cell infiltration into murine SCCVII tumour by photofrin-based photodynamic therapy. Br. J. Cancer 1995, 71, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.-H.; Baumann, H.; Tracy, E.; Wang, Y.; Hutson, A.; Rose-John, S.; Henderson, B.W. Interleukin-6 trans signalling enhances photodynamic therapy by modulating cell cycling. Br. J. Cancer 2007, 97, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- CM, B.; JB, M.; SS, E.; SO, G. IL-17 promotes neutrophil entry into tumor-draining lymph nodes following induction of sterile inflammation. J. Immunol. 2013, 191, 4348–4357. [Google Scholar] [CrossRef] [Green Version]
- De Vree, W.; Fontijne-Dorsman, A.N.R.D.; Koster, J.F.; Sluiter, W. Photodynamic treatment of human endothelial cells promotes the adherence of neutrophils in vitro. Br. J. Cancer 1996, 73, 1335. [Google Scholar] [CrossRef] [Green Version]
- Kousis, P.C.; Henderson, B.W.; Maier, P.G.; Gollnick, S.O. Photodynamic Therapy Enhancement of Antitumor Immunity Is Regulated by Neutrophils. Cancer Res. 2007, 67, 10501–10510. [Google Scholar] [CrossRef] [Green Version]
- Korbelik, M.; Sun, J. Photodynamic therapy-generated vaccine for cancer therapy. Cancer Immunol. Immunother. 2006, 55, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Kabingu, E.; Vaughan, L.; Owczarczak, B.; Ramsey, K.; Gollnick, S. CD8+ T cell-mediated control of distant tumours following local photodynamic therapy is independent of CD4+ T cells and dependent on natural killer cells. Br. J. Cancer 2007, 96, 1839–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Yin, G.; Le, V.; Zhang, A.; Chen, S.; Liang, X.; Liu, J. Photodynamic-therapy Activates Immune Response by disrupting Immunity Homeostasis of Tumor Cells, which Generates Vaccine for Cancer Therapy. Int. J. Biol. Sci. 2016, 12, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollnick, S.O.; Brackett, C.M. Enhancement of anti-tumor immunity by photodynamic therapy. Immunol. Res. 2010, 46, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ji, J.; Zhang, H.; Fan, Z.; Zhang, L.; Shi, L.; Zhou, F.; Chen, W.R.; Wang, H.; Wang, X. Stimulation of dendritic cells by DAMPs in ALA-PDT treated SCC tumor cells. Oncotarget 2015, 6, 44688–44702. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Fan, Z.; Zhou, F.; Wang, X.; Shi, L.; Zhang, H.; Wang, P.; Yang, D.; Zhang, L.; Chen, W.R.; et al. Improvement of DC vaccine with ALA-PDT induced immunogenic apoptotic cells for skin squamous cell carcinoma. Oncotarget 2015, 6, 17135–17146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, P.; Wang, X.; Shi, L.; Fan, Z.; Zhang, G.; Yang, D.; Bahavar, C.F.; Zhou, F.; Chen, W.R.; et al. Antitumor Effects of DC Vaccine With ALA-PDT-Induced Immunogenic Apoptotic Cells for Skin Squamous Cell Carcinoma in Mice. Technol. Cancer Res. Treat. 2018, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Baert, T.; Garg, A.D.; Vindevogel, E.; Van Hoylandt, A.; Verbist, G.; Agostinis, P.; Vergote, I.; Coosemans, A.N.; Agonistinis, P. In Vitro Generation of Murine Dendritic Cells for Cancer Immunotherapy: An Optimized Protocol. Anticancer Res. 2016, 36, 5793–5801. [Google Scholar] [CrossRef] [Green Version]
- Trempolec, N.; Doix, B.; Degavre, C.; Brusa, D.; Bouzin, C.; Riant, O.; Feron, O. Photodynamic therapy-based dendritic cell vaccination suited to treat peritoneal mesothelioma. Cancers 2020, 12, 545. [Google Scholar] [CrossRef] [Green Version]
- Jalili, A.; Makowski, M.; Switaj, T.; Nowis, D.; Wilczynski, G.M.; Wilczek, E.; Chorazy-Massalska, M.; Radzikowska, A.; Maslinski, W.; Biały, L.; et al. Effective photoimmunotherapy of murine colon carcinoma induced by the combination of photodynamic therapy and dendritic cells. Clin. Cancer Res. 2004, 10, 4498–4508. [Google Scholar] [CrossRef] [Green Version]
- Mroz, P.; Szokalska, A.; Wu, M.X.; Hamblin, M.R. Photodynamic Therapy of Tumors Can Lead to Development of Systemic Antigen-Specific Immune Response. PLoS ONE 2010, 5, e15194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Zhu, G.; Wang, S.; Yu, G.; Yang, Z.; Lin, L.; Zhou, Z.; Liu, Y.; Dai, Y.; Zhang, F.; et al. In Situ Dendritic Cell Vaccine for Effective Cancer Immunotherapy. ACS Nano 2019, 13, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Kleinovink, J.W.; van Driel, P.B.; Snoeks, T.J.A.; Prokopi, N.; Fransen, M.F.; Javier Cruz, L.; Mezzanotte, L.; Chan, A.; Lowik, C.W.G.M.; Ossendorp, F. Combination of photodynamic therapy and specific immunotherapy efficiently eradicates established tumors. Clin. Cancer Res. 2016, 22, 1459–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroz, P.; Vatansever, F.; Muchowicz, A.; Hamblin, M.R. Photodynamic therapy of murine mastocytoma induces specific immune responses against the cancer/testis antigen P1A. Cancer Res. 2013, 73, 6462–6470. [Google Scholar] [CrossRef] [Green Version]
- Canti, G.L.; Lattuada, D.; Nicolin, A.; Taroni, P.; Valentini, G.; Cubeddu, R. Immunopharmacology studies on photosensitizers used in photodynamic therapy. Photodyn. Ther. Cancer 1994, 2078, 268–275. [Google Scholar] [CrossRef]
- Kleinovink, J.W.; Fransen, M.F.; Löwik, C.W.; Ossendorp, F. Photodynamic-Immune Checkpoint Therapy Eradicates Local and Distant Tumors by CD8 þ T Cells. Cancer Immunol. Res. 2017, 5, 832–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbelik, M.; Dougherty, G.J. Photodynamic Therapy-Mediated Immune Response against Subcutaneous Mouse Tumors; American Association for Cancer Research: Philadelphia, PA, USA, 1999; Volume 59. [Google Scholar]
- Preise, D.; Oren, R.; Glinert, I.; Kalchenko, V.; Jung, S.; Scherz, A.; Salomon, Y. Systemic antitumor protection by vascular-targeted photodynamic therapy involves cellular and humoral immunity. Cancer Immunol. Immunother. 2009, 58, 71–84. [Google Scholar] [CrossRef]
- Korbelik, M.; Cecic, I. Contribution of myeloid and lymphoid host cells to the curative outcome of mouse sarcoma treatment by photodynamic therapy. Cancer Lett. 1999, 137, 91–98. [Google Scholar] [CrossRef]
- Korbelik, M.; Krosl, G.; Krosl, J.; Dougherty, G.J. The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res. 1996, 56, 5647–5652. [Google Scholar]
- Thong, P.S.P.; Ong, K.W.; Goh, N.S.G.; Kho, K.W.; Manivasager, V.; Bhuvaneswari, R.; Olivo, M.; Soo, K.C. Photodynamic-therapy-activated immune response against distant untreated tumours in recurrent angiosarcoma. Lancet Oncol. 2007, 8, 950–952. [Google Scholar] [CrossRef]
- Abdel-Hady, E.S.; Martin-Hirsch, P.; Duggan-Keen, M.; Stern, P.L.; Moore, J.V.; Corbitt, G.; Kitchener, H.C.; Hampson, I.N. Immunological and viral factors associated with the response of vulval intraepithelial neoplasia to photodynamic therapy. Cancer Res. 2001, 61, 192–196. [Google Scholar] [PubMed]
