
A New Challenge for the Old Excipient Calcium Carbonate: To Improve the Dissolution Rate of Poorly Soluble Drugs
Abstract
:1. Introduction
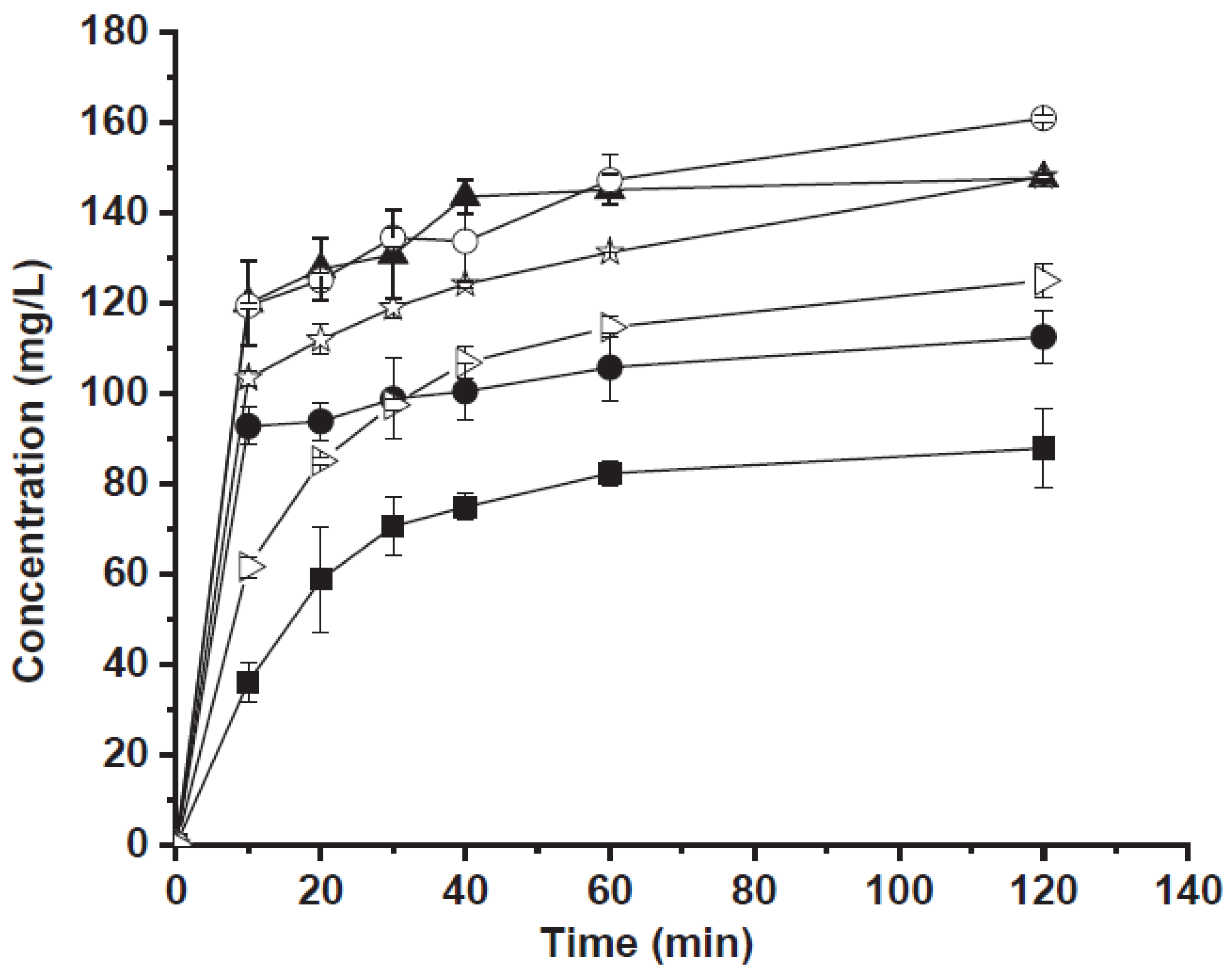
2. CaCO3 as an Excipient for Dissolution Improvement of Poorly Water-Soluble Drugs
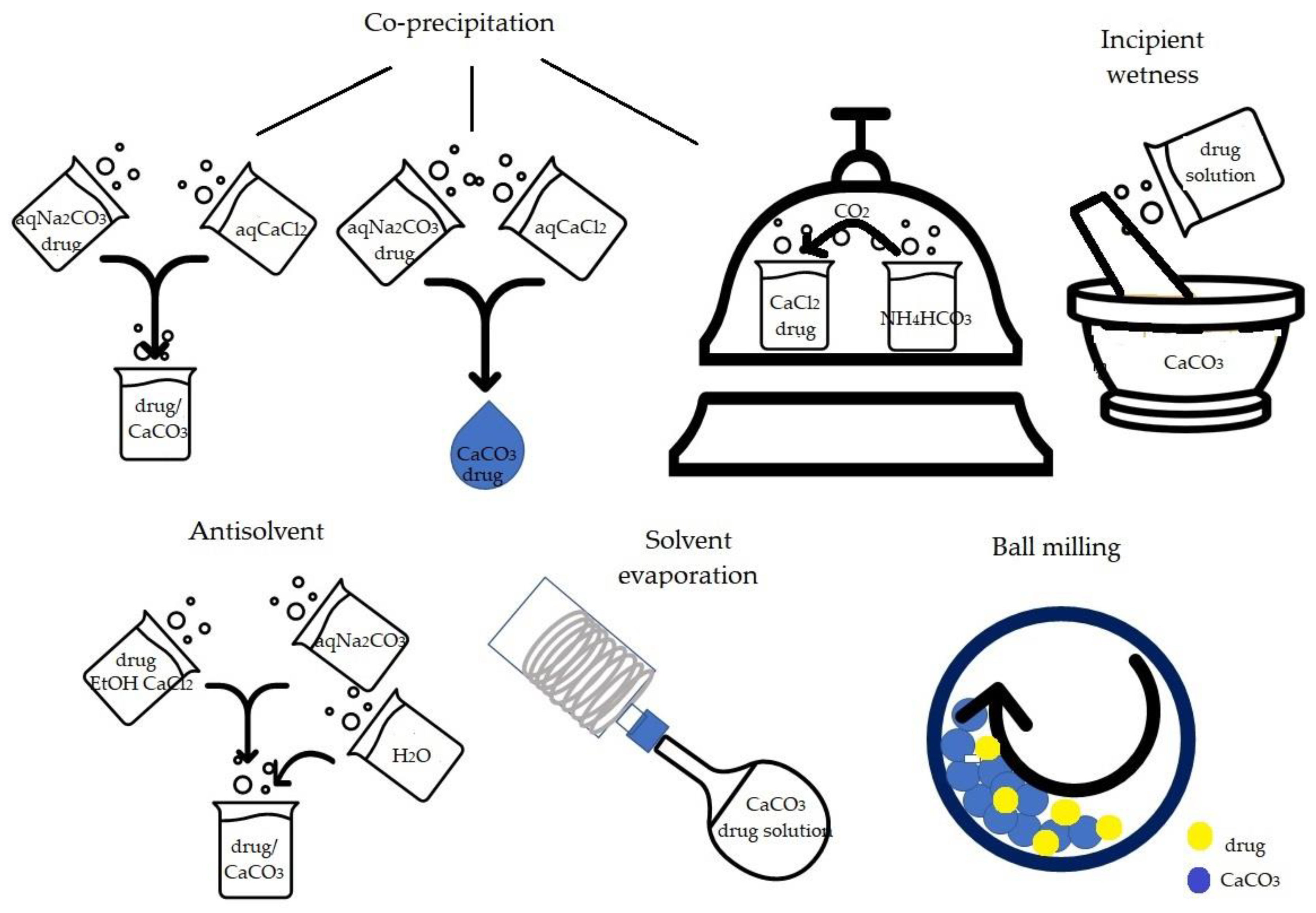
2.1. Ball Milling
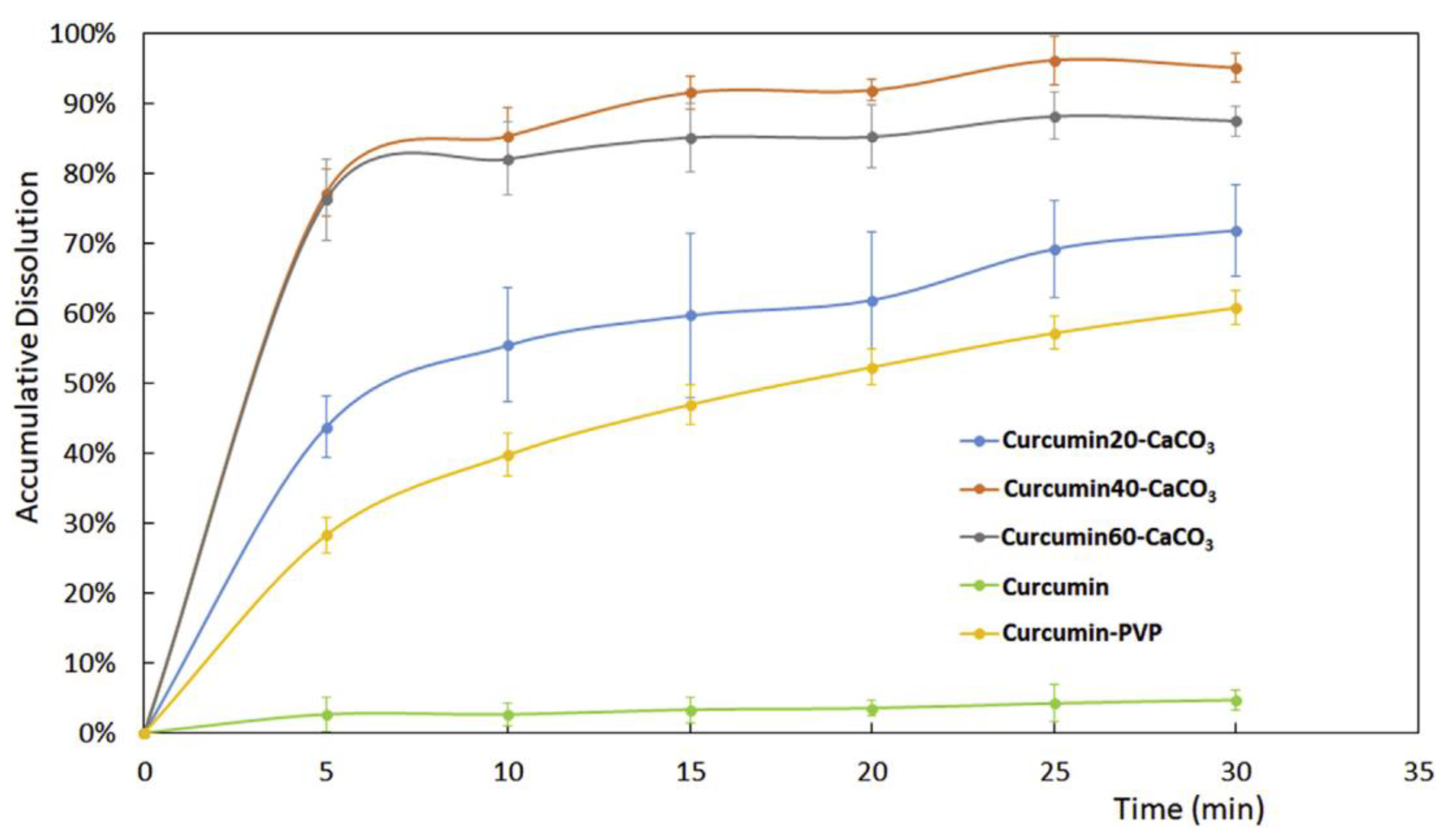
2.2. Co-Precipitation
2.3. Solvent Evaporation Procedure
2.4. Antisolvent Procedure
3. Porous CaCO3
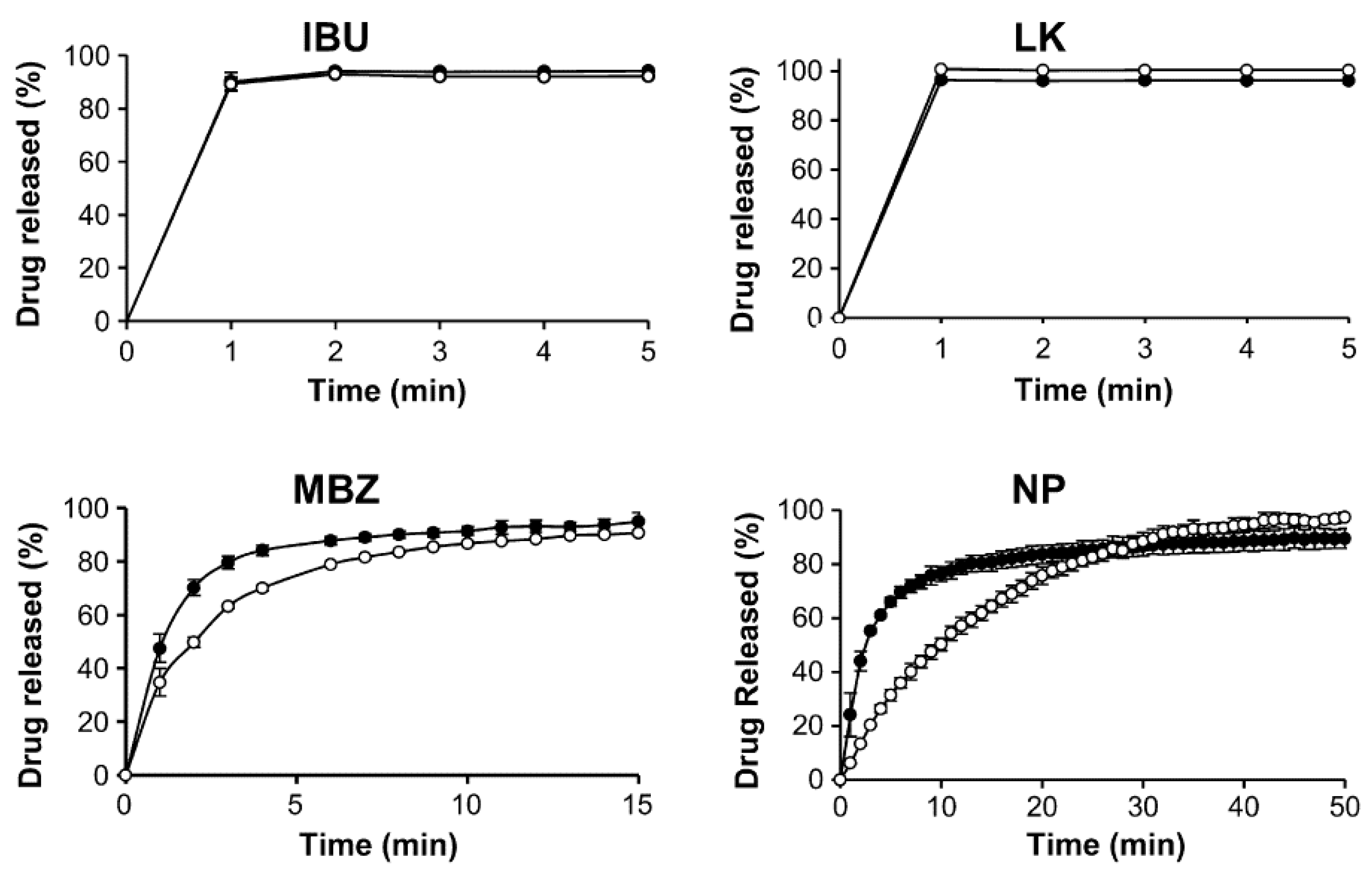
4. Porous Functionalized CaCO3 (FCC)
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R.A. Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horter, D.; Dressman, J.B. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv. Drug Del. Rev. 2001, 46, 75–87. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Chandel, A.K.S. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Controll. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Costantino, U.; Ambrogi, V.; Nocchetti, M.; Perioli, L. Hydrotalcite-like compounds: Versatile layered hosts of molecular anions with biological activity. Micropor. Mesopor. Mater. 2008, 107, 149–160. [Google Scholar] [CrossRef]
- Maleki, A.; Kettiger, H.; Schoubben, A.; Rosenholm, J.M.; Ambrogi, V.; Hamidi, M. Mesoporous silica materials: From physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. J. Controll. Release 2017, 262, 329–347. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef]
