
Tyrosine Kinase Inhibitors for Glioblastoma Multiforme: Challenges and Opportunities for Drug Delivery

Abstract
:1. Introduction
2. Pathophysiology of GBM and Role of TKs
2.1. Pathophysiology of GBM
2.1.1. p53 and PTEN Pathway
2.1.2. Isocitrate Dehydrogenase (IDH) Pathway
2.1.3. Retinoblastoma (RB) Pathway
2.1.4. Histone H3 Pathway
2.1.5. Interleukins (ILs) Pathway
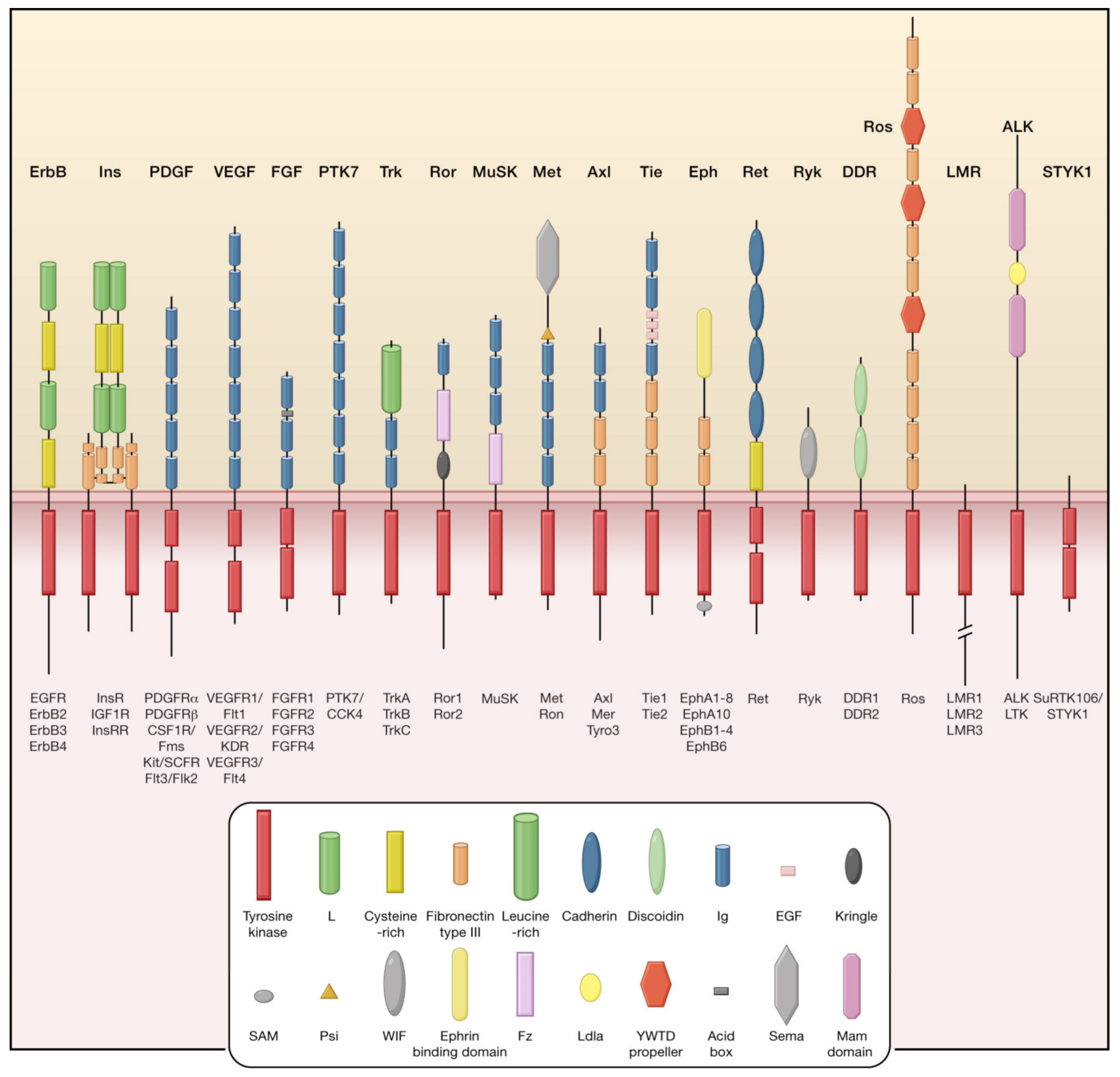
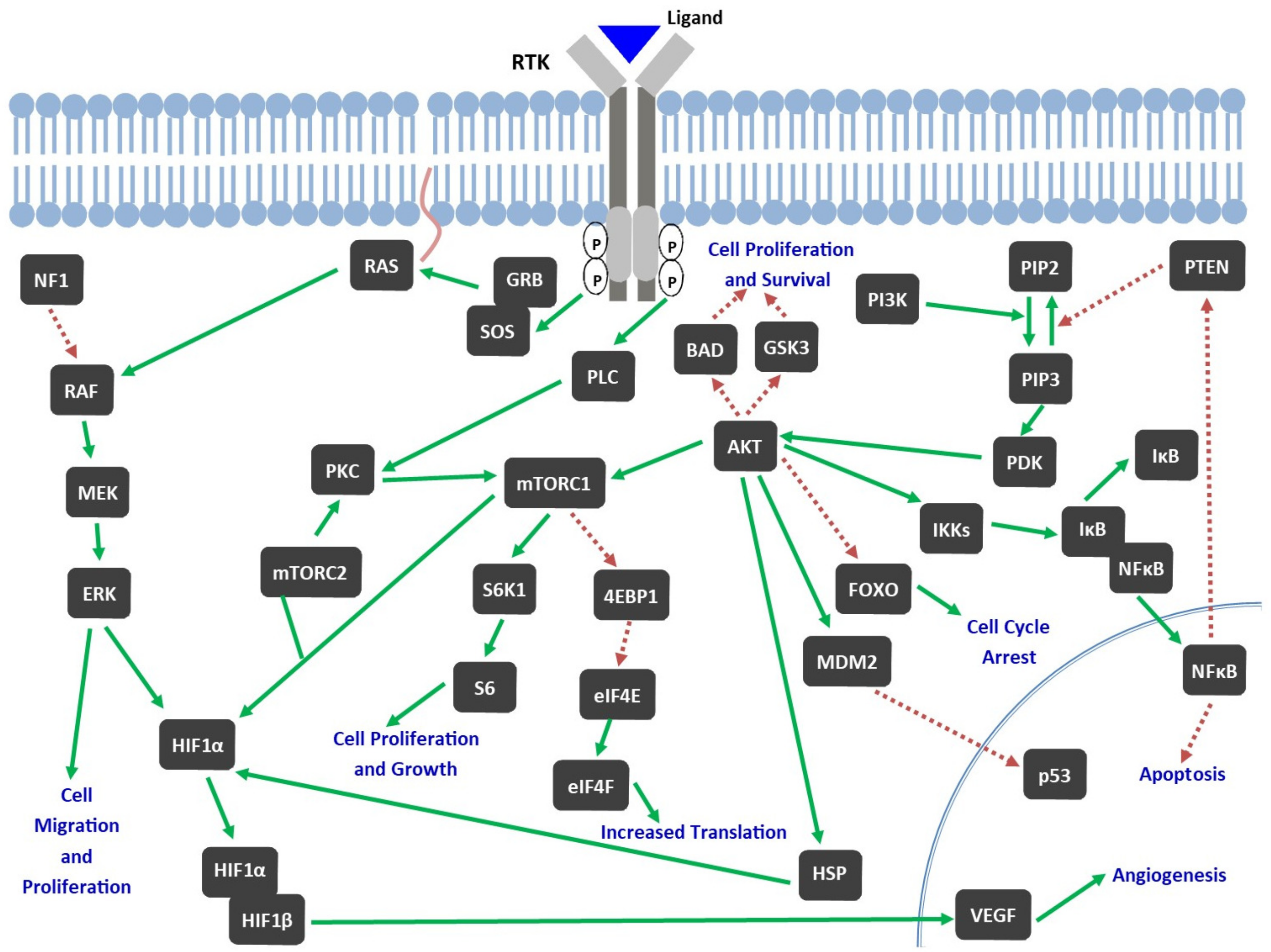
2.2. Role of TKs in Pathophysiology of GBM
3. Preclinical GBM Models
4. Tyrosine Kinase Inhibitors (TKIs)
5. Clinical Trials of TKIs in Treatment of GBM and Disappointing Outcomes
6. Pediatric High-Grade Gliomas (pHGGS)
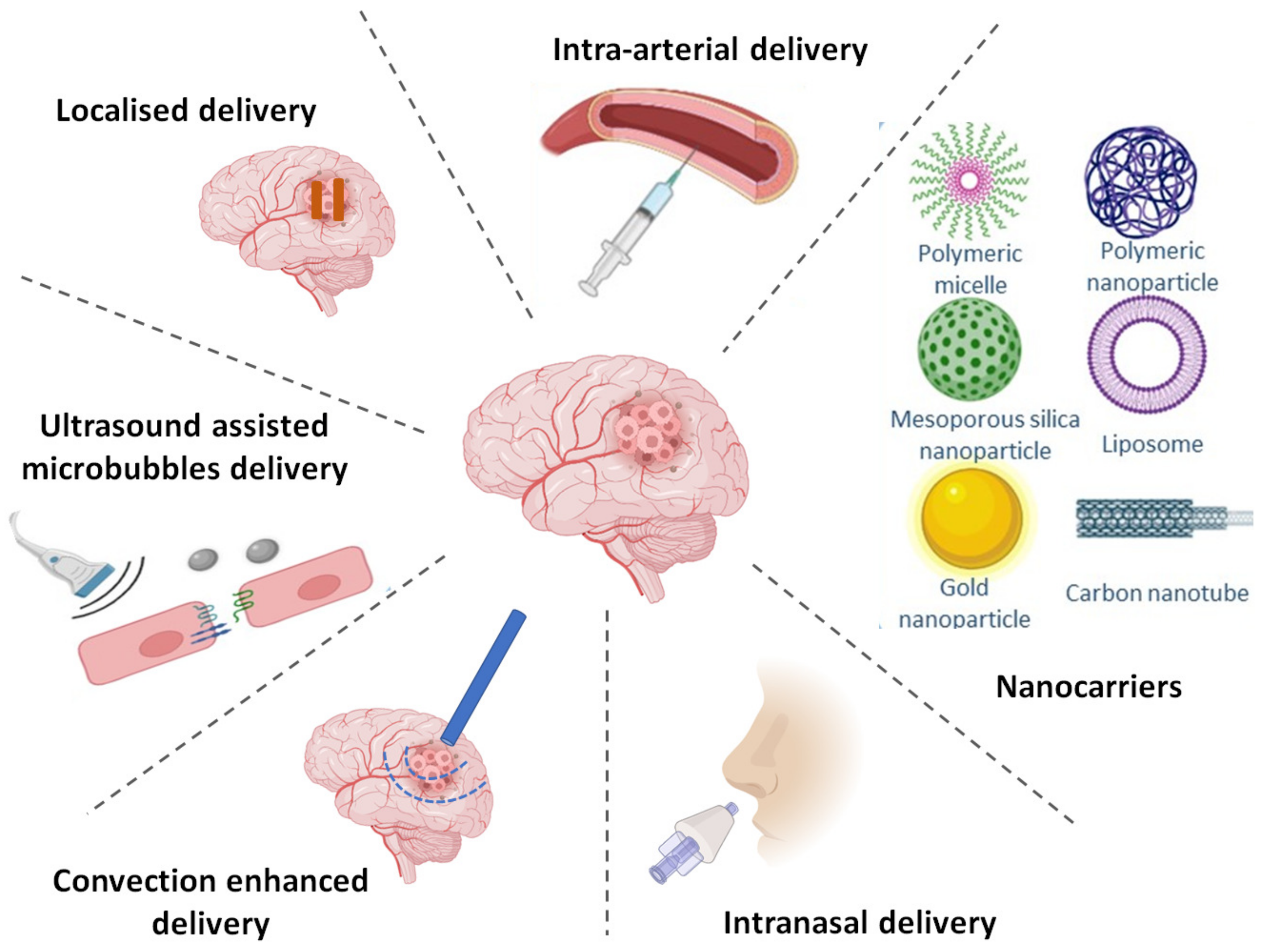
7. Strategies to Achieve Targeted Delivery of TKIs
7.1. Altering BBB Permeability
7.1.1. Chemical Disruption
7.1.2. Focused Ultrasound
7.1.3. Inhibition of Efflux Transporters
7.1.4. Chemical Modification of Molecules
7.1.5. Intra-Arterial Drug Delivery
7.2. Bypassing BBB via Alternate Routes
7.2.1. Direct Injection
7.2.2. Implantable Polymeric Systems
7.2.3. Convection Enhanced Delivery (CED)
7.2.4. Intranasal Delivery
7.3. BBB Crossing via Nanotechnology
7.3.1. Liposomes
7.3.2. Polymeric Nanoparticles
7.3.3. Polymeric Micelles
7.3.4. Albumin Nanoparticles
7.3.5. Inorganic Nanocarriers
7.3.6. Other Nanocarriers

{kind=link}
{kind=link}
{kind=link}
| Formulation | Drug/Targeting Ligand | Particle size | Cell line/Model Used | Outcomes/References |
|---|---|---|---|---|
| Actively targeted nanoparticles | ||||
| Ultra-small fluorescent core–shell silica NPs | Dasatinib; cyclic-arginine-glycine-aspartic acid peptide | 6–7 nm | TS543 neurosphere cultures Genetically engineered mouse model of glioblastoma |
|
| Polymeric NPs based on PLGA-b-PEG-COOH | Dactolisib; Gint4.T aptamer | - | U87MG GBM cells Nude mice bearing intracranial U87MG tumor xenografts |
|
| PEG-b-poly(ε-caprolactone-co-dithiolane trimethylene carbonate)-mefenamate micelles | Sorafenib; Apo-lipoprotein E peptide | 24 nm | U-87 MG cells U-87 MG-Luc tumor-bearing mice |
|
| PEGylated bilirubin NPs | Cediranib and paclitaxel; D-T7 peptide | Cediranib NPs: 71.5 nm Paclitaxel NPs: 77.2 nm | C6 cell line C6 glioma bearing mice |
|
| Liposomes composed of DOTAP, DOPE, Cholesterol and DSPE-PEG2000 | Doxorubicin and Erlotinib; Transferrin, cell penetrating peptide (PFVYLI) | 161.90 ± 4.60 nm | U87, bEnd.3 and glial cell lines |
|
| Passively targeted nanoparticles | ||||
| Lipid nanocapsules | Sorafenib | 54 ± 1 nm | U87MG cell line Orthotopic U87MG glioblastoma model |
|
| Poly(styrene-co-maleic acid) micelles | Crizotinib and Dasatinib | Crizotinib micelles: 121 nm Dasatinib micelles: 89.14 nm | Various cell lines; U87 and NZG1003 3D spheroids; Female C57BL/6 mice, inoculated with GL261 GBM tumor mass SC |
|
| Human serum albumin nanoparticles | Ibrutinib and Hydroxychloroquine | 160.1 ± 0.7 nm | C6-luc cells Orthotopic glioma xenograft developed by intracranial transplantation of C6-luc cells in mice |
|
8. Conclusions and Perspectives
Funding
Data Availability Statement
Conflicts of Interest
References
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G.S.; Lyutfi, E.; Georgieva, R.; Georgiev, R.; Dzhenkov, D.L.; Petkova, L.; Ivanov, B.D.; Kaprelyan, A.; Ghenev, P. Reclassification of Glioblastoma Multiforme According to the 2021 World Health Organization Classification of Central Nervous System Tumors: A Single Institution Report and Practical Significance. Cureus 2022, 14, e21822. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Castro, L.N.; Wesseling, P. The cIMPACT-NOW updates and their significance to current neuro-oncology practice. Neuro-Oncol. Pract. 2021, 8, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, P.; Arozarena, I. Tyrosine Kinase Inhibitors in Adult Glioblastoma: An (Un)Closed Chapter? Cancers 2021, 13, 5799. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Pazdur, R. Food and Drug Administration Drug approval summary: Temozolomide plus radiation therapy for the treatment of newly diagnosed glioblastoma multiforme. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6767–6771. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br. J. Cancer 2021, 124, 697–709. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Davis, M.E.; Elzinga, G.; Butowski, N.; Tran, D.; Villano, J.L.; DiMeglio, L.; Davies, A.M.; Wong, E.T. Characterization and management of dermatologic adverse events with the NovoTTF-100A System, a novel anti-mitotic electric field device for the treatment of recurrent glioblastoma. Semin. Oncol. 2014, 41 (Suppl 4), S1–S14. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.A.; Rauschkolb, P.K. Tumor treating fields for glioblastoma: Should it or will it ever be adopted? Curr. Opin. Neurol. 2019, 32, 857–863. [Google Scholar] [CrossRef]
- Foroughi-Nia, B.; Barar, J.; Memar, M.Y.; Aghanejad, A.; Davaran, S. Progresses in polymeric nanoparticles for delivery of tyrosine kinase inhibitors. Life Sci. 2021, 278, 119642. [Google Scholar] [CrossRef]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Hamada, J. Mechanism of chemoresistance against tyrosine kinase inhibitors in malignant glioma. Brain Tumor Pathol. 2014, 31, 198–207. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol.J Hematol Oncol 2020, 13, 143. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, E731. [Google Scholar] [CrossRef] [Green Version]