- Natali, P.G.; Bigotti, A.; Nicotra, M.R.; Viora, M.; Manfredi, D.; Ferrone, S. Distribution of human Class I (HLA-A,B,C) histocompatibility antigens in normal and malignant tissues of nonlymphoid origin. Cancer Res. 1984, 44, 4679–4687. [Google Scholar] [PubMed]
- Garrido, F.; Cabrera, T.; Concha, A.; Glew, S.; Ruiz-Cabello, F.; Stern, P.L. Natural history of HLA expression during tumour development. Immunol. Today 1993, 14, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; Fuks, Z.; Drobnjak, M.; Moreno, C.; Eisenbach, L.; Feldman, M. Expression of HLA-A,B,C antigens on primary and metastatic tumor cell populations of human carcinomas. Cancer Res. 1991, 51, 6372–6380. [Google Scholar]
- Rocha, L.B.; Gomes-da-Silva, L.C.; Dąbrowski, J.M.; Arnaut, L.G. Elimination of primary tumours and control of metastasis with rationally designed bacteriochlorin photodynamic therapy regimens. Eur. J. Cancer 2015, 51, 1822–1830. [Google Scholar] [CrossRef]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Xu, L.; Wang, C.; Yang, R.; Zhuang, Q.; Han, X.; Dong, Z.; Zhu, W.; Peng, R.; Liu, Z. Near-Infrared-Triggered Photodynamic Therapy with Multitasking Upconversion Nanoparticles in Combination with Checkpoint Blockade for Immunotherapy of Colorectal Cancer. ACS Nano 2017, 11, 4463–4474. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Hu, C.; Cai, L.; Pang, M. A covalent organic framework as a nanocarrier for synergistic phototherapy and immunotherapy. J. Mater. Chem. B 2020, 8, 5451. [Google Scholar] [CrossRef]
- Cai, Z.; Xin, F.; Wei, Z.; Wu, M.; Lin, X.; Du, X.; Chen, G.; Zhang, D.; Zhang, Z.; Liu, X.; et al. Photodynamic Therapy Combined with Antihypoxic Signaling and CpG Adjuvant as an In Situ Tumor Vaccine Based on Metal–Organic Framework Nanoparticles to Boost Cancer Immunotherapy. Adv. Healthc. Mater. 2020, 9, 1900996. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, L.; Wang, C.; Han, Y.; Lu, Y.; Liu, J.; Hu, X.; Yao, T.; Lin, Y.; Liang, S.; et al. Tumor-Targeted Drug and CpG Delivery System for Phototherapy and Docetaxel-Enhanced Immunotherapy with Polarization toward M1-Type Macrophages on Triple Negative Breast Cancers. Adv. Mater. 2019, 31, 1904997. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, F.; Deng, H.; Lin, L.; Wang, S.; Kang, F.; Yu, G.; Lau, J.; Tian, R.; Zhang, M.; et al. Smart Nanovesicle-Mediated Immunogenic Cell Death through Tumor Microenvironment Modulation for Effective Photodynamic Immunotherapy. ACS Nano 2020, 14, 620–631. [Google Scholar] [CrossRef]
- Ferreira, D.P.; Conceic, D.S.; Fernandes, F.; Sousa, T.; Calhelha, R.C.; Ferreira, I.C.F.R.; Santos, P.F.; Ferreira, L.F.V.; Lisboa, D.; Pais, A.R. Characterization of a Squaraine/Chitosan System for Photodynamic Therapy of Cancer. J. Phys. Chem. B 2016, 120, 1212–1220. [Google Scholar] [CrossRef]
- Mfouo-Tynga, I.; Houreld, N.N.; Abrahamse, H. Characterization of a multiple particle delivery complex and determination of cellular photodamage in skin fibroblast and breast cancer cell lines. J. Biophotonics 2017, 11, e201700077. [Google Scholar] [CrossRef] [PubMed]
- Bharathiraja, S.; Moorthy, M.S.; Manivasagan, P.; Seo, H.; Lee, K.D.; Oh, J. Chlorin e6 conjugated silica nanoparticles for targeted and effective photodynamic therapy. Photodiagnosis Photodyn. Ther. 2017, 19, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Pramual, S.; Lirdprapamongkol, K.; Svasti, J.; Bergkvist, M.; Jouan-Hureaux, V.; Arnoux, P.; Frochot, C.; Barberi-Heyob, M.; Niamsiri, N. Polymer-lipid-PEG hybrid nanoparticles as photosensitizer carrier for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2017, 173, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Bharathiraja, S.; Manivasagan, P.; Moorthy, M.S.; Bui, N.Q.; Lee, K.D.; Oh, J. Chlorin e6 conjugated copper sulfide nanoparticles for photodynamic combined photothermal therapy. Photodiagnosis Photodyn. Ther. 2017, 19, 128–134. [Google Scholar] [CrossRef]
- Wu, J.; Han, H.; Jin, Q.; Li, Z.; Li, H.; Ji, J. Design and Proof of Programmed 5-Aminolevulinic Acid Prodrug Nanocarriers for Targeted Photodynamic Cancer Therapy. ACS Appl. Mater. Interfaces 2017, 9, 14596–14605. [Google Scholar] [CrossRef]
- Penon, O.; Marín, M.J.; Russell, D.A.; Pérez-García, L. Water soluble, multifunctional antibody-porphyrin gold nanoparticles for targeted photodynamic therapy. J. Colloid Interface Sci. 2017, 496, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Montanha, M.C.; Silva, L.L.; Pangoni, F.B.B.; Cesar, G.B.; Gonçalves, R.S.; Caetano, W.; Hioka, N.; Tominaga, T.T.; Consolaro, M.E.L.; Diniz, A.; et al. Response surface method optimization of a novel Hypericin formulation in P123 micelles for colorectal cancer and antimicrobial photodynamic therapy. J. Photochem. Photobiol. B Biol. 2017, 170, 247–255. [Google Scholar] [CrossRef]
- Liu, K.; Xing, R.; Zou, Q.; Ma, G.; Mçhwald, H.; Yan, X. Assembled Nanodrugs Simple Peptide-Tuned Self-Assembly of Photosensitizers towards Anticancer Photodynamic Therapy. Angewandte. Angew. Chem.-Int. Ed. 2016, 55, 3036–3039. [Google Scholar] [CrossRef]
- Camerin, M.; Moreno, M.; Marín, M.J.; Schofield, C.L.; Chambrier, I.; Cook, M.J.; Coppellotti, O.; Jori, G.; Russell, D.A. Delivery of a hydrophobic phthalocyanine photosensitizer using PEGylated gold nanoparticle conjugates for the in vivo photodynamic therapy of amelanotic melanoma. Photochem. Photobiol. Sci. 2016, 15, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-X.; Jia, H.-R.; Chen, Z.; Wu, F.-G. Photosensitizer (PS)/polyhedral oligomeric silsesquioxane (POSS)-crosslinked nanohybrids for enhanced imaging-guided photodynamic cancer therapy. Nanoscale 2017, 9, 12874–12884. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.R.; Jiang, Y.W.; Zhu, Y.X.; Li, Y.H.; Wang, H.Y.; Han, X.; Yu, Z.W.; Gu, N.; Liu, P.; Chen, Z.; et al. Plasma membrane activatable polymeric nanotheranostics with self-enhanced light-triggered photosensitizer cellular influx for photodynamic cancer therapy. J. Control. Release 2017, 255, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Yu, Y.; Zhong, F.; Gao, M.; Sun, T.; Liu, J.; Zhang, H.; Qian, H.; Tao, W.; Yang, X. Design of tumor acidity-responsive sheddable nanoparticles for fluorescence/magnetic resonance imaging-guided photodynamic therapy. Theranostics 2017, 7, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, F.; Ren, C.; Yang, L.; Liu, J.; Cheng, Z.; Chu, L.; Liu, J. Targeted Chemo-Photodynamic Combination Platform Based on the DOX Prodrug Nanoparticles for Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2017, 9, 13016–13028. [Google Scholar] [CrossRef]