- García-Arieta, A. Interactions between active pharmaceutical ingredients and excipients affecting bioavailability: Impact on bioequivalence. Eur. J. Pharm. Sci. 2014, 65, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press: London, UK; American Pharmacists Association: Wanshington, DC, USA, 2009; pp. 506–509. [Google Scholar]
- Fausett, H.; Gayser, C.; Dash, A.K. Evaluation of Quick Disintegrating Calcium Carbonate Tablets. AAPS PharmSciTech 2000, 1, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razali, S.; Wong, T.W. Design of superdisintegrant- and effervescent agent-less dispersible fast-release melt pellets. Powder Technol. 2013, 235, 289–298. [Google Scholar] [CrossRef]
- Addadi, L.; Raz, S.; Weiner, S. Taking advantage of disorder: Amorphous calcium carbonate and its roles in biomineralization. Adv. Mat. 2003, 15, 959–970. [Google Scholar] [CrossRef]
- Tan, C.; Dima, C.; Huang, M.; Assadpour, E.; Wang, J.; Sun, B.; Kharazmi, M.S.; Jafari, S.M. Advanced CaCO3-derived delivery systems for bioactive compounds. Adv. Colloid Interface Sci. 2022, 309, 102791. [Google Scholar] [CrossRef] [PubMed]
- Trofimov, A.D.; Ivanova, A.A.; Zyuzin, M.V.; Timin, A.S. Porous Inorganic Carriers Based on Silica, Calcium Carbonate and Calcium Phosphate for Controlled/Modulated Drug Delivery: Fresh Outlook and Future Perspectives. Pharmaceutics 2018, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Cao, L.; Parakhonskiy, B.V.; Skirtach, A.G. Hard, Soft, and Hard-and-Soft Drug Delivery Carriers Based on CaCO3 and Alginate Biomaterials: Synthesis, Properties, Pharmaceutical Applications. Pharmaceutics 2022, 14, 909. [Google Scholar] [CrossRef]