- Ahluwalia, M.S.; Becker, K.; Levy, B.P. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors for Central Nervous System Metastases from Non-Small Cell Lung Cancer. The Oncologist 2018, 23, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.S.; Muresanu, D.F.; Castellani, R.J.; Nozari, A.; Lafuente, J.V.; Tian, Z.R.; Sahib, S.; Bryukhovetskiy, I.; Bryukhovetskiy, A.; Buzoianu, A.D.; et al. Pathophysiology of blood-brain barrier in brain tumor. Novel therapeutic advances using nanomedicine. Int. Rev. Neurobiol. 2020, 151, 1–66. [Google Scholar] [CrossRef]
- Zhao, Y.; Bilal, M.; Raza, A.; Khan, M.I.; Mehmood, S.; Hayat, U.; Hassan, S.T.S.; Iqbal, H.M.N. Tyrosine kinase inhibitors and their unique therapeutic potentialities to combat cancer. Int. J. Biol. Macromol. 2021, 168, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Degl’Innocenti, A.; di Leo, N.; Ciofani, G. Genetic Hallmarks and Heterogeneity of Glioblastoma in the Single-Cell Omics Era. Adv. Ther. 2020, 3, 1900152. [Google Scholar] [CrossRef] [PubMed]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, D.; Mikkelsen, T.; Schwechheimer, K.; Rosenblum, M.L.; Cavanee, W.; Vogelstein, B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992, 355, 846–847. [Google Scholar] [CrossRef] [PubMed]
- O’Campo, J.; Uyehara-Lock, J.; Aldape, K.D.; Burton, E.C.; Lamborn, K.R.; Prados, M.; Berger, M.; Forsyth, P.; Scott, J.; Passe, S.; et al. Aberrant p53, mdm2, and proliferation differ in glioblastomas from long-term compared with typical survivors. Clin. Cancer Res. 2002, 8, 180–187. [Google Scholar]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.-J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Kloosterhof, N.K.; Bralten, L.B.C.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 Mutations Are Early Events in the Development of Astrocytomas and Oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Krell, D.; Assoku, M.; Galloway, M.; Mulholland, P.; Tomlinson, I.; Bardella, C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH mutations in glioblastoma. PloS ONE 2011, 6, e19868. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Wang, J.Y.J. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1094–1099. [Google Scholar] [CrossRef]
- Chkheidze, R.; Raisanen, J.; Gagan, J.; Richardson, T.E.; Pinho, M.C.; Raj, K.; Achilleos, M.; Slepicka, C.; White, C.L.; Evers, B.M.; et al. Alterations in the RB Pathway With Inactivation of RB1 Characterize Glioblastomas With a Primitive Neuronal Component. J. Neuropathol. Exp. Neurol. 2021, 80, 1092–1098. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Lu, V.M.; Akinduro, O.O.; Daniels, D.J. H3K27M mutation in adult cerebellar glioblastoma. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2020, 71, 316–317. [Google Scholar] [CrossRef]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor-tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Maire, C.L.; Ligon, K.L. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro-Oncology 2014, 16 (Suppl. 8), viii1–viii6. [Google Scholar] [CrossRef]
- Yamazaki, H.; Ohba, Y.; Tamaoki, N.; Shibuya, M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jpn. J. Cancer Res. Gann 1990, 81, 773–779. [Google Scholar] [CrossRef]
- Daneshimehr, F.; Barabadi, Z.; Abdolahi, S.; Soleimani, M.; Verdi, J.; Ebrahimi-Barough, S.; Ai, J. Angiogenesis and Its Targeting in Glioblastoma with Focus on Clinical Approaches. Cell J. 2022, 24, 555–568. [Google Scholar] [CrossRef]
- Ahmad, A.; Nawaz, M.I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J. Cell. Biochem. 2022, 132, 1938–1965. [Google Scholar] [CrossRef]
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker? Ups. J. Med. Sci. 2014, 119, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.; Cilibrasi, C.; Chen, J.; Shah, K.; Messuti, E.; Mazarakis, N.K.; Stebbing, J.; Critchley, G.; Song, E.; Simon, T.; et al. PDGF-R inhibition induces glioblastoma cell differentiation via DUSP1/p38MAPK signalling. Oncogene 2022, 41, 2749–2763. [Google Scholar] [CrossRef]
- Zhang, Y.; Farenholtz, K.E.; Yang, Y.; Guessous, F.; diPierro, C.G.; Calvert, V.S.; Deng, J.; Schiff, D.; Xin, W.; Lee, J.K.; et al. Hepatocyte growth factor sensitizes brain tumors to c-MET kinase inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1433–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Rhun, E.; Chamberlain, M.C.; Zairi, F.; Delmaire, C.; Idbaih, A.; Renaud, F.; Maurage, C.A.; Grégoire, V. Patterns of response to crizotinib in recurrent glioblastoma according to ALK and MET molecular profile in two patients. CNS Oncol. 2015, 4, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.S.; Gross, J.L.; Herblin, W.F.; Reilly, T.M.; LaSala, P.A.; Alterman, R.L.; Moskal, J.R.; Kornblith, P.L.; Dexter, D.L. Basic fibroblast growth factor-like activity and receptors are expressed in a human glioma cell line. Cancer Res. 1990, 50, 2524–2529. [Google Scholar]