- Li, X.; Gao, M.; Xin, K.; Zhang, L.; Ding, D.; Kong, D.; Wang, Z.; Shi, Y.; Kiessling, F.; Lammers, T.; et al. Singlet oxygen-responsive micelles for enhanced photodynamic therapy. J. Control. Release 2017, 260, 12–21. [Google Scholar] [CrossRef]
- Feng, L.; Tao, D.; Dong, Z.; Chen, Q.; Chao, Y.; Liu, Z.; Chen, M. Near-infrared light activation of quenched liposomal Ce6 for synergistic cancer phototherapy with effective skin protection. Biomaterials 2017, 127, 13–24. [Google Scholar] [CrossRef]
- Hussain, A.F.; Kampmeier, F.; Von Felbert, V.; Merk, H.; Tur, M.K.; Barth, S. SNAP-Tag Technology Mediates Site Specific Conjugation of Antibody Fragments with a Photosensitizer and Improves Target Specific Phototoxicity in Tumor Cells. Bioconjug. Chem. 2011, 22, 2487–2495. [Google Scholar] [CrossRef]
- Von Felbert, V.; Bauerschlag, D.; Maass, N.; Bräutigam, K.; Meinhold, I.; Mira, H.; Stefan, W.; Ahmad, B.; Hussain, F. A specific photoimmunotheranostics agent to detect and eliminate skin cancer cells expressing EGFR. J. Cancer Res. Clin. Oncol. 2016, 142, 1003–1011. [Google Scholar] [CrossRef]
- Jankun, J. Protein-based nanotechnology: Antibody conjugated with photosensitizer in targeted anticancer photoimmunotherapy. Int. J. Oncol. 2011, 39, 949–953. [Google Scholar] [CrossRef]
- Savellano, M.D.; Hasan, T. Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem. Photobiol. 2003, 77, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Savellano, M.D.; Hasan, T. Photochemical Targeting of Epidermal Growth Factor Receptor: A Mechanistic Study. Clin. Cancer Res. 2005, 11, 1658–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, Y.; Elrington, S.A.; Hasan, T. A new nanoconstruct for epidermal growth factor receptor-targeted photo-immunotherapy of ovarian cancer. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, B.Q.; Abu-yousif, A.O.; Palanisami, A.; Rizvi, I.; Zheng, X.; Mai, Z.; Anbil, S.; Sears, R.B.; Mensah, L.B.; Goldschmidt, R.; et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc. Natl. Acad. Sci. USA 2014, 111, E933–E942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasu, N.; Yamada, H.; Shibaguchi, H.; Kuroki, M.; Kuroki, M. Potent and specific antitumor effect of CEA-targeted photoimmunotherapy. Int. J. Cancer 2014, 135, 28907. [Google Scholar] [CrossRef]
- Nakajima, T.; Sano, K.; Choyke, P.; Kobayashi, H. Improving the efficacy of Photoimmunotherapy (PIT) using a cocktail of antibody conjugates in a multiple antigen tumor model. Theranostics 2013, 3, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Kim, J.; Kim, D.H.; Hwang, H.S.; Na, K. Multifunctional trastuzumab-chlorin e6 conjugate for the treatment of HER2-positive human breast cancer. Biomater. Sci. 2018, 6, 1217–1226. [Google Scholar] [CrossRef]
- Moore, L.S.; de Boer, E.; Warram, J.M.; Tucker, M.D.; Carroll, W.R.; Korb, M.L.; Brandwein-Gensler, M.S.; van Dam, G.M.; Rosenthal, E.L. Photoimmunotherapy of residual disease after incomplete surgical resection in head and neck cancer models. Cancer Med. 2016, 5, 1526–1534. [Google Scholar] [CrossRef] [Green Version]
- Yaghini, E.; Dondi, R.; Tewari, K.M.; Loizidou, M.; Eggleston, I.M.; MacRobert, A.J. Endolysosomal targeting of a clinical chlorin photosensitiser for light-triggered delivery of nano-sized medicines. Sci. Rep. 2017, 7, 6059. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhao, Y.; Mao, C.; Kong, Y.; Ming, X. RGD-Modified Albumin Nanoconjugates for Targeted Delivery of a Porphyrin Photosensitizer. Mol. Pharm. 2017, 14, 2793–2804. [Google Scholar] [CrossRef]
- Li, K.; Qiu, L.; Liu, Q.; Lv, G.; Zhao, X.; Wang, S.; Lin, J. Conjugate of biotin with silicon(IV) phthalocyanine for tumor-targeting photodynamic therapy. J. Photochem. Photobiol. B Biol. 2017, 174, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Q.; Zhang, Y.; Shi, H.; Liu, H.; Guo, W.; Ma, Y.; Huang, W.; Hong, Z. Folic Acid–Conjugated Pyropheophorbide a as the Photosensitizer Tested for In Vivo Targeted Photodynamic Therapy. J. Pharm. Sci. 2017, 106, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Gao, D.; Gao, L.; Lai, J.; Zhang, C.; Zhao, Y.; Zhong, L.; Jia, B.; Wang, F.; Chen, X.; et al. Inhibiting Metastasis and Preventing Tumor Relapse by Triggering Host Immunity with Tumor-Targeted Photodynamic Therapy Using Photosensitizer-Loaded Functional Nanographenes. ACS Nano 2017, 11, 10147–10158. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Cerio, R.J.; Roberts, J.N.; Thompson, C.D.; Pinos, E.d.L.; Lowy, D.R.; Schiller, J.T. Human papillomavirus capsids preferentially bind and infect tumor cells. Int. J. Cancer 2016, 138, 901. [Google Scholar] [CrossRef] [Green Version]
- Knappe, M.; Bodevin, S.; Selinka, H.-C.; Spillmann, D.; Streeck, R.E.; Chen, X.S.; Lindahl, U.; Sapp, M. Surface-exposed Amino Acid Residues of HPV16 L1 Protein Mediating Interaction with Cell Surface Heparan Sulfate. J. Biol. Chem. 2007, 282, 27913–27922. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of Heparan Sulfate in Attachment to and Infection of the Murine Female Genital Tract by Human Papillomavirus. J. Virol. 2009, 83, 2067. [Google Scholar] [CrossRef] [Green Version]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential Roles for Soluble Virion-Associated Heparan Sulfonated Proteoglycans and Growth Factors in Human Papillomavirus Infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef]
- Kines, R.C.; Varsavsky, I.; Choudhary, S.; Bhattacharya, D.; Spring, S.; McLaughlin, R.; Kang, S.J.; Grossniklaus, H.E.; Vavvas, D.; Monks, S.; et al. An Infrared Dye–Conjugated Virus-like Particle for the Treatment of Primary Uveal Melanoma. Mol. Cancer Ther. 2018, 17, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Kines, R.C.; Thompson, C.D.; Spring, S.; Li, Z.; de Los Pinos, E.; Monks, S.; Schiller, J.T. Virus-Like Particle–Drug Conjugates Induce Protective, Long-lasting Adaptive Antitumor Immunity in the Absence of Specifically Targeted Tumor Antigens. Cancer Immunol. Res. 2021, 9, 693–706. [Google Scholar] [CrossRef]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer. J. Biol. Chem. 2014, 289, 3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Mathivanan, S.; Fahner, C.J.; Reid, G.E.; Simpson, R.J. ExoCarta 2012: Database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012, 40, D1241. [Google Scholar] [CrossRef] [Green Version]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Gezer, U.; Özgür, E.; Cetinkaya, M.; Isin, M.; Dalay, N. Long non-coding RNAs with low expression levels in cells are enriched in secreted exosomes. Cell Biol. Int. 2014, 38, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell–derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003. [Google Scholar] [CrossRef] [Green Version]