- Jimoh, O.A.; Ariffin, K.S.; Hussin, H.B.; Temitope, A.E. Synthesis of precipitated calcium carbonate: A review. Carbonates Evaporites 2018, 33, 331–346. [Google Scholar] [CrossRef]
- Niu, Y.Q.; Liu, J.H.; Aymonier, C.; Fermani, S.; Kralj, D.; Falini, G.; Zhou, C.H. Calcium carbonate: Controlled synthesis, surface functionalization, and nanostructured materials. Chem. Soc. Rev. 2022, 51, 7883–7943. [Google Scholar] [CrossRef]
- Muhammad Mailafiya, M.; Abubakar, K.; Danmaigoro, A.; Musa Chiroma, S.; Bin Abdul Rahim, E.; Aris Mohd Moklas, M.; Abu Bakar Zakaria, Z. Cockle Shell-Derived Calcium Carbonate (Aragonite) Nanoparticles: A Dynamite to Nanomedicine. Appl. Sci. 2019, 9, 2897. [Google Scholar] [CrossRef]
- Trushina, D.B.; Borodina, T.N.; Belyakov, S.; Antipina, M.N. Calcium carbonate vaterite particles for drug delivery: Advances and challenges. Mater. Today Adv. 2022, 14, 100214. [Google Scholar] [CrossRef]
- Fu, J.; Leo, C.P.; Show, P.L. Recent advances in the synthesis and applications of pH-responsive CaCO3. Biochem. Eng. J. 2022, 187, 108446. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Vikulina, A.S.; Volodkin, D. CaCO3 crystals as versatile carriers for controlled delivery of antimicrobials. J. Controll. Rel. 2020, 328, 470–489. [Google Scholar] [CrossRef]
- Basria, R.S.; Mydin, M.N.; Zahidi, I.N.M.; Ishak, N.N.; Ghazali, N.S.S.N.; Moshawih, S.; Siddiquee, S. Potential of Calcium Carbonate Nanoparticles for Therapeutic Applications. Mal. J. Med. Health Sci. 2018, 14, 201–206. [Google Scholar]