- Loilome, W.; Joshi, A.D.; ap Rhys, C.M.J.; Piccirillo, S.; Vescovi, A.L.; Angelo, V.L.; Gallia, G.L.; Riggins, G.J. Glioblastoma cell growth is suppressed by disruption of Fibroblast Growth Factor pathway signaling. J. Neurooncol. 2009, 94, 359–366. [Google Scholar] [CrossRef]
- Tirrò, E.; Massimino, M.; Romano, C.; Martorana, F.; Pennisi, M.S.; Stella, S.; Pavone, G.; Di Gregorio, S.; Puma, A.; Tomarchio, C.; et al. Prognostic and Therapeutic Roles of the Insulin Growth Factor System in Glioblastoma. Front. Oncol. 2020, 10, 612385. [Google Scholar] [CrossRef]
- Maris, C.; D’Haene, N.; Trépant, A.-L.; Le Mercier, M.; Sauvage, S.; Allard, J.; Rorive, S.; Demetter, P.; Decaestecker, C.; Salmon, I. IGF-IR: A new prognostic biomarker for human glioblastoma. Br. J. Cancer 2015, 113, 729–737. [Google Scholar] [CrossRef]
- Lebedev, T.D.; Vagapova, E.R.; Astashkova, O.O.; Spirin, P.V.; Prassolov, V.S. Inhibition of Non-Receptor Tyrosine Kinase JAK2 Reduces Neuroblastoma Cell Growth and Enhances the Action of Doxorubicin. Mol. Biol. (Mosk.) 2020, 54, 293–299. [Google Scholar] [CrossRef]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC Kinase in Glioblastoma News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef]
- Alphandéry, E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Cismowski, M.J. Tyrosine Kinase Inhibitors. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. ISBN 978-0-08-055232-3. [Google Scholar]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Oertel, S.; Krempien, R.; Lindel, K.; Zabel, A.; Milker-Zabel, S.; Bischof, M.; Lipson, K.E.; Peschke, P.; Debus, J.; Abdollahi, A.; et al. Human glioblastoma and carcinoma xenograft tumors treated by combined radiation and imatinib (Gleevec). Strahlenther. Onkol. Organ Dtsch. Rontgengesellschaft Al 2006, 182, 400–407. [Google Scholar] [CrossRef]
- Schneider, H.; Szabo, E.; Machado, R.A.C.; Broggini-Tenzer, A.; Walter, A.; Lobell, M.; Heldmann, D.; Süssmeier, F.; Grünewald, S.; Weller, M. Novel TIE-2 inhibitor BAY-826 displays in vivo efficacy in experimental syngeneic murine glioma models. J. Neurochem. 2017, 140, 170–182. [Google Scholar] [CrossRef]
- Piao, Y.; Park, S.Y.; Henry, V.; Smith, B.D.; Tiao, N.; Flynn, D.L.; de Groot, J.F. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neuro-Oncology 2016, 18, 1230–1241. [Google Scholar] [CrossRef] [Green Version]
- Ballard, P.; Yates, J.W.T.; Yang, Z.; Kim, D.-W.; Yang, J.C.-H.; Cantarini, M.; Pickup, K.; Jordan, A.; Hickey, M.; Grist, M.; et al. Preclinical Comparison of Osimertinib with Other EGFR-TKIs in EGFR-Mutant NSCLC Brain Metastases Models, and Early Evidence of Clinical Brain Metastases Activity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. CR 2019, 38, 219. [Google Scholar] [CrossRef]
- Yun, J.; Hong, M.H.; Kim, S.-Y.; Park, C.-W.; Kim, S.; Yun, M.R.; Kang, H.N.; Pyo, K.-H.; Lee, S.S.; Koh, J.S.; et al. YH25448, an Irreversible EGFR-TKI with Potent Intracranial Activity in EGFR Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2575–2587. [Google Scholar] [CrossRef] [Green Version]
- Stea, B.; Falsey, R.; Kislin, K.; Patel, J.; Glanzberg, H.; Carey, S.; Ambrad, A.A.; Meuillet, E.J.; Martinez, J.D. Time and dose-dependent radiosensitization of the glioblastoma multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor ZD1839 (‘Iressa’). Cancer Lett. 2003, 202, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Vengoji, R.; Macha, M.A.; Nimmakayala, R.K.; Rachagani, S.; Siddiqui, J.A.; Mallya, K.; Gorantla, S.; Jain, M.; Ponnusamy, M.P.; Batra, S.K.; et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J. Exp. Clin. Cancer Res. CR 2019, 38, 266. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Fang, D.-M.; Zhou, X.-L.; Gao, F. Natural products as important tyrosine kinase inhibitors. Eur. J. Med. Chem. 2019, 182, 111664. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bailey, C.P.; Sadighi, Z.; Zaky, W.; Chandra, J. Pediatric high-grade glioma: Aberrant epigenetics and kinase signaling define emerging therapeutic opportunities. J. Neurooncol. 2020, 150, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Patel, M.; Peereboom, D.M. Role of tyrosine kinase inhibitors in the management of high-grade gliomas. Expert Rev. Anticancer Ther. 2011, 11, 1739–1748. [Google Scholar] [CrossRef]
- Marona, P.; Górka, J.; Kwapisz, O.; Jura, J.; Rys, J.; Hoffman, R.M.; Miekus, K. Resistance to tyrosine kinase inhibitors promotes renal cancer progression through MCPIP1 tumor-suppressor downregulation and c-Met activation. Cell Death Dis. 2022, 13, 814. [Google Scholar] [CrossRef]
- Abshire, C.; Murad, H.Y.; Arora, J.S.; Liu, J.; Mandava, S.H.; John, V.T.; Khismatullin, D.B.; Lee, B.R. Focused Ultrasound-Triggered Release of Tyrosine Kinase Inhibitor From Thermosensitive Liposomes for Treatment of Renal Cell Carcinoma. J. Pharm. Sci. 2017, 106, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Portnow, J.; Badie, B.; Markel, S.; Liu, A.; D’Apuzzo, M.; Frankel, P.; Jandial, R.; Synold, T.W. A neuropharmacokinetic assessment of bafetinib, a second generation dual BCR-Abl/Lyn tyrosine kinase inhibitor, in patients with recurrent high-grade gliomas. Eur. J. Cancer Oxf. Engl. 1990 2013, 49, 1634–1640. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.; Fiorelli, R.; Bao, X.; Pennington-Krygier, C.; Derogatis, A.; Kim, S.; Yoo, W.; Li, J.; Sanai, N. A Phase 0 Trial of Ceritinib in Patients with Brain Metastases and Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 289–297. [Google Scholar] [CrossRef]