- Millard, M.; Yakavetsa, I.; Piffoux, M.; Brun, A.; Gazeau, F.; Guigner, J.M.; Jasniewski, J.; Lassalle, H.P.; Wilhelm, C.; Bezdetnaya, L. mTHPC-loaded extracellular vesicles outperform liposomal and free mTHPC formulations by an increased stability, drug delivery efficiency and cytotoxic effect in tridimensional model of tumors. Drug Deliv. 2018, 25, 1790–1801. [Google Scholar] [CrossRef] [Green Version]
- Millard, M.; Posty, S.; Piffoux, M.; Jasniewski, J.; Lassalle, H.P.; Yakavets, I.; Gazeau, F.; Wilhelm, C.; Silva, A.K.A.; Bezdetnaya, L. mTHPC-Loaded Extracellular Vesicles Significantly Improve mTHPC Diffusion and Photodynamic Activity in Preclinical Models. Pharmaceutics 2020, 12, 676. [Google Scholar] [CrossRef] [PubMed]
- Gazeau, F.; Pocard, M.; Pinto, A.; Marangon, I.; Méreaux, J.; Nicolás-Boluda, A.; Lavieu, G.; Wilhelm, C.; Sarda-Mantel, L.; Silva, A.K.A. Immune reprogramming precision photodynamic therapy of peritoneal metastasis by scalable stem-cell-derived extracellular vesicles. ACS Nano 2021, 15, 3251–3263. [Google Scholar] [CrossRef]
- Ding, J.; Lu, G.; Nie, W.; Huang, L.-L.; Zhang, Y.; Fan, W.; Wu, G.; Liu, H.; Xie, H.-Y.; Ding, J.; et al. Self-Activatable Photo-Extracellular Vesicle for Synergistic Trimodal Anticancer Therapy. Adv. Mater. 2021, 33, 2005562. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.; Huis In’t Veld, R.V.; Jorquera-Cordero, C.; Chan, A.B.; Ossendorp, F.; Cruz, L.J. Zinc-Phthalocyanine-Loaded Extracellular Vesicles Increase Efficacy and Selectivity of Photodynamic Therapy in Co-Culture and Preclinical Models of Colon Cancer. Pharmaceutics 2021, 13, 1547. [Google Scholar] [CrossRef]
- Xia, F.; Hou, W.; Liu, Y.; Wang, W.; Han, Y.; Yang, M.; Zhi, X.; Li, C.; Qi, D.; Li, T.; et al. Cytokine induced killer cells-assisted delivery of chlorin e6 mediated self-assembled gold nanoclusters to tumors for imaging and immuno-photodynamic therapy. Biomaterials 2018, 170, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mallidi, S.; Anbil, S.; Bulin, A.L.; Obaid, G.; Ichikawa, M.; Hasan, T. Beyond the Barriers of Light Penetration: Strategies, Perspectives and Possibilities for Photodynamic Therapy. Theranostics 2016, 6, 2458. [Google Scholar] [CrossRef] [Green Version]
- Mou, J.; Lin, T.; Huang, F.; Chen, H.; Shi, J. Biomaterials Black titania-based theranostic nanoplatform for single NIR laser induced dual-modal imaging-guided PTT/PDT. Biomaterials 2016, 84, 13–24. [Google Scholar] [CrossRef]
- Yang, T.; Liu, L.; Deng, Y.; Guo, Z.; Zhang, G.; Ge, Z.; Ke, H.; Chen, H. Ultrastable Near-Infrared Conjugated-Polymer Nanoparticles for Dually Photoactive Tumor Inhibition. Adv. Mater. 2017, 29, 487. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Wang, P.; Jiang, D.; Ban, D.; Zhong, N.; Jiang, G.; Li, H.; Hu, Z.; Xiao, J.; et al. NaYbF4 nanoparticles as near infrared light excited inorganic photosensitizers for deep penetration photodynamic therapy. Nanoscale 2017, 9, 2706–2710. [Google Scholar] [CrossRef]
- Mou, J.; Lin, T.; Huang, F.; Shi, J.; Chen, H. A new green titania with enhanced NIR absorption for mitochondria-targeted cancer therapy. Theranostics 2017, 7, 1531–1542. [Google Scholar] [CrossRef]
- Cherrick, G.R.; Stein, S.W.; Leevy, C.M.; Davidson, C.S. Indocyanine green: Observations on its physical properties, plasma decay, and hepatic extraction. J. Clin. Investig. 1960, 39, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Bejjanki, N.K.; Miao, X.; Weng, H.; Li, Q.; Zhang, J.; Liu, T.; Vannam, R.; Xie, M. Synthesis and Photothermal Effects of Intracellular Aggregating Nanodrugs Targeting Nasopharyngeal Carcinoma. Front. Bioeng. Biotechnol. 2021, 9, 730925. [Google Scholar] [CrossRef] [PubMed]
- Dash, B.; Das, S.; Chen, J. Photosensitizer-Functionalized Nanocomposites for Light-Activated Cancer Theranostics. Int. J. Mol. Sci. 2021, 22, 6658. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, S.; Yu, F.; Lv, J.; Zhao, R.; Hu, F.; Yuan, H. Dual-Channel Theranostic System for Quantitative Self-Indication and Low-Temperature Synergistic Therapy of Cancer. Small 2021, 17, e2007953. [Google Scholar] [CrossRef] [PubMed]
- Shirata, C.; Kaneko, J.; Inagaki, Y.; Kokudo, T.; Sato, M.; Kiritani, S.; Akamatsu, N.; Arita, J.; Sakamoto, Y.; Hasegawa, K.; et al. Near-infrared photothermal/photodynamic therapy with indocyanine green induces apoptosis of hepatocellular carcinoma cells through oxidative stress. Sci. Reports 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Tamai, K.; Mizushima, T.; Wu, X.; Inoue, A.; Ota, M.; Yokoyama, Y.; Miyoshi, N.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; et al. Photodynamic Therapy Using Indocyanine Green Loaded on Super Carbonate Apatite as Minimally Invasive Cancer Treatment. Mol. Cancer Ther. 2018, 17, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadel, M.; Samy, N.; Nasr, M.; Alyoussef, A.A. Topical colloidal indocyanine green-mediated photodynamic therapy for treatment of basal cell carcinoma. Pharm. Dev. Technol. 2016, 22, 545–550. [Google Scholar] [CrossRef]
- Arnaut, L.G.; Pereira, M.M.; Dąbrowski, J.M.; Silva, E.F.F.; Schaberle, F.A.; Abreu, A.R.; Rocha, L.B.; Barsan, M.M.; Urbańska, K.; Stochel, G.; et al. Photodynamic Therapy Efficacy Enhanced by Dynamics: The Role of Charge Transfer and Photostability in the Selection of Photosensitizers. Chem.–A Eur. J. 2014, 20, 5346–5357. [Google Scholar] [CrossRef] [PubMed]
- Karwicka, M.; Pucelik, B.; Gonet, M.; Elas, M.; Dąbrowski, J.M. Effects of Photodynamic Therapy with Redaporfin on Tumor Oxygenation and Blood Flow in a Lung Cancer Mouse Model. Sci. Rep. 2019, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Gomes-da-Silva, L.C.; Zhao, L.; Arnaut, L.G.; Kroemer, G.; Kepp, O. Redaporfin induces immunogenic cell death by selective destruction of the endoplasmic reticulum and the Golgi apparatus. Oncotarget 2018, 9, 31169. [Google Scholar] [CrossRef]
- Santos, L.L.; Oliveira, J.; Monteiro, E.; Santos, J.; Sarmento, C. Treatment of Head and Neck Cancer with Photodynamic Therapy with Redaporfin: A Clinical Case Report. Case Rep. Oncol. 2018, 11, 769. [Google Scholar] [CrossRef]
- Chatterjee, D.K.; Yong, Z. Upconverting nanoparticles as nanotransducers for photodynamic therapy in cancer cells. Nanomedicine 2008, 3, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Tao, H.; Cheng, L.; Liu, Z. Near-infrared light induced in vivo photodynamic therapy of cancer based on upconversion nanoparticles. Biomaterials 2011, 32, 6145–6154. [Google Scholar] [CrossRef] [PubMed]