- Fadia, P.; Tyagi, ·S.; Bhagat, S.; Nair, A.; Panchal, P.; Dave, H.; Dang, S.; Singh, S. Calcium carbonate nano- and microparticles: Synthesis methods and biological applications. 3 Biotech 2021, 11, 457. [Google Scholar] [CrossRef]
- Boldyrev, V.V.; Shakhtshneider, T.P.; Chizhik, S.A. On the mechanism of solubilization of drugs in the presence of poorly soluble additives. Int. J. Pharm. 2005, 295, 177–182. [Google Scholar] [CrossRef]
- Maver, U.; Bele, M.; Jamnik, J.; Gaberšček, M.; Planinšek, O. A fast and simple method for preparation of calcium carbonate–drug composites or fast drug release. Mater. Res. Bull. 2013, 48, 137–145. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, W.; Hu, H.; Ni, X.; Ni, S.; Xu, Y.; Yang, l.; Xu, D. Co-precipitation of calcium carbonate and curcumin in an ethanol medium as a novel approach for curcumin dissolution enhancement. J. Drug Del. Sci. Tech. 2019, 51, 397–402. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Carazo, E.; Albertini, B.; Passerini, N.; Perissutti, B.; Cerezo, P.; Viseras, C.; Hernández-Laguna, A.; Aguzzi, C.; Sainz-Díaz, C.I. Conformational polymorphic changes in the crystal structure of the chiral antiparasitic drug praziquantel and interactions with calcium carbonate. Eur. J. Pharm. Biopharm. 2018, 132, 180–191. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Sánchez-Espejo, R.; Albertini, B.; Passerini, N.; Cerezo, P.; Viseras, C.; Sainz-Díaz, C.I. Ground Calcium Carbonate as a Low Cost and Biosafety Excipient for Solubility and Dissolution Improvement of Praziquantel. Pharmaceutics 2019, 11, 533. [Google Scholar] [CrossRef] [Green Version]