- Agarwal, S.; Sane, R.; Gallardo, J.L.; Ohlfest, J.R.; Elmquist, W.F. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J. Pharmacol. Exp. Ther. 2010, 334, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Kort, A.; Durmus, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Brain and Testis Accumulation of Regorafenib is Restricted by Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm. Res. 2015, 32, 2205–2216. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Bruin, M.A.C.; Gan, C.; Lebre, M.C.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of tivozanib is restricted by ABCB1 (P-glycoprotein) and ABCG2 (breast cancer resistance protein) in mice. Int. J. Pharm. 2020, 581, 119277. [Google Scholar] [CrossRef]
- Salmaso, S.; Mastrotto, F.; Roverso, M.; Gandin, V.; De Martin, S.; Gabbia, D.; De Franco, M.; Vaccarin, C.; Verona, M.; Chilin, A.; et al. Tyrosine kinase inhibitor prodrug-loaded liposomes for controlled release at tumor microenvironment. J. Control. Release Off. J. Control. Release Soc. 2021, 340, 318–330. [Google Scholar] [CrossRef]
- Gandin, V.; Ferrarese, A.; Dalla Via, M.; Marzano, C.; Chilin, A.; Marzaro, G. Targeting kinases with anilinopyrimidines: Discovery of N-phenyl-N’-[4-(pyrimidin-4-ylamino)phenyl]urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily. Sci. Rep. 2015, 5, 16750. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, T.T.; Gerstner, E.R.; Ye, X.; Desideri, S.; Duda, D.G.; Peereboom, D.; Lesser, G.J.; Chowdhary, S.; Wen, P.Y.; Grossman, S.; et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-β tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro-Oncology 2017, 19, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, 3326. [Google Scholar] [CrossRef] [Green Version]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef]
- Brown, N.; McBain, C.; Nash, S.; Hopkins, K.; Sanghera, P.; Saran, F.; Phillips, M.; Dungey, F.; Clifton-Hadley, L.; Wanek, K.; et al. Multi-Center Randomized Phase II Study Comparing Cediranib plus Gefitinib with Cediranib plus Placebo in Subjects with Recurrent/Progressive Glioblastoma. PloS ONE 2016, 11, e0156369. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, C.R.; Rath, P.; Oyinlade, O.; Lopez, H.; Mughal, S.; Xia, S.; Li, Y.; Kaur, H.; Zhou, X.; Ahmed, A.K.; et al. Crizotinib and erlotinib inhibits growth of c-Met+/EGFRvIII+ primary human glioblastoma xenografts. Clin. Neurol. Neurosurg. 2018, 171, 26–33. [Google Scholar] [CrossRef]
- Sautter, L.; Hofheinz, R.; Tuettenberg, J.; Grimm, M.; Vajkoczy, P.; Groden, C.; Schmieder, K.; Hochhaus, A.; Wenz, F.; Giordano, F.A. Open-Label Phase II Evaluation of Imatinib in Primary Inoperable or Incompletely Resected and Recurrent Glioblastoma. Oncology 2020, 98, 16–22. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Papadantonakis, N.; Alva Venur, V.; Schilero, C.; Peereboom, D.M.; Stevens, G.; Rosenfeld, S.; Vogelbaum, M.A.; Elson, P.; Nixon, A.B.; et al. Phase II trial of dovitinib in recurrent glioblastoma. J. Clin. Oncol. 2015, 33, 2050. [Google Scholar] [CrossRef]
- Hutterer, M.; Nowosielski, M.; Haybaeck, J.; Embacher, S.; Stockhammer, F.; Gotwald, T.; Holzner, B.; Capper, D.; Preusser, M.; Marosi, C.; et al. A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (SURGE 01-07). Neuro-Oncology 2014, 16, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, A.D.; Schiff, D.; Ahluwalia, M.S.; Lesser, G.J.; Nayak, L.; Lee, E.Q.; Rinne, M.L.; Muzikansky, A.; Dietrich, J.; Purow, B.; et al. Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J. Neurooncol. 2015, 121, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Holland, J.; et al. Phase II study of cabozantinib in patients with progressive glioblastoma: Subset analysis of patients naive to antiangiogenic therapy. Neuro-Oncology 2018, 20, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchelor, T.T.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Plotkin, S.R.; Gerstner, E.; Eichler, A.F.; Drappatz, J.; Hochberg, F.H.; Benner, T.; et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2817. [Google Scholar] [CrossRef]
- Yung, W.K.A.; Vredenburgh, J.J.; Cloughesy, T.F.; Nghiemphu, P.; Klencke, B.; Gilbert, M.R.; Reardon, D.A.; Prados, M.D. Safety and efficacy of erlotinib in first-relapse glioblastoma: A phase II open-label study. Neuro-Oncology 2010, 12, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, F.M.; Lamborn, K.R.; Robins, H.I.; Mehta, M.P.; Chang, S.M.; Butowski, N.A.; Deangelis, L.M.; Abrey, L.E.; Zhang, W.-T.; Prados, M.D.; et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro-Oncology 2010, 12, 855–861. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef] [Green Version]
- Sepúlveda-Sánchez, J.M.; Vaz, M.Á.; Balañá, C.; Gil-Gil, M.; Reynés, G.; Gallego, Ó.; Martínez-García, M.; Vicente, E.; Quindós, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro-Oncology 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2015, 17, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II Study of Bevacizumab in Combination with Sorafenib in Recurrent Glioblastoma (N0776): A North Central Cancer Treatment Group Trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef]