- Eitao, W.; Uang, D.; Hang, Y.; Iu, Y.; Ueqing, Y.; Ian, Z. Real-Time Monitoring of Singlet Oxygen and Oxygen Partial Pressure During the Deep Photodynamic Therapy In Vitro. Ann. Biomed. Eng. 2016, 44, 2737–2745. [Google Scholar] [CrossRef]
- Wang, A.; Jin, W.; Chen, E.; Zhou, J.; Zhou, L.; Wei, S. Drug delivery function of carboxymethyl-β-cyclodextrin modified upconversion nanoparticles for adamantine phthalocyanine and their NIR-triggered cancer treatment. Dalt. Trans. 2016, 45, 3853–3862. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, R.; Yang, L.; Shi, J.; Liu, Z.; Hu, Y.; Wang, Y.; Liu, S.; Gan, Y. Design and Synthesis of Core−Shell−Shell Upconversion Nanoparticles for NIR-Induced Drug Release, Photodynamic Therapy, and Cell Imaging. ACS Appl. Mater. Interfaces 2016, 8, 4416–4423. [Google Scholar] [CrossRef]
- Han, R.; Shi, J.; Liu, Z.; Wang, H.; Wang, Y. Fabrication of Mesoporous-Silica-Coated Upconverting Nanoparticles with Ultrafast Photosensitizer Loading and 808 nm NIR-Light-Triggering Capability for Photodynamic Therapy. Chem. Asian J. 2017, 12, 2197–2201. [Google Scholar] [CrossRef]
- Chang, Y.; Li, X.; Zhang, L.; Xia, L.; Liu, X.; Li, C.; Zhang, Y.; Tu, L.; Xue, B.; Zhao, H.; et al. Precise Photodynamic Therapy of Cancer via Subcellular Dynamic Tracing of Dual-loaded Upconversion Nanophotosensitizers. Sci. Rep. 2017, 7, 45633. [Google Scholar] [CrossRef] [Green Version]
- Kiew, L.V.; Cheah, H.Y.; Voon, S.H.; Gallon, E.; Movellan, J.; Ng, K.H.; Alpugan, S.; Lee, H.B.; Dumoulin, F.; Vicent, M.J.; et al. Near-infrared activatable phthalocyanine-poly-L-glutamic acid conjugate: Increased cellular uptake and light-dark toxicity ratio toward an effective photodynamic cancer therapy. Nanomedicine 2017, 13, 1447–1458. [Google Scholar] [CrossRef]
- Hou, B.; Zheng, B.; Yang, W.; Dong, C.; Wang, H.; Chang, J. Construction of near infrared light triggered nanodumbbell for cancer photodynamic therapy. J. Colloid Interface Sci. 2017, 494, 363–372. [Google Scholar] [CrossRef]
- Liu, X.; Fan, Z.; Zhang, L.; Jin, Z.; Yan, D.; Zhang, Y.; Li, X.; Tu, L.; Xue, B.; Chang, Y.; et al. Bcl-2 inhibitor uploaded upconversion nanophotosensitizers to overcome the photodynamic therapy resistance of cancer through adjuvant intervention strategy. Biomaterials 2017, 144, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, P.; Sun, M.; Bi, H.; Liu, B.; Yang, D.; Gai, S.; He, F.; Lin, J. Highly Emissive Dye-Sensitized Upconversion Nanostructure for Dual-Photosensitizer Photodynamic Therapy and Bioimaging. ACS Nano 2017, 11, 4133–4144. [Google Scholar] [CrossRef]
- Xu, J.; Gulzar, A.; Liu, Y.; Bi, H.; Gai, S.; Liu, B.; Yang, D.; He, F.; Yang, P. Integration of IR-808 Sensitized Upconversion Nanostructure and MoS2 Nanosheet for 808 nm NIR Light Triggered Phototherapy and Bioimaging. Small 2017, 13, 1701841. [Google Scholar] [CrossRef]
- Chu, Z.; Tian, T.; Tao, Z.; Yang, J.; Chen, B.; Chen, H.; Wang, W.; Yin, P.; Xia, X.; Wang, H.; et al. Upconversion nanoparticles@AgBiS2 core-shell nanoparticles with cancer-cell-specific cytotoxicity for combined photothermal and photodynamic therapy of cancers. Bioact. Mater. 2022, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, G.; Wang, Y.; Zhao, Y.; Jiang, H.; Han, Y.; Yang, P. Multiple imaging and excellent anticancer efficiency of an upconverting nanocarrier mediated by single near infrared light. Nanoscale 2017, 9, 4759–4769. [Google Scholar] [CrossRef] [PubMed]
- Kolemen, S.; Ozdemir, T.; Lee, D.; Kim, G.M.; Karatas, T.; Yoon, J.; Akkaya, E.U. Remote-Controlled Release of Singlet Oxygen by the Plasmonic Heating of Endoperoxide-Modified Gold Nanorods: Towards a Paradigm Change in Photodynamic Therapy. Angew. Chem.-Int. Ed. 2016, 55, 3606–3610. [Google Scholar] [CrossRef] [PubMed]
- Gilson, R.C.; Black, K.C.L.; Lane, D.D.; Achilefu, S. Hybrid TiO2–Ruthenium Nano-photosensitizer Synergistically Produces Reactive Oxygen Species in both Hypoxic and Normoxic Conditions. Angew. Chem.-Int. Ed. 2017, 56, 10717–10720. [Google Scholar] [CrossRef] [PubMed]
- Lameijer, L.N.; Ernst, D.; Hopkins, S.L.; Meijer, M.S.; Askes, S.H.C.; Le Dévédec, S.E.; Bonnet, S. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem.-Int. Ed. 2017, 56, 11549–11553. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.-W.; Li, B.; Li, C.-X.; Fan, J.-X.; Lei, Q.; Li, C.; Xu, Z.; Zhang, X.-Z. Carbon-Dot-Decorated Carbon Nitride Nanoparticles for Enhanced Photodynamic Therapy against Hypoxic Tumor via Water Splitting. ACS Nano 2016, 10, 8715–8722. [Google Scholar] [CrossRef]
- Kim, J.; Cho, H.R.; Jeon, H.; Kim, D.; Song, C.; Lee, N.; Choi, S.H.; Hyeon, T. Continuous O2-Evolving MnFe2O4 Nanoparticle-Anchored Mesoporous Silica Nanoparticles for Efficient Photodynamic Therapy in Hypoxic Cancer. J. Am. Chem. Soc. 2017, 139, 10992–10995. [Google Scholar] [CrossRef]
- Ren, H.; Liu, J.; Li, Y.; Wang, H.; Ge, S.; Yuan, A.; Hu, Y.; Wu, J. Oxygen self-enriched nanoparticles functionalized with erythrocyte membranes for long circulation and enhanced phototherapy. Acta Biomater. 2017, 59, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Cheng, Y.; Huang, S.; Zhi, F.; Yuan, A.; Hu, Y.; Wu, J. Overcome the limitation of hypoxia against photodynamic therapy to treat cancer cells by using perfluorocarbon nanodroplet for photosensitizer delivery. Biochem. Biophys. Res. Commun. 2017, 487, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Bi, H.; Wang, Z.; Li, C.; Wang, C.; Xu, J.; Yang, D.; He, F.; Gai, S.; Yang, P. O2-Generating Metal-Organic Framework-Based Hydrophobic Photosensitizer Delivery System for Enhanced Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 36347–36358. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Li, J.; Peng, Z.-H.; Sheinin, Y.; Zhou, J.; Oupický, D. Tumor-Penetrating Nanoparticles for Enhanced Anticancer Activity of Combined Photodynamic and Hypoxia-Activated Therapy. ACS Nano 2017, 11, 2227–2238. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, X.; Yao, C.; Wang, W.; Zhao, M.; El-Toni, A.M.; Zhang, F. Orthogonal near-infrared upconversion co-regulated site-specific O2 delivery and photodynamic therapy for hypoxia tumor by using red blood cell microcarriers. Biomaterials 2017, 125, 90–100. [Google Scholar] [CrossRef]
- Gong, H.; Chao, Y.; Xiang, J.; Han, X.; Song, G.; Feng, L.; Liu, J.; Yang, G.; Chen, Q.; Liu, Z. Hyaluronidase To Enhance Nanoparticle-Based Photodynamic Tumor Therapy. Nano Lett. 2016, 16, 2512–2521. [Google Scholar] [CrossRef]