- Di Marzio, L.; Borrego- Sánchez, A.; Felaco, M.; Pacinelli, M.E.; Gómez-Morales, J.; d’Avanzo, N.; Sainz-Díaz, C.I.; Celia, C.; Viseras, C. Praziquantel-loaded calcite crystals: Synthesis, physicochemical characterization, and biopharmaceutical properties of inorganic biomaterials for drug delivery. J. Drug Del. Sci. Tech. 2022, 68, 103021. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, J.; Fu, M.; Zeng, J.; Omari-Siaw, E.; Yu, J.; Xu, X.M. Calcium Carbonate Nanoparticles Templated by Mixed Polymeric Micelles: Characterization, In Vitro Drug Release and Oral Bioavailability in Beagle Dogs. Lat. Am. J. Pharm. 2014, 33, 1106–1113. [Google Scholar]
- Donnadio, A.; Corneli, C.; Ricci, P.; Bini, M.; Ambrogi, V. Use of calcium carbonate as an excipient for release of poorly water soluble drugs: The case of carbamazepine. Int. J. Pharm. 2020, 589, 119860. [Google Scholar] [CrossRef] [PubMed]
- Forsgren, J.; Andersson, M.; Nilsson, P.; Mihranyan, A. Mesoporous Calcium Carbonate as a Phase Stabilizer of Amorphous Celecoxib—An Approach to Increase the Bioavailability of Poorly Soluble Pharmaceutical Substances. Adv. Healthc. Mater. 2013, 2, 1469–1476. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, P.; Bajnóczi, É.G.; Neagu, A.; Tai, C.-W.; Persson, I.; Strømme, M.; Cheung, O. Amorphous Calcium Carbonate Constructed from Nanoparticle Aggregates with Unprecedented Surface Area and Mesoporosity. ACS Appl. Mater. Interfaces 2018, 10, 21556–21564. [Google Scholar] [CrossRef]
- Preisig, D.; Haid, D.; Varum, F.J.O.; Bravo, R.; Alles, R.; Huwyler, J.; Puchkov, M. Drug loading into porous calcium carbonate microparticles by solvent evaporation. Eur. J. Pharm. Biopharm. 2014, 87, 548–558. [Google Scholar] [CrossRef]