- Duerinck, J.; Du Four, S.; Vandervorst, F.; D’Haene, N.; Le Mercier, M.; Michotte, A.; Van Binst, A.M.; Everaert, H.; Salmon, I.; Bouttens, F.; et al. Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. J. Neurooncol. 2016, 128, 147–155. [Google Scholar] [CrossRef]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.-M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J. Immunother. Cancer 2020, 8, e001146. [Google Scholar] [CrossRef]
- Lee, E.Q.; Kaley, T.J.; Duda, D.G.; Schiff, D.; Lassman, A.B.; Wong, E.T.; Mikkelsen, T.; Purow, B.W.; Muzikansky, A.; Ancukiewicz, M.; et al. A Multicenter, Phase II, Randomized, Noncomparative Clinical Trial of Radiation and Temozolomide with or without Vandetanib in Newly Diagnosed Glioblastoma Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3610–3618. [Google Scholar] [CrossRef] [Green Version]
- La Madrid, A.M.; Kieran, M.W. Epigenetics in Clinical Management of Children and Adolescents with Brain Tumors. Curr. Cancer Drug Targets 2018, 18, 57–64. [Google Scholar] [CrossRef]
- Pötschke, R.; Gielen, G.; Pietsch, T.; Kramm, C.; Klusmann, J.-H.; Hüttelmaier, S.; Kühnöl, C.D. Musashi1 enhances chemotherapy resistance of pediatric glioblastoma cells in vitro. Pediatr. Res. 2020, 87, 669–676. [Google Scholar] [CrossRef]
- Glod, J.; Rahme, G.J.; Kaur, H.; Raabe, E.H.; Hwang, E.I.; Israel, M.A. Pediatric Brain Tumors: Current Knowledge and Therapeutic Opportunities. J. Pediatr. Hematol. Oncol. 2016, 38, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Fangusaro, J.; Warren, K.E. Unclear standard of care for pediatric high grade glioma patients. J. Neurooncol. 2013, 113, 341–342. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef] [Green Version]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Fan, W.; Lau, J.; Deng, L.; Shen, Z.; Chen, X. Emerging blood-brain-barrier-crossing nanotechnology for brain cancer theranostics. Chem. Soc. Rev. 2019, 48, 2967–3014. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, E.; Choi, P.J.; Denny, W.A.; Jose, J.; Dragunow, M.; Park, T.I.-H. The Use of Heptamethine Cyanine Dyes as Drug-Conjugate Systems in the Treatment of Primary and Metastatic Brain Tumors. Front. Oncol. 2021, 11, 654921. [Google Scholar] [CrossRef]
- Zeng, L.; Zou, L.; Yu, H.; He, X.; Cao, H.; Zhang, Z.; Yin, Q.; Zhang, P.; Gu, W.; Chen, L.; et al. Treatment of Malignant Brain Tumor by Tumor-Triggered Programmed Wormlike Micelles with Precise Targeting and Deep Penetration. Adv. Funct. Mater. 2016, 26, 4201–4212. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- El Kheir, W.; Marcos, B.; Virgilio, N.; Paquette, B.; Faucheux, N.; Lauzon, M.-A. Drug Delivery Systems in the Development of Novel Strategies for Glioblastoma Treatment. Pharmaceutics 2022, 14, 1189. [Google Scholar] [CrossRef]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood-Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef]
- Ningaraj, N.S.; Rao, M.K.; Black, K.L. Adenosine 5′-triphosphate-sensitive Potassium Channel-mediated Blood-Brain Tumor Barrier Permeability Increase in a Rat Brain Tumor Model. Cancer Res 2003, 63, 8899–8911. [Google Scholar]
- Ningaraj, N.S.; Rao, M.; Black, K.L. Calcium-dependent potassium channels as a target protein for modulation of the blood-brain tumor barrier. Drug News Perspect. 2003, 16, 291–298. [Google Scholar] [CrossRef]
- Kast, R.E.; Focosi, D. Three paths to better tyrosine kinase inhibition behind the blood-brain barrier in treating chronic myelogenous leukemia and glioblastoma with imatinib. Transl. Oncol. 2010, 3, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-L.; Fan, C.-H.; Ting, C.-Y.; Yeh, C.-K. Combining microbubbles and ultrasound for drug delivery to brain tumors: Current progress and overview. Theranostics 2014, 4, 432–444. [Google Scholar] [CrossRef]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef]
- Goutal, S.; Gerstenmayer, M.; Auvity, S.; Caillé, F.; Mériaux, S.; Buvat, I.; Larrat, B.; Tournier, N. Physical blood-brain barrier disruption induced by focused ultrasound does not overcome the transporter-mediated efflux of erlotinib. J. Control. Release Off. J. Control. Release Soc. 2018, 292, 210–220. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Abate, C.; Rolando, B.; Battaglia, L.; Gazzano, E.; Colombino, E.; Costamagna, C.; Annovazzi, L.; Mellai, M.; Berardi, F.; et al. Validation of Thiosemicarbazone Compounds as P-Glycoprotein Inhibitors in Human Primary Brain-Blood Barrier and Glioblastoma Stem Cells. Mol. Pharm. 2019, 16, 3361–3373. [Google Scholar] [CrossRef]
- Lemos, C.; Jansen, G.; Peters, G.J. Drug transporters: Recent advances concerning BCRP and tyrosine kinase inhibitors. Br. J. Cancer 2008, 98, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Krchniakova, M.; Skoda, J.; Neradil, J.; Chlapek, P.; Veselska, R. Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. Int. J. Mol. Sci. 2020, 21, 3157. [Google Scholar] [CrossRef]