- Chen, B.; Crane, C.; He, C.; Gondek, D.; Agharkar, P.; Savellano, M.; Hoopes, P.; Pogue, B. Disparity between prostate tumor interior versus peripheral vasculature in response to verteporfin-mediated vascular-targeting therapy. Int. J. Cancer 2008, 123, 695–701. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; van Beijnum, J.; van Berkel, M.; van den Bergh, H.; Griffioen, A. Vascular regrowth following photodynamic therapy in the chicken embryo chorioallantoic membrane. Angiogenesis 2010, 13, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Moshfeghi, D.; Kaiser, P.; Grossniklaus, H.; Sternberg, P.; Sears, J.; Johnson, M.; Ratliff, N.; Branco, A.; Blumenkranz, M.; Lewis, H. Clinicopathologic study after submacular removal of choroidal neovascular membranes treated with verteporfin ocular photodynamic therapy. Am. J. Ophthalmol. 2003, 135, 343–350. [Google Scholar] [CrossRef]
- Kraus, D.; Palasuberniam, P.; Chen, B. Targeting Phosphatidylinositol 3-Kinase Signaling Pathway for Therapeutic Enhancement of Vascular-Targeted Photodynamic Therapy. Mol. Cancer Ther. 2017, 16, 2422–2431. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Yin, Y.; Wang, Y.; Zhang, B.; Tan, P.; Jiang, T.; Mei, H.; Deng, J.; Wang, H.; Guo, T.; et al. A tissue factor-cascade-targeted strategy to tumor vasculature: A combination of EGFP-EGF1 conjugation nanoparticles with photodynamic therapy. Oncotarget 2017, 8, 32212–32227. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Cheng, J.; Xu, J.; Ruf, W.; Lockwood, C.J. Tissue factor is an angiogenic-specific receptor for factor VII-targeted immunotherapy and photodynamic therapy. Angiogenesis 2017, 20, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yin, Y.; Jin, W.; Zhang, B.; Yan, H.; Mei, H.; Wang, H.; Guo, T.; Shi, W.; Hu, Y. Tissue Factor-Targeted “O2-Evolving” Nanoparticles for Photodynamic Therapy in Malignant Lymphoma. Front. Oncol. 2020, 10, 2220. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Tao, Z.; Yang, H.; Fan, Q.; Wei, D.; Wan, L.; Lu, X. PDGFRβ-specific affibody-directed delivery of a photosensitizer, IR700, is efficient for vasculartargeted photodynamic therapy of colorectal cancer. Drug Deliv. 2017, 24, 1818–1830. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Xu, S.; Wang, N.; Jiang, H.; Huang, Y.; Jin, X.; Xue, B.; Zhang, C.; Zhu, X. Prodrug-embedded angiogenic vessel-targeting nanoparticle: A positive feedback amplifier in hypoxia-induced chemo-photo therapy. Biomaterials 2017, 144, 188–198. [Google Scholar] [CrossRef]
- Kim, K.; Zhang, H.; La Rosa, S.; Jebiwott, S.; Desai, P.; Kimm, S.; Scherz, A.; O’Donoghue, J.A.; Weber, W.A.; Coleman, J.A. Bombesin Antagonist-Based Radiotherapy of Prostate Cancer Combined with WST-11 Vascular Targeted Photodynamic Therapy. Clin. Cancer Res. 2017, 23, 3343–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abiraj, K.; Mansi, R.; Tamma, M.; Fani, M.; Forrer, F.; Nicolas, G.; Cescato, R.; Reubi, J.; Maecke, H. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 2011, 52, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg, H.; Philippou, Y.; Magnussen, A.; Tullis, I.; Bridges, E.; Chatrian, A.; Lefebvre, J.; Tam, K.; Murphy, E.; Rittscher, J.; et al. Tumour irradiation combined with vascular-targeted photodynamic therapy enhances antitumour effects in pre-clinical prostate cancer. Br. J. Cancer 2021, 125, 534–546. [Google Scholar] [CrossRef]
- Peng, Y.; He, G.; Tang, D.; Xiong, L.; Wen, Y.; Miao, X.; Hong, Z.; Yao, H.; Chen, C.; Yan, S.; et al. Lovastatin Inhibits Cancer Stem Cells and Sensitizes to Chemo- and Photodynamic Therapy in Nasopharyngeal Carcinoma. J. Cancer 2017, 8, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Chen, H.; Tham, H.P.; Zeng, S.; Phua, F.; Liu, J.-G.; Zhao, Y. Cyclometalated Iridium(III)-Complex-Based Micelles for Glutathione- Responsive Targeted Chemotherapy and Photodynamic Therapy. ACS Appl. Mater. Interfaces 2017, 9, 27553–27562. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Z.; Wang, Y.; Zhu, H.; Li, F.; Shen, Y.; Guo, S. A new NIR-triggered doxorubicin and photosensitizer indocyanine green co-delivery system for enhanced multidrug resistant cancer treatment through simultaneous chemo/photothermal/photodynamic therapy. Acta Biomater. 2017, 59, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Flak, D.; Yate, L.; Nowaczyk, G.; Jurga, S. Hybrid ZnPc@TiO2 nanostructures for targeted photodynamic therapy, bioimaging and doxorubicin delivery. Mater. Sci. Eng. C 2017, 78, 1072–1085. [Google Scholar] [CrossRef]
- Lee, Y.; Ma, Y. Synthesis, characterization, and biological verification of anti-HER2 indocyanine green–doxorubicin-loaded polyethyleneimine-coated perfluorocarbon double nanoemulsions for targeted photochemotherapy of breast cancer cells. J. Nanobiotechnology 2017, 15, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lin, H.; Tong, R.; An, N.; Qu, F. Near-infrared light-mediated DOX-UCNPs@mHTiO2 nanocomposite for chemo/photodynamic therapy and imaging. Colloids Surf. B Biointerfaces 2017, 154, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-González, R.; Milán, P.; Bresolí-Obach, R.; Stockert, J.C.; Villanueva, A.; Cañete, M.; Nonell, S. Photodynamic Synergistic Effect of Pheophorbide a and Doxorubicin in Combined Treatment against Tumoral Cells. Cancers 2017, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.-L.; Song, M.-R.; Yuan, G.-K.; Ye, H.-N.; Tian, Y.; Huang, M.-D.; Xue, J.-P.; Zhang, Z.-H.; Liu, J.-Y. A Molecular Combination of Zinc(II) Phthalocyanine and Tamoxifen Derivative for Dual Targeting Photodynamic Therapy and Hormone Therapy. J. Med. Chem. 2017, 60, 6693–6703. [Google Scholar] [CrossRef]
- Zhu, K.; Liu, G.; Hu, J.; Liu, S. Near-Infrared Light-Activated Photochemical Internalization of Reduction-Responsive Polyprodrug Vesicles for Synergistic Photodynamic Therapy and Chemotherapy. Biomacromolecules 2017, 18, 2571–2582. [Google Scholar] [CrossRef]
- Hu, F.; Yuan, Y.; Mao, D.; Wu, W.; Liu, B. Smart activatable and traceable dual-prodrug for image-guided combination photodynamic and chemo-therapy. Biomaterials 2017, 144, 53–59. [Google Scholar] [CrossRef]
- Yue, C.; Yang, Y.; Song, J.; Alfranca, G.; Zhang, C.; Zhang, Q.; Yin, T.; Pan, F.; de la Fuente, J.M.; Cui, D. Mitochondria-targeting near-infrared light-triggered thermosensitive liposomes for localized photothermal and photodynamic ablation of tumors combined with chemotherapy. Nanoscale 2017, 9, 11103–11118. [Google Scholar] [CrossRef]