- Johnson, M.L.; Noreland, D.; Gane, P.; Schoelkopf, J.; Ridgway, C.; Fureby, A.M. Porous calcium carbonate as a carrier material to increase the dissolution rate of poorly soluble flavouring compounds. Food Funct. 2017, 8, 1627–1640. [Google Scholar] [CrossRef]
- Liu, J.; Rades, T.; Tho, I.; Kissi, E.O. Functionalised calcium carbonate as a coformer to stabilize amorphous drugs by mechanochemical activation, Eur. J. Pharm. Biopharm. 2020, 155, 22–28. [Google Scholar] [CrossRef]
- Kim, D.; Kim, Y.; Tin, Y.Y.; Soe, M.T.P.; Ko, B.; Park, S. Recent Technologies for Amorphization of Poorly Water-Soluble Drugs. Pharmaceutics 2021, 13, 1318. [Google Scholar] [CrossRef]
- Ambrogi, V.; Perioli, L.; Marmottini, F.; Accorsi, O.; Pagano, C.; Ricci, M.; Rossi, C. Role of mesoporous silicates on carbamazepine dissolution rate enhancement. Microporous Mesoporous Mater. 2008, 113, 445–452. [Google Scholar] [CrossRef]
- Sliwinska-Bartkowiak, M.; Dudziak, G.; Gras, R.; Sikorski, R.; Radhakrishnan, R.; Gubbins, K.E. Freezing behavior in porous glasses and MCM-41. Colloid. Surf. A 2001, 523, 187–188. [Google Scholar] [CrossRef]
- Stirnimann, T.; Maiuta, N.D.; Gerard, D.E.; Alles, R.; Huwyler, J.; Puchkov, M. Functionalized calcium carbonate as a novel pharmaceutical excipient for the preparation of orally dispersible tablets. Pharm. Res. 2013, 30, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, C.J.; Gane, P.A.; Schoelkopf, J. Modified calcium carbonate coatings with rapid absorption and extensive liquid uptake capacity. Colloids Surf., Physicochem. Eng. Asp. 2004, 236, 91–102. [Google Scholar] [CrossRef]