- Rice, A.; Liu, Y.; Michaelis, M.L.; Himes, R.H.; Georg, G.I.; Audus, K.L. Chemical modification of paclitaxel (Taxol) reduces P-glycoprotein interactions and increases permeation across the blood-brain barrier in vitro and in situ. J. Med. Chem. 2005, 48, 832–838. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.J.; Park, T.I.; Cooper, E.; Dragunow, M.; Denny, W.A.; Jose, J. Heptamethine Cyanine Dye Mediated Drug Delivery: Hype or Hope. Bioconjug. Chem. 2020, 31, 1724–1739. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.J.; Cooper, E.; Schweder, P.; Mee, E.; Faull, R.; Denny, W.A.; Dragunow, M.; Park, T.I.-H.; Jose, J. The synthesis of a novel Crizotinib heptamethine cyanine dye conjugate that potentiates the cytostatic and cytotoxic effects of Crizotinib in patient-derived glioblastoma cell lines. Bioorg. Med. Chem. Lett. 2019, 29, 2617–2621. [Google Scholar] [CrossRef] [PubMed]
- Haumann, R.; Videira, J.C.; Kaspers, G.J.L.; van Vuurden, D.G.; Hulleman, E. Overview of Current Drug Delivery Methods Across the Blood-Brain Barrier for the Treatment of Primary Brain Tumors. CNS Drugs 2020, 34, 1121–1131. [Google Scholar] [CrossRef]
- Yu, F.; Asghar, S.; Zhang, M.; Zhang, J.; Ping, Q.; Xiao, Y. Local strategies and delivery systems for the treatment of malignant gliomas. J. Drug Target. 2019, 27, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Bastiancich, C.; Danhier, P.; Préat, V.; Danhier, F. Anticancer drug-loaded hydrogels as drug delivery systems for the local treatment of glioblastoma. J. Control. Release Off. J. Control. Release Soc. 2016, 243, 29–42. [Google Scholar] [CrossRef]
- Attenello, F.J.; Mukherjee, D.; Datoo, G.; McGirt, M.J.; Bohan, E.; Weingart, J.D.; Olivi, A.; Quinones-Hinojosa, A.; Brem, H. Use of Gliadel (BCNU) wafer in the surgical treatment of malignant glioma: A 10-year institutional experience. Ann. Surg. Oncol. 2008, 15, 2887–2893. [Google Scholar] [CrossRef]
- Sabel, M.; Giese, A. Safety profile of carmustine wafers in malignant glioma: A review of controlled trials and a decade of clinical experience. Curr. Med. Res. Opin. 2008, 24, 3239–3257. [Google Scholar] [CrossRef]
- Bastiancich, C.; Malfanti, A.; Préat, V.; Rahman, R. Rationally designed drug delivery systems for the local treatment of resected glioblastoma. Adv. Drug Deliv. Rev. 2021, 177, 113951. [Google Scholar] [CrossRef]
- McCrorie, P.; Mistry, J.; Taresco, V.; Lovato, T.; Fay, M.; Ward, I.; Ritchie, A.A.; Clarke, P.A.; Smith, S.J.; Marlow, M.; et al. Etoposide and olaparib polymer-coated nanoparticles within a bioadhesive sprayable hydrogel for post-surgical localised delivery to brain tumours. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 2020, 157, 108–120. [Google Scholar] [CrossRef]
- Tosi, U.; Kommidi, H.; Bellat, V.; Marnell, C.S.; Guo, H.; Adeuyan, O.; Schweitzer, M.E.; Chen, N.; Su, T.; Zhang, G.; et al. Real-Time, in Vivo Correlation of Molecular Structure with Drug Distribution in the Brain Striatum Following Convection Enhanced Delivery. ACS Chem. Neurosci. 2019, 10, 2287–2298. [Google Scholar] [CrossRef]
- Souweidane, M.M.; Kramer, K.; Pandit-Taskar, N.; Zhou, Z.; Haque, S.; Zanzonico, P.; Carrasquillo, J.A.; Lyashchenko, S.K.; Thakur, S.B.; Donzelli, M.; et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: A single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018, 19, 1040–1050. [Google Scholar] [CrossRef]
- Tosi, U.; Souweidane, M. Convection Enhanced Delivery for Diffuse Intrinsic Pontine Glioma: Review of a Single Institution Experience. Pharmaceutics 2020, 12, 660. [Google Scholar] [CrossRef]
- D’Amico, R.S.; Aghi, M.K.; Vogelbaum, M.A.; Bruce, J.N. Convection-enhanced drug delivery for glioblastoma: A review. J. Neurooncol. 2021, 151, 415–427. [Google Scholar] [CrossRef]
- Bruinsmann, F.A.; Richter Vaz, G.; de Cristo Soares Alves, A.; Aguirre, T.; Raffin Pohlmann, A.; Stanisçuaski Guterres, S.; Sonvico, F. Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules 2019, 24, 4312. [Google Scholar] [CrossRef] [Green Version]
- Upadhaya, P.G.; Pulakkat, S.; Patravale, V.B. Nose-to-brain delivery: Exploring newer domains for glioblastoma multiforme management. Drug Deliv. Transl. Res. 2020, 10, 1044–1056. [Google Scholar] [CrossRef]
- Ferraris, C.; Cavalli, R.; Panciani, P.P.; Battaglia, L. Overcoming the Blood–Brain Barrier: Successes and Challenges in Developing Nanoparticle-Mediated Drug Delivery Systems for the Treatment of Brain Tumours. Int. J. Nanomedicine 2020, 15, 2999–3022. [Google Scholar] [CrossRef]
- Moradpour, Z.; Barghi, L. Novel Approaches for Efficient Delivery of Tyrosine Kinase Inhibitors. J. Pharm. Pharm. Sci. Publ. Can. Soc. Pharm. Sci. Soc. Can. Sci. Pharm. 2019, 22, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Lou, B.; van Steenbergen, M.J.; Sijbrandi, N.J.; Hennink, W.E.; Kok, R.J. Polymeric Micelles Employing Platinum(II) Linker for the Delivery of the Kinase Inhibitor Dactolisib. Part. Part. Syst. Charact. 2019, 36, 1900236. [Google Scholar] [CrossRef]
- Mojarad-Jabali, S.; Farshbaf, M.; Walker, P.R.; Hemmati, S.; Fatahi, Y.; Zakeri-Milani, P.; Sarfraz, M.; Valizadeh, H. An update on actively targeted liposomes in advanced drug delivery to glioma. Int. J. Pharm. 2021, 602, 120645. [Google Scholar] [CrossRef]