- Yang, X.; Xue, X.; Luo, Y.; Lin, T.; Zhang, H.; Lac, D.; Xiao, K.; He, Y.; Jia, B.; Lam, K.S.; et al. Sub-100 nm, long tumor retention SN-38-loaded photonic micelles for tri-modal cancer therapy. J. Control. Release 2017, 261, 297–306. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Li, Y.; Wang, P.; Feng, X.; Liu, Q.; Yan, F.; Zheng, H. Sensitivity to antitubulin chemotherapeutics is potentiated by a photoactivable nanoliposome. Biomaterials 2017, 141, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wu, B.; Hu, X.; Xing, D. NIR-triggered high-efficient photodynamic and chemo-cascade therapy using caspase-3 responsive functionalized upconversion nanoparticles. Biomaterials 2017, 141, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, Y.; Chen, Z.; Geng, Y.; Xie, X.; Shen, X.; Li, T.; Li, S.; Wu, C.; Liu, Y. Chemo-photodynamic combined gene therapy and dual-modal cancer imaging achieved by pH-responsive alginate/chitosan multilayer-modified magnetic mesoporous silica nanocomposites. Biomater. Sci. 2017, 5, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-S.; Peng, Y.-B.; Yao, M.; Teng, J.-P.; Ni, D.; Zhu, Z.-J.; Zhuang, B.-F.; Yang, Z.-Y. Cisplatin and photodynamic therapy exert synergistic inhibitory effects on small-cell lung cancer cell viability and xenograft tumor growth. Biochem. Biophys. Res. Commun. 2017, 487, 567–572. [Google Scholar] [CrossRef]
- Chen, J.; Cheng, F.; Luo, D.; Huang, J.; Ouyang, J.; Nezamzadeh-Ejhieh, A.; Khan, M.S.; Liu, J.; Peng, Y. Recent advances in Ti-based MOFs in biomedical applications. Dalt. Trans. 2022, 51, 14817–14832. [Google Scholar] [CrossRef]
- Qiu, Y.; Tan, G.; Fang, Y.; Liu, S.; Zhou, Y.; Kumar, A.; Trivedi, M.; Liu, D.; Liu, J. Biomedical applications of metal–organic framework (MOF)-based nano-enzymes. New J. Chem. 2021, 45, 20987–21000. [Google Scholar] [CrossRef]
- Qin, L.; Liang, F.; Li, Y.; Wu, J.; Guan, S.; Wu, M.; Xie, S.; Luo, M.; Ma, D. A 2D Porous Zinc-Organic Framework Platform for Loading of 5-Fluorouracil. Inorganics 2022, 10, 202. [Google Scholar] [CrossRef]
- Ding, Q.; Xu, Z.; Zhou, L.; Rao, C.; Li, W.; Muddassir, M.; Sakiyama, H.; Li, B.; Ouyang, Q.; Liu, J. A multimodal Metal-Organic framework based on unsaturated metal site for enhancing antitumor cytotoxicity through Chemo-Photodynamic therapy. J. Colloid Interface Sci. 2022, 621, 180–194. [Google Scholar] [CrossRef]
- Tran, T.H.; Nguyen, H.T.; Van Le, N.; Tran, T.T.P.; Lee, J.S.; Ku, S.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Engineering of multifunctional temperature-sensitive liposomes for synergistic photothermal, photodynamic, and chemotherapeutic effects. Int. J. Pharm. 2017, 528, 692–704. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, F.; Shao, D.; Chang, Z.; Wang, L.; Hu, H.; Zheng, X.; Li, X.; Chen, F.; Tu, Z.; et al. Janus Nanobullets Combine Photodynamic Therapy and Magnetic Hyperthermia to Potentiate Synergetic Anti-Metastatic Immunotherapy. Adv. Sci. 2019, 6, 1901690. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yang, H.; Chen, X.; Gao, J.; Duan, Y.; Wei, D.; Zhang, J.; Ge, K.; Liang, X.J.; Huang, Y.; et al. Nano-herb medicine and PDT induced synergistic immunotherapy for colon cancer treatment. Biomaterials 2021, 269, 120654. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Elsen, S.; Krysko, D.V.; Vandenabeele, P.; De Witte, P.; Agostinis, P. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 2015, 6, 26841–26860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, N.; Jung, H.; Kang, M.; Lee, J.; Song, J.; Geuk, H.; Bae, Y.; Lim, D. Photodynamic therapy-mediated DC immunotherapy is highly effective for the inhibition of established solid tumors. Cancer Lett. 2012, 324, 58–65. [Google Scholar] [CrossRef]
- Jin, Y.; Guan, Z.; Wang, X.; Wang, Z.; Zeng, R.; Xu, L.; Cao, P. ALA-PDT promotes HPV-positive cervical cancer cells apoptosis and DCs maturation via miR-34a regulated HMGB1 exosomes secretion. Photodiagnosis. Photodyn. Ther. 2018, 24, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Merchant, S. Photodynamic therapy—Generated cancer vaccine elicits acute phase and hormonal response in treated mice. Cancer Immunol. Immunother. 2012, 61, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Banáth, J.; Saw, K.M.; Zhang, W.; Ĝiplys, E. Calreticulin as Cancer Treatment Adjuvant: Combination with Photodynamic Therapy and Photodynamic Therapy-Generated Vaccines. Front. Oncol. 2015, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.S.; Kim, H.; Oh, J.E.; Jung, H.E.; Lee, Y.S.; Park, J.-H.; Lee, H.K. Intratumoral depletion of regulatory T cells using CD25-targeted photodynamic therapy in a mouse melanoma model induces anti-tumoral immune responses. Oncotarget 2017, 8, 47440–47453. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Wang, L.; Tian, Y.; Guan, X.; Liu, Q.; Li, S.; Qin, X.; Yang, H.; Liu, Y. “Triple-Punch” Anticancer Strategy Mediated by Near-Infrared Photosensitizer/CpG Oligonucleotides Dual-Dressed and Mitochondria-Targeted Nanographene. ACS Appl. Mater. Interfaces 2018, 10, 6942–6955. [Google Scholar] [CrossRef]
- Song, W.; Kuang, J.; Li, C.-X.; Zhang, M.; Zheng, D.; Zeng, X.; Liu, C.; Zhang, X.-Z. Enhanced Immunotherapy Based on Photodynamic Therapy for Both Primary and Lung Metastasis Tumor Eradication. ACS Nano 2018, 12, 1978–1989. [Google Scholar] [CrossRef]
- Lu, K.; He, C.; Guo, N.; Chan, C.; Ni, K.; Weichselbaum, R.R.; Lin, W. Chlorin-Based Nanoscale Metal-Organic Framework Systemically Rejects Colorectal Cancers via Synergistic Photodynamic Therapy and Checkpoint Blockade Immunotherapy HHS Public Access. J. Am. Chem. Soc. 2016, 138, 12502–12510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shaughnessy, M.J.; Murray, K.S.; La Rosa, S.P.; Budhu, S.; Merghoub, T.; Somma, A.; Monette, S.; Kim, K.; Corradi, R.B.; Scherz, A.; et al. Systemic Antitumor Immunity by PD-1/PD-L1 Inhibition Is Potentiated by Vascular-Targeted Photodynamic Therapy of Primary Tumors. Clin. Cancer Res. 2018, 24, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Chan, C.; Guo, N.; Han, W.; Weichselbaum, R.R.; Lin, W. Photodynamic Therapy Mediated by Nontoxic Core–Shell Nanoparticles Synergizes with Immune Checkpoint Blockade To Elicit Antitumor Immunity and Antimetastatic Effect on Breast Cancer HHS Public Access. J. Am. Chem. Soc. 2016, 138, 16686–16695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Yang, J.; Ji, J.; Wang, P.; Zhang, L.; Yan, G.; Wu, Y.; Chen, Q.; Liu, J.; Zhang, G.; et al. PD-L1 blockade potentiates the antitumor effects of ALA-PDT and optimizes the tumor microenvironment in cutaneous squamous cell carcinoma. Oncoimmunology 2022, 11, 2061396. [Google Scholar] [CrossRef]