- Levy, C.L.; Matthews, G.P.; Laudone, G.M.; Gribble, C.M.; Turner, A.; Ridgway, C.J.; Gerard, D.E.; Schoelkopf, J.; Gane, P.A.C. Diffusion and Tortuosity in Porous Functionalized Calcium Carbonate. Ind. Eng. Chem. Res. 2015, 54, 9938–9947. [Google Scholar] [CrossRef]
- Stirnimann, T.; Atria, S.; Schoelkopf, J.; Gane, P.A.C.; Alles, R.; Huwyler, J.; Puchkov, M. Compaction of functionalized calcium carbonate, a porous and crystalline microparticulate material with a lamellar surface. Int. J. Pharm. 2014, 466, 266–275. [Google Scholar] [CrossRef]
- Eberle, V.A.; Schoelkopf, J.; Gane, P.A.C.; Alles, R.; Huwyler, J.; Puchkov, M. Floating gastroretentive drug delivery systems: Comparison of experimental and simulated dissolution profiles and floatation behavior. Eur. J. Pharm. Sci. 2014, 58, 34–43. [Google Scholar] [CrossRef]
- Preisig, D.; Roth, R.; Tognola, S.; Varum, F.J.O.; Bravo, R.; Cetinkaya, Y.; Huwyler, J.; Puchkov, M. Mucoadhesive microparticles for local treatment of gastrointestinal diseases. Eur. J. Pharm. Biopharm. 2016, 105, 156–165. [Google Scholar] [CrossRef]
- Roth, R.; Schoelkopf, J.; Huwyler, J.; Puchkov, M. Functionalized calcium carbonate microparticles for the delivery of proteins. Eur. J. Pharm. Biopharm. 2018, 122, 96–103. [Google Scholar] [CrossRef]
- Available online: https://www.pharmaexcipients.com/wp-content/uploads/2020/11/COG-FLY-Omyapharm-lowres-A4_EN.pdf (accessed on 13 January 2023).