- Pandey, N.; Anastasiadis, P.; Carney, C.P.; Kanvinde, P.P.; Woodworth, G.F.; Winkles, J.A.; Kim, A.J. Nanotherapeutic treatment of the invasive glioblastoma tumor microenvironment. Adv. Drug Deliv. Rev. 2022, 188, 114415. [Google Scholar] [CrossRef] [PubMed]
- Ganipineni, L.P.; Danhier, F.; Préat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release Off. J. Control. Release Soc. 2018, 281, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomedicine 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Lakkadwala, S.; Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf. B Biointerfaces 2019, 173, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Wang, M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer 2013, 13, 267. [Google Scholar] [CrossRef] [Green Version]
- Benezra, M.; Hambardzumyan, D.; Penate-Medina, O.; Veach, D.R.; Pillarsetty, N.; Smith-Jones, P.; Phillips, E.; Ozawa, T.; Zanzonico, P.B.; Longo, V.; et al. Fluorine-labeled dasatinib nanoformulations as targeted molecular imaging probes in a PDGFB-driven murine glioblastoma model. Neoplasia 2012, 14, 1132–1143. [Google Scholar] [CrossRef] [Green Version]
- Rehman, U.; Parveen, N.; Sheikh, A.; Abourehab, M.A.S.; Sahebkar, A.; Kesharwani, P. Polymeric nanoparticles-siRNA as an emerging nano-polyplexes against ovarian cancer. Colloids Surf. B Biointerfaces 2022, 218, 112766. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Khan, A.M.; Ahmad, F.J.; Panda, A.K.; Talegaonkar, S. Investigation of imatinib loaded surface decorated biodegradable nanocarriers against glioblastoma cell lines: Intracellular uptake and cytotoxicity studies. Int. J. Pharm. 2016, 507, 61–71. [Google Scholar] [CrossRef]
- Monaco, I.; Camorani, S.; Colecchia, D.; Locatelli, E.; Calandro, P.; Oudin, A.; Niclou, S.; Arra, C.; Chiariello, M.; Cerchia, L.; et al. Aptamer Functionalization of Nanosystems for Glioblastoma Targeting through the Blood-Brain Barrier. J. Med. Chem. 2017, 60, 4510–4516. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov. Today 2003, 8, 1112–1120. [Google Scholar] [CrossRef]
- Xu, W.; Ye, C.; Qing, X.; Liu, S.; Lv, X.; Wang, W.; Dong, X.; Zhang, Y. Multi-target tyrosine kinase inhibitor nanoparticle delivery systems for cancer therapy. Mater. Today Bio 2022, 16, 100358. [Google Scholar] [CrossRef]
- Wei, J.; Xia, Y.; Meng, F.; Ni, D.; Qiu, X.; Zhong, Z. Small, Smart, and LDLR-Specific Micelles Augment Sorafenib Therapy of Glioblastoma. Biomacromolecules 2021, 22, 4814–4822. [Google Scholar] [CrossRef]
- Nehoff, H.; Parayath, N.N.; McConnell, M.J.; Taurin, S.; Greish, K. A combination of tyrosine kinase inhibitors, crizotinib and dasatinib for the treatment of glioblastoma multiforme. Oncotarget 2015, 6, 37948–37964. [Google Scholar] [CrossRef] [Green Version]
- Greish, K.; Jasim, A.; Parayath, N.; Abdelghany, S.; Alkhateeb, A.; Taurin, S.; Nehoff, H. Micellar formulations of Crizotinib and Dasatinib in the management of glioblastoma multiforme. J. Drug Target. 2018, 26, 692–708. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release Off. J. Control. Release Soc. 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Yang, Z.; Du, Y.; Lei, L.; Xia, X.; Wang, X.; Tong, F.; Li, Y.; Gao, H. Co-delivery of ibrutinib and hydroxychloroquine by albumin nanoparticles for enhanced chemotherapy of glioma. Int. J. Pharm. 2023, 630, 122436. [Google Scholar] [CrossRef]
- Zhou, X.; Shi, K.; Hao, Y.; Yang, C.; Zha, R.; Yi, C.; Qian, Z. Advances in nanotechnology-based delivery systems for EGFR tyrosine kinases inhibitors in cancer therapy. Asian J. Pharm. Sci. 2020, 15, 26–41. [Google Scholar] [CrossRef]
- Juthani, R.; Madajewski, B.; Yoo, B.; Zhang, L.; Chen, P.-M.; Chen, F.; Turker, M.Z.; Ma, K.; Overholtzer, M.; Longo, V.A.; et al. Ultrasmall Core-Shell Silica Nanoparticles for Precision Drug Delivery in a High-Grade Malignant Brain Tumor Model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Poland, C.A.; Duffin, R.; Kinloch, I.; Maynard, A.; Wallace, W.A.H.; Seaton, A.; Stone, V.; Brown, S.; MacNee, W.; Donaldson, K. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat. Nanotechnol. 2008, 3, 423–428. [Google Scholar] [CrossRef]
- Moore, T.L.; Grimes, S.W.; Lewis, R.L.; Alexis, F. Multilayered polymer-coated carbon nanotubes to deliver dasatinib. Mol. Pharm. 2014, 11, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliou, B.; Thomas, O.; Lautram, N.; Clavreul, A.; Hureaux, J.; Urban, T.; Benoit, J.-P.; Lagarce, F. Development and in vitro evaluation of a novel lipid nanocapsule formulation of etoposide. Eur. J. Pharm. Sci. 2013, 50, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavreul, A.; Roger, E.; Pourbaghi-Masouleh, M.; Lemaire, L.; Tétaud, C.; Menei, P. Development and characterization of sorafenib-loaded lipid nanocapsules for the treatment of glioblastoma. Drug Deliv. 2018, 25, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Su, D.; Yang, Y.; Qin, L.; Hu, C.; Liu, R.; Zhou, Y.; Yang, C.; Yang, X.; Wang, G.; et al. D-T7 Peptide-Modified PEGylated Bilirubin Nanoparticles Loaded with Cediranib and Paclitaxel for Antiangiogenesis and Chemotherapy of Glioma. ACS Appl. Mater. Interfaces 2019, 11, 176–186. [Google Scholar] [CrossRef]