- Lan, G.; Ni, K.; Xu, Z.; Veroneau, S.S.; Song, Y.; Lin, W. Nanoscale Metal−Organic Framework Overcomes Hypoxia for Photodynamic Therapy Primed Cancer Immunotherapy. J. Am. Chem. Soc. 2018, 140, 5670–5673. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xu, L.; Xu, J.; Zhang, R.; Song, G.; Chao, Y.; Feng, L.; Han, F.; Dong, Z.; Li, B.; et al. Smart Nanoreactors for pH-Responsive Tumor Homing, Mitochondria-Targeting, and Enhanced Photodynamic- Immunotherapy of Cancer. Nano Lett. 2018, 18, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Mei, Q.; Wang, P.; Miao, P.; Dong, W.-F.; Li, L. Light-triggered multifunctional nanoplatform for efficient cancer photo-immunotherapy. J. Nanobiotechnol. 2022, 20, 181. [Google Scholar] [CrossRef]
- Hao, Y.; Chung, C.K.; Yu, Z.; Huis In’T Veld, R.V.; Ossendorp, F.A.; Ten Dijke, P.; Cruz, L.J. Combinatorial Therapeutic Approaches with Nanomaterial-Based Photodynamic Cancer Therapy. Pharmaceutics 2022, 14, 120. [Google Scholar] [CrossRef]
- Gunaydin, G.; Gedik, M.E.; Ayan, S. Photodynamic Therapy for the Treatment and Diagnosis of Cancer—A Review of the Current Clinical Status. Front. Chem. 2021, 9, 608. [Google Scholar] [CrossRef]






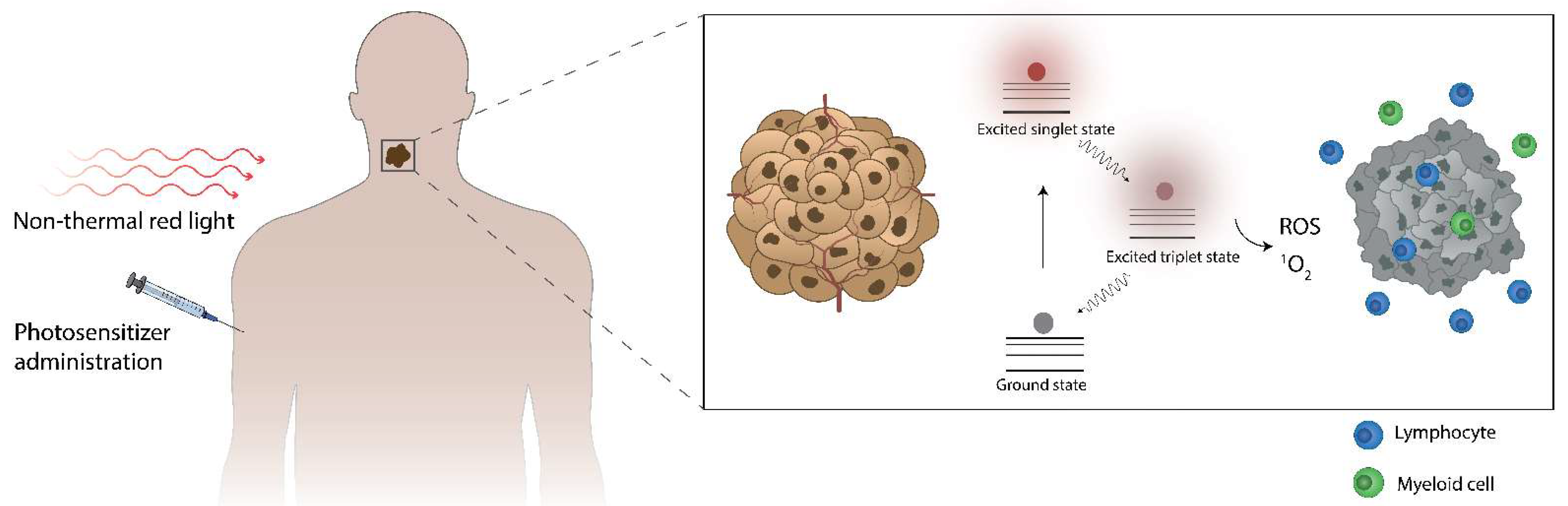{kind=link}
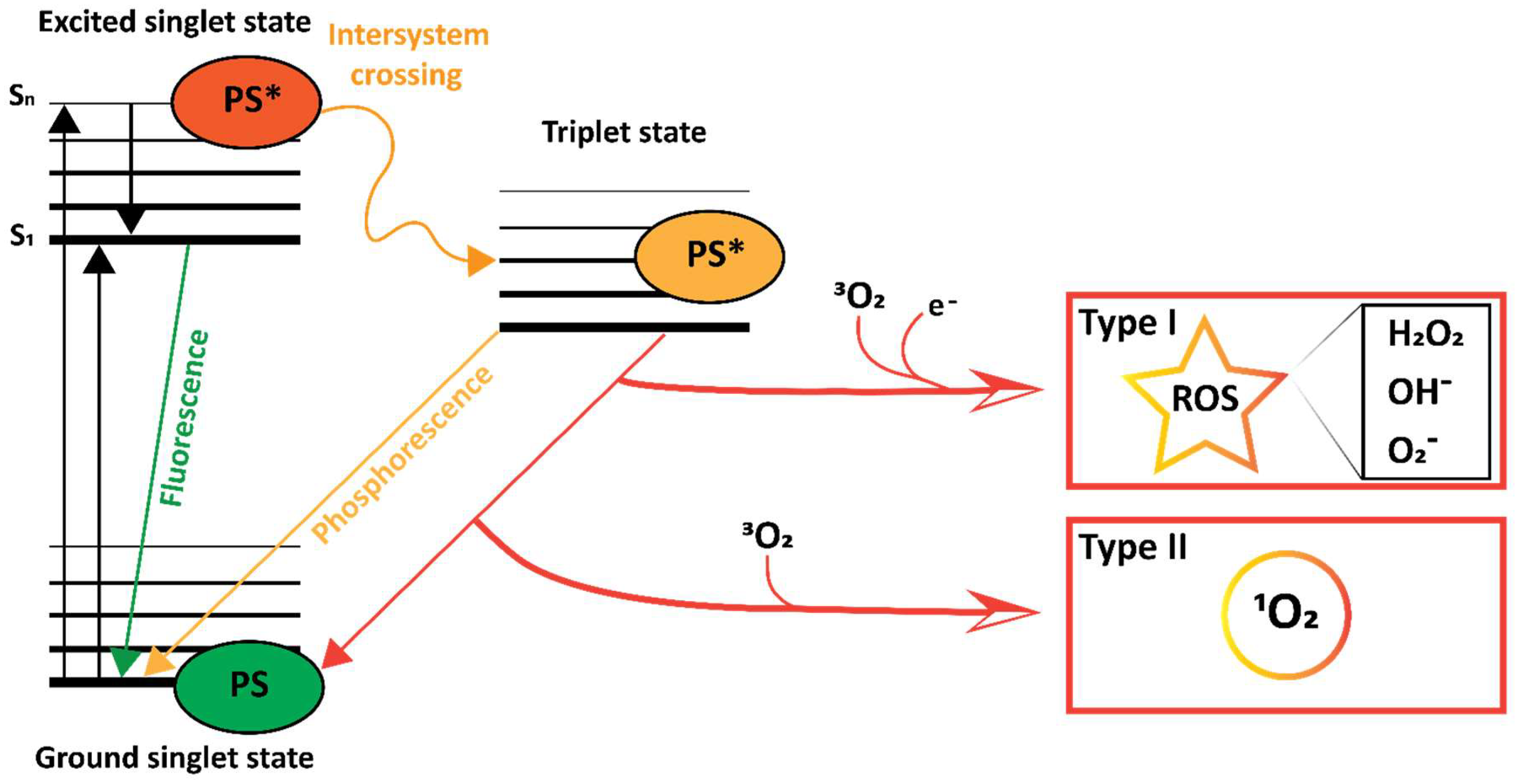{kind=link}
{kind=link}
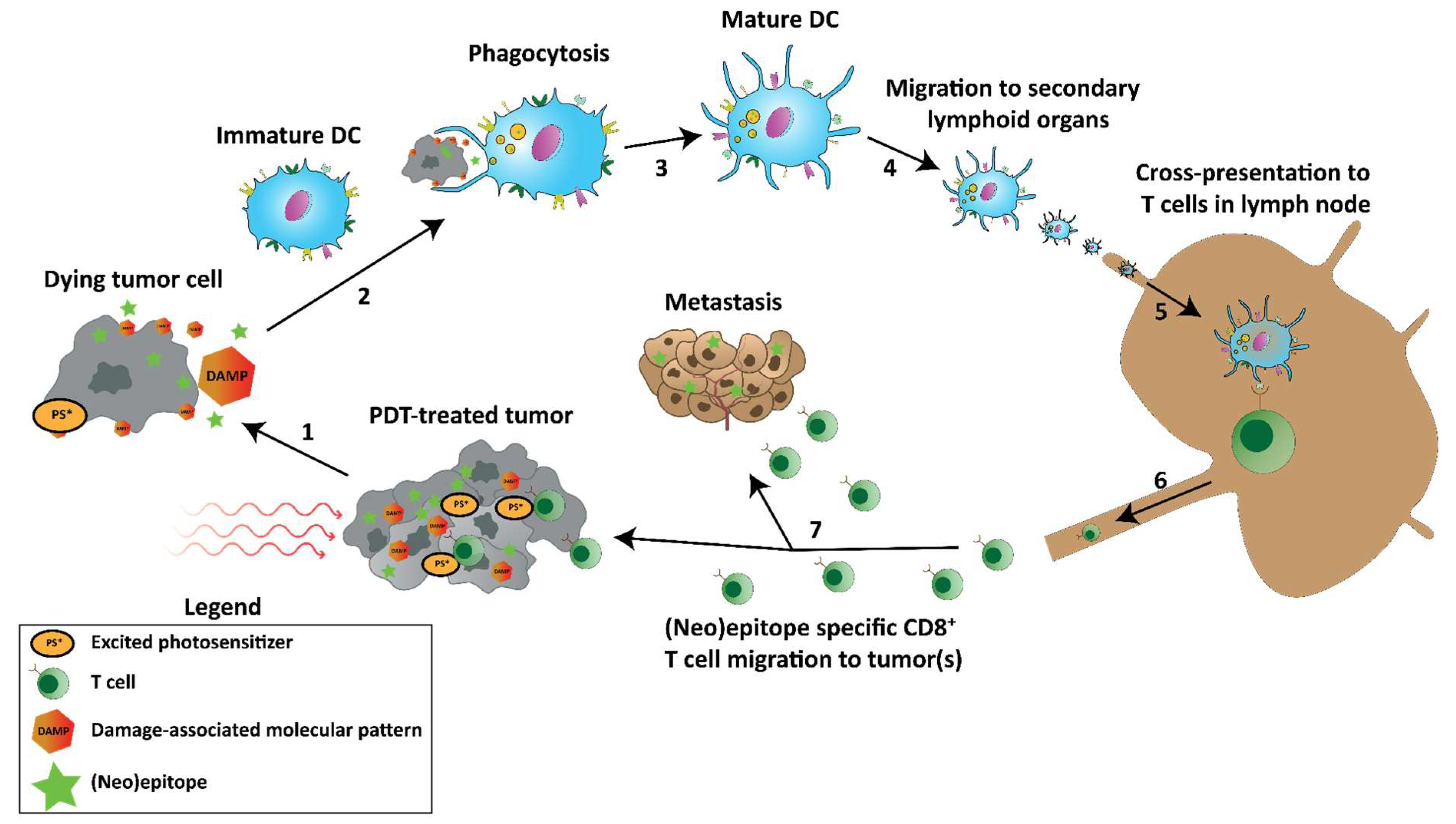{kind=link}
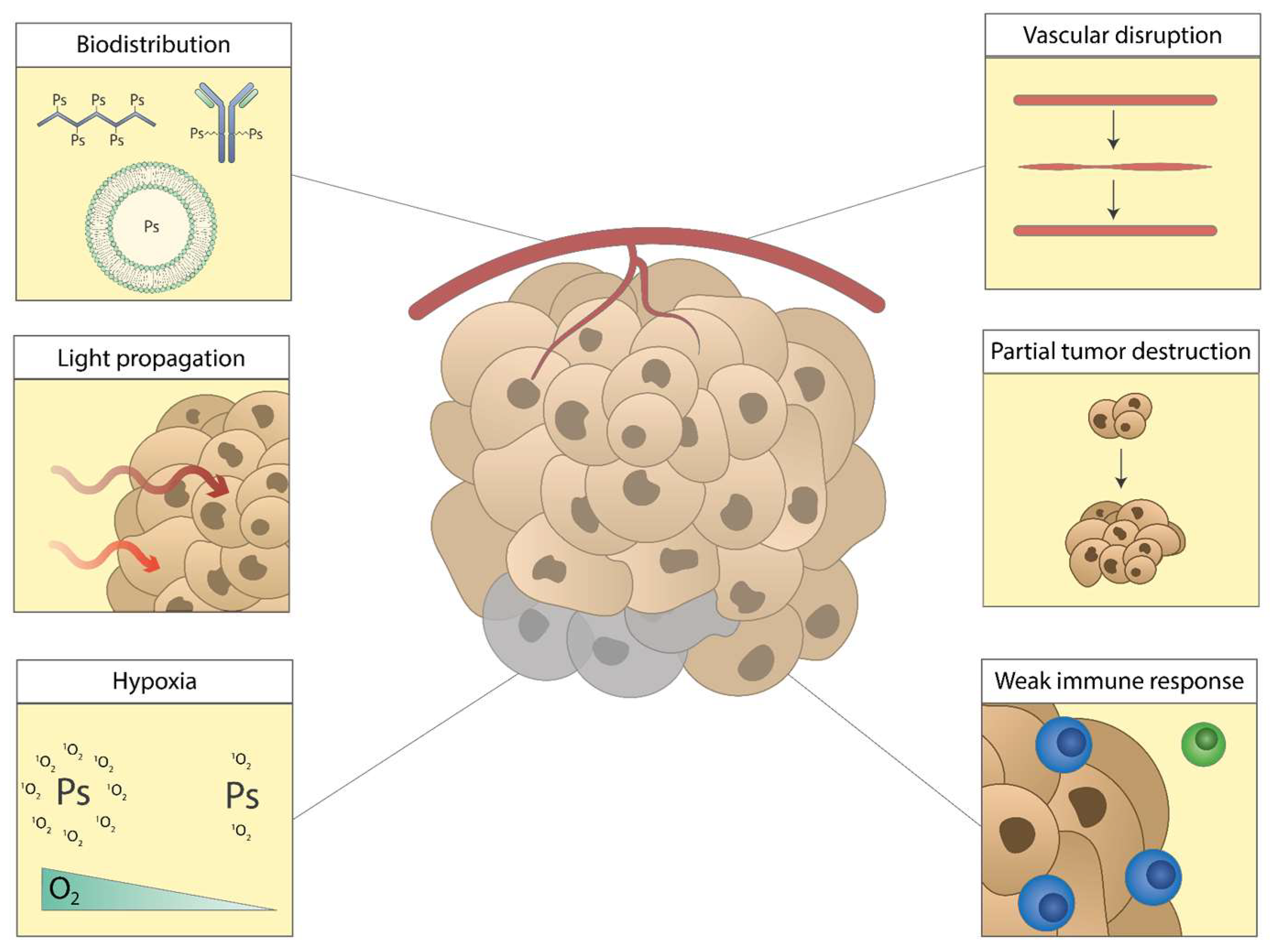{kind=link}
| Sensitizer (Brand Name) | Approval | Indication | Wavelength (nm) |
|---|---|---|---|
| 5-aminolevulinic acid/5-ALA (Ameluz) (Levulan) | Worldwide | Mild to moderate actinic keratosis | 635 |
| Bremachlorin (Radachlorin) | Russia, Belarus | Non-small cell lung cancer, bladder, cutaneous lesions | 662 |
| Hexaminolevulinate hydrochloride (Cysview) | Europe, USA, Canada | Bladder cancer detection | 360–450 |
| Methyl aminolevulinate (Metvix) (Metvixia) | Worldwide | Non-hyperkeratotic actinic keratosis and basal cell carcinoma | 570–670 |
| Porfimer sodium (Photofrin) | Worldwide | Esophageal cancer, Barrett’s Esophagus, non-small cell lung cancer | 630 |
| Redaporfin (LUZ11) | Orphan status in Europe | Biliary tract cancer | 749 |
| Synthetic hypericin (SGX301) | Orphan status in Europe, conditional FDA | Early-stage cutaneous T-cell lymphoma | 570–650 |
| Talaporfin sodium/NPe6 (Laserphyrin) | Japan | Lung cancer | 664 |
| Temoporfin/mTHPC (Foscan) | Europe | Advanced Head and neck cancer | 652 |
| Verteporfin (Visudyne) | Worldwide | Age-related macular degeneration | 690 |
| WST-11/padeliporfin (TOOKAD) | Europe | Prostatic Neoplasms | 753 |
| Challenge | Strategy to Overcome | Advantages | Disadvantages |
|---|---|---|---|
| Sensitizer biodistribution | Nanoparticle encapsulation | Control over pharmacokinetics, possibility of adding targeting moieties. | The enhanced-permeability and retention effect is much less pronounced in humans. |
| Antibody conjugation | Specific targeting of tumor epitopes, adoption of antibody biodistribution. | Lack of truly specific tumor targets. | |
| Peptide association | Targeting of tumor present ligands, adoption of peptide association. | Lack of truly specific ligands in the tumor. | |
| EV incorporation | Enhanced biodistribution of the PS, enhanced antitumor efficacy depending on the EV origin. | Large scale production is challenging. Restricted to the use of cell lines. | |
| Immune cells | Tunable distribution based on cell type and immunological state. Possibility to simultaneously use immune cells for therapy. | Restricted to distribution and functionality of immune cells in use. Distribution of PS to tumor cells required following death of carrier cells. | |
| Light propagation | NIR absorbing sensitizers | Increased penetration depth of light used for PDT. | High fluence required due to a reduced energy of therapeutic NIR light. |
| Upconversion Nanoparticles | Increased penetration depth of light used for PDT. Possibility to co-encapsulate additional therapeutic agents. | High fluence required due to a reduced energy of therapeutic NIR light. | |
| Hypoxia | O2-generating strategies | Possibility to increase ROS quantum yields after PDT. | Requires the use of carrier systems for O2-generating agents. |
| Hypoxia-responsive prodrugs | Drug selectivity to hypoxic areas in the body, such as the tumor. | Restricted to certain prodrugs that are hypoxia-responsive. | |
| O2 tumor diffusion | Increased availability of O2 for PDT throughout the tumor. | Requires PS or PDT protocols that can be directed to the ECM. | |
| Vascular disruption | Tumor vasculature disrupting agents | Enhanced tumor vasculature disruption. | Risk of adverse events of vasculature-disrupting agents. |
| Tumor vasculature targeting | Enhanced tumor vasculature disruption. | Risk of adverse events due to vasculature-destruction. | |
| VTP to enhance combination treatments | Increased potential for synergy with additional agent due to vasculature disruption. | Efficacy depending on ability of VTP to sufficiently disrupt the vasculature. | |
| Partial tumor destruction | Combination with chemotherapy | Increased antitumor efficacy. | Associated with a higher risk of adverse events. |
| Combination with other neoplastic agents | Increased antitumor efficacy. | Depending on the agent used, but often associated with increased risk of adverse events. | |
| Insufficient PDT-induced immune response | PDT-generated or enhanced tumor vaccines | Possibility to generate in situ vaccinations, or to enhance vaccination efficacy. Possibility to affect metastatic tumors. | Efficacy of the treatment is dependent on the ability of PDT to induce a pro-inflammatory environment in the tumor. |
| Combination with immunostimulatory agents | Increased antitumor efficacy, possibility to generate an in situ vaccination. Possibility to affect metastatic tumors. | Certain immunostimulatory compounds require encapsulation to prevent adverse events. | |
| Combination with immune checkpoint inhibition | Increased antitumor efficacy. Possibility to affect metastatic tumors. | Increased risk of adverse events |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huis in ‘t Veld, R.V.; Heuts, J.; Ma, S.; Cruz, L.J.; Ossendorp, F.A.; Jager, M.J. Current Challenges and Opportunities of Photodynamic Therapy against Cancer. Pharmaceutics 2023, 15, 330. https://doi.org/10.3390/pharmaceutics15020330
Huis in ‘t Veld RV, Heuts J, Ma S, Cruz LJ, Ossendorp FA, Jager MJ. Current Challenges and Opportunities of Photodynamic Therapy against Cancer. Pharmaceutics. 2023; 15(2):330. https://doi.org/10.3390/pharmaceutics15020330
Chicago/Turabian StyleHuis in ‘t Veld, Ruben V., Jeroen Heuts, Sen Ma, Luis J. Cruz, Ferry A. Ossendorp, and Martine J. Jager. 2023. "Current Challenges and Opportunities of Photodynamic Therapy against Cancer" Pharmaceutics 15, no. 2: 330. https://doi.org/10.3390/pharmaceutics15020330