- Trushina, D.B.; Bukreeva, T.V.; Kovalchuk, M.V.; Antipina, M.N. CaCO3 vaterite microparticles for biomedical and personal care applications. Mat. Sci. Eng. C 2014, 45, 644–658. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | CaCO3 Form | Procedure | Drug Loading (% w/w a Unless Differently Indicated) | Drug/CaCO3 Physical Solid State | Ref. b |
|---|---|---|---|---|---|
| Sulfathiazole | calcite | Ball milling | 72 (2:1 M/M) | Mechano-composite sulfathiazole-CaCO3 (no physico- chemical characterization) | [27] |
| Naproxen | calcite and vaterite forms | Co-precipitation c | 3.4 | - | [28] |
| Curcumin | calcite | Co-precipitation | 15.8–42.6 | Amorphous form At higher drug loadings small cuboid crystals | [29] |
| Praziquantel | calcite | Solvent evaporation | 16.67 | Polymorphic forms | [30,31,32] |
| Silybin | NanoCaCO3 d (calcite) | (1) Preparation of CaCO3 nanoparticles templated by polymeric micelles (2) Silybin adsorption | 50 | - | [33] |
| Carbamazepine | Calcite | Antisolvent (Ethanol/water) Ball milling | 10–70 | polymorphic form polymorphic form | [34] |
| Celecoxib | Vaterite mesoporous CaCO3 amorphous mesoporous CaCO3 | One pot with CaCO3 preparation (Methanol) Solvent evaporation (Ethanol) | 48–25 (V/V) 14.49 | Amorphous form Amorphous form | [35] [36] |
| Itraconazole | Amorphous mesoporous | Solvent evaporation Dichloromethane | 25.58 | Amorphous form | [36] |
| Metronidazole benzoate | Porous FCC e | Solvent evaporation (Acetone) | 25–50 | Amorphous form | [37] |
| Ibuprofen | Porous FCC e | Solvent evaporation (Acetone) | 25–50 | Amorphous form | [37] |
| Losartan potassium f | Porous FCC | Solvent evaporation (Methanol) | 25–50 | Amorphous form | [37] |
| Nifedipine | Porous FCC e | Solvent evaporation (Acetone) | 25–50 | Amorphous form | [37] |
| L-carvone | Porous FCC e | Incipient wetness (L-carvone oil without solvent) | 1.3–35 | - | [38] |
| Vanillin f | Porous FCC e | Incipient wetness (Acetone) Melting | 1.3–35 | Amorphous/crystalline depending on the drug loading and FCCs | [38] |
| Curcumin | Porous FCC e | Incipient wetness (Acetone) Melting | 1.3–35 | Amorphous form | [38] |
| Carvedilol | Porous FCC e | Ball milling | 10–90 | Amorphous form | [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrogi, V. A New Challenge for the Old Excipient Calcium Carbonate: To Improve the Dissolution Rate of Poorly Soluble Drugs. Pharmaceutics 2023, 15, 300. https://doi.org/10.3390/pharmaceutics15010300
Ambrogi V. A New Challenge for the Old Excipient Calcium Carbonate: To Improve the Dissolution Rate of Poorly Soluble Drugs. Pharmaceutics. 2023; 15(1):300. https://doi.org/10.3390/pharmaceutics15010300
Chicago/Turabian StyleAmbrogi, Valeria. 2023. "A New Challenge for the Old Excipient Calcium Carbonate: To Improve the Dissolution Rate of Poorly Soluble Drugs" Pharmaceutics 15, no. 1: 300. https://doi.org/10.3390/pharmaceutics15010300