- Power, E.A.; Rechberger, J.S.; Gupta, S.; Schwartz, J.D.; Daniels, D.J.; Khatua, S. Drug delivery across the blood-brain barrier for the treatment of pediatric brain tumors—An update. Adv. Drug Deliv. Rev. 2022, 185, 114303. [Google Scholar] [CrossRef]



| Drug (Dosing) | Clinical Indication and Target of TKI | Clinical Trial | Outcomes of Trial (References) |
|---|---|---|---|
| Imatinib (600 mg/day) | Chronic myeloid leukemia, gastrointestinal tumor Bcr-Abl, KIT, PDGFR | Phase II trial for primary inoperable or incompletely resected and recurrent GBM |
|
| Imatinib mesylate (oral dose of 600 mg/day) in combination with hydroxyurea (oral dose of 500 mg twice daily) vs. hydroxyurea alone (500 mg 3 times daily) | - | Phase III study in patients with temozolomide resistant progressive glioblastoma |
|
| Gefitinib (initial oral dose of 500 mg/day, escalated to 750 mg and then 1000 mg in case patient received enzyme-inducing drugs or dexamethasone) | Non-small cell lung cancer EGFR | Phase II trial for GBM at first recurrence |
|
| Cediranib (oral dose of 30 mg/day) and gefitinib (oral dose of 500 mg/day) vs. cediranib and placebo | - | Phase II trial for recurrent or progressive GBM |
|
| Dovitinib (oral dose of 500 mg/day for 5 days, 2 days off weekly on a 28-day cycle) | FGFR, VEGFR, PDGFR | Phase II trial for relapsed or progressive GBM |
|
| Sunitinib (oral dose of 37.5 mg/day to start, escalation to 50 mg/day or reduction to 25 or 12.5 mg/day depending on the toxicities) | Gastrointestinal tumor, renal cell carcinoma, pancreatic neuroendocrine tumor KIT, PDGFR, VEGFR1-2, FLT3 | Phase II trial for first recurrence of primary GBM |
|
| Nintedanib (oral dose of 200 mg twice a day) | PDGFR, FGFR, VEGFR | Phase II trial for recurrent high-grade gliomas |
|
| Cabozantinib (Starting oral dose of 140 mg/day considered to be high and then reduced to 100 mg/day) | Progressive metastatic medullary thyroid cancer, renal cell carcinoma, hepatocellular carcinoma previously treated with sorafenib VEGFR1-2, Met, ROS1, RET, AXL, NTRK, KIT | Phase II trial for recurrent or refractory GBM naïve to prior antiangiogenic therapy |
|
| Cediranib (Initial treatment with 45 mg/day, followed by stepwise dose reduction in patients with dose-limiting toxicities) | VEGFR1-3, KIT, PDGFR | Phase II trial for recurrent GBM |
|
| Erlotinib (oral dose of 150 mg/day for patients not on drugs that increase CYP3A4 activity and 300 mg/day for patients on drugs that increase CYP3A4 activity, followed by dose escalation) | Non-small cell lung cancer EGFR | Phase II trial for first relapse GBM |
|
| Pazopanib (oral dose of 800 mg/day) | Renal cell carcinoma, soft tissue sarcoma VEGFR, PDGFR, KIT | Phase II trial for recurrent GBM |
|
| Cediranib (oral dose of 30 mg/day) monotherapy and cediranib (oral dose of 20 mg/day) combination with lomustine (oral dose of 110 mg/m2 once every 6 weeks) versus lomustine alone | VEGFR1-3, KIT, PDGFR | Phase III trial for recurrent GBM |
|
| Dacomitinib (oral dose of 45 mg/day) | Non-small cell lung cancer EGFR, HER2 | Phase II trial for recurrent GBM and EGFR amplification with or without variant III (EGFRvIII) deletion |
|
| Afatinib (initiated at 20 mg/day and escalated to 40 and 50 mg/day) with or without temozolomide (75 mg/m2 for 21 days every 28-day cycle) vs. temozolomide monotherapy | Non-small cell lung cancer, squamous cell carcinoma of lung EGFR, HER2 | Phase I/II trial for recurrent GBM |
|
| Bevacizumab (5 mg/kg intravenously every 2 weeks) alone and in combination with sorafenib (200 mg twice a day for 1–5 days a week then modified to 200 mg/day because of toxicities) | Renal cell carcinoma, hepatocellular carcinoma, differentiated thyroid cancer VEGFR1-3, TIE2, PDGFR, FGFR, BRAF, CRAF, KIT, FLT-3 | Phase II trial for recurrent GBM |
|
| Axitinib (treatment initiated at oral dose of 5 mg twice daily and adjusted according to toxicity) or bevacizumab or lomustine | Renal cell carcinoma VEGFR1-3, PDGFR, KIT, FLT-3 | Phase II trial for recurrent GBM |
|
| Axitinib (started at oral dose of 5 mg twice daily)Avelumab (10 mg/kg intravenously over 60 min every 2 weeks) | - | Phase II trial for recurrent GBM |
|
| Radiation plus temozolomide with or without vandetanib (100 mg/day 5–7 days prior to radiation) | Unresectable or metastatic medullary thyroid cancer EGFR, VEGFR2-3, RET | Phase II trial for newly diagnosed GBM |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brar, H.K.; Jose, J.; Wu, Z.; Sharma, M. Tyrosine Kinase Inhibitors for Glioblastoma Multiforme: Challenges and Opportunities for Drug Delivery. Pharmaceutics 2023, 15, 59. https://doi.org/10.3390/pharmaceutics15010059
Brar HK, Jose J, Wu Z, Sharma M. Tyrosine Kinase Inhibitors for Glioblastoma Multiforme: Challenges and Opportunities for Drug Delivery. Pharmaceutics. 2023; 15(1):59. https://doi.org/10.3390/pharmaceutics15010059
Chicago/Turabian StyleBrar, Harpinder K., Jiney Jose, Zimei Wu, and Manisha Sharma. 2023. "Tyrosine Kinase Inhibitors for Glioblastoma Multiforme: Challenges and Opportunities for Drug Delivery" Pharmaceutics 15, no. 1: 59. https://doi.org/10.3390/pharmaceutics15